")
Back to Journals » International Journal of Nanomedicine » Volume 20
Preparation, and ex vivo and in vivo Characterization of Favipiravir-Loaded Aspasomes and Niosomes for Nose-to-Brain Administration
Authors Salamah M, Volk B, Lekli I, Bak I, Gyöngyösi A, Kozma G , Kónya Z , Szalenkó-Tőkés Á, Kiricsi Á, Rovó L, Balogh-Weiser D, Zupkó I, Csóka I, Katona G , Balogh GT
Received 13 February 2025
Accepted for publication 5 May 2025
Published 22 May 2025 Volume 2025:20 Pages 6489—6514
DOI https://doi.org/10.2147/IJN.S518486
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Maryana Salamah,1,2 Balázs Volk,3 István Lekli,4 István Bak,4 Alexandra Gyöngyösi,4 Gábor Kozma,5 Zoltán Kónya,5 Ágnes Szalenkó-Tőkés,6 Ágnes Kiricsi,6 László Rovó,6 Diána Balogh-Weiser,7,8 István Zupkó,2 Ildikó Csóka,1 Gábor Katona,1,* György Tibor Balogh9,10,*
1Institute of Pharmaceutical Technology and Regulatory Affairs, Faculty of Pharmacy, University of Szeged, Szeged, Hungary; 2Institute of Pharmacodynamics and Biopharmacy, Faculty of Pharmacy, University of Szeged, Szeged, Hungary; 3Directorate of Drug Substance Development, Egis Pharmaceuticals Plc., Budapest, Hungary; 4Department of Pharmacology, Faculty of Pharmacy, University of Debrecen, Debrecen, Hungary; 5Faculty of Science and Informatics, Department of Applied & Environmental Chemistry, University of Szeged, Szeged, Hungary; 6Department of Oto-Rhino-Laryngology and Head-Neck Surgery, University of Szeged, Szeged, Hungary; 7Department of Organic Chemistry and Technology, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Budapest, Hungary; 8Department of Physical Chemistry and Materials Science, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Budapest, Hungary; 9Department of Pharmaceutical Chemistry, Semmelweis University, Budapest, Hungary; 10Center for Pharmacology and Drug Research & Development, Department of Pharmaceutical Chemistry, Semmelweis University, Budapest, Hungary
*These authors contributed equally to this work
Correspondence: Gábor Katona, Institute of Pharmaceutical Technology and Regulatory Affairs, Faculty of Pharmacy, University of Szeged, Eötvös Str. 6, Szeged, H-6720, Hungary, Email [email protected] György Tibor Balogh, Department of Pharmaceutical Chemistry, Semmelweis University, Hőgyes Endre Str. 9, Budapest, H-1092, Hungary, Email [email protected]
Purpose: The present study aimed to develop and compare the intranasal applicability of favipiravir-loaded aspasomes (FAV-ASPs) using film hydration method, and favipiravir-loaded niosomes (FAV-NIOs) using ethanol injection method.
Methods: The FAV-ASP and FAV-NIO formulations were characterized according to nanoparticulate characteristics (DLS, drug loading, drug encapsulation efficacy, droplet size distribution), drug release and permeability behavior.
Results: The optimized FAV-ASP formulation (FAV-ASP8) consisted of FAV, ascorbyl palmitate, Span® 60 and cholesterol (30:25:25:50 w/w) with nano-scale size range (292.06 ± 2.10 nm), narrow polydispersity index (PDI) value (0.36 ± 0.03), adequate zeta potential (− 74.73 ± 3.28 mV) and acceptable encapsulation efficiency (55.33 ± 0.41%). The optimized FAV-NIO formulation (FAV-NIO9) contained FAV, Span® 60 and cholesterol (30:30:40 w/w) with nano-scale size range (167.13 ± 1.60 nm), narrow PDI value (0.07 ± 0.01), adequate zeta potential (− 27.1 ± 1.24 mV) and acceptable encapsulation efficiency (51.30 ± 0.69%). FAV-ASP8 and FAV-NIO9 were suitable for spraying into the nasal cavity (droplet size distribution < 200 μm). In vitro drug release and permeability studies demonstrated enhanced solubility and increased blood–brain barrier (BBB) permeability of FAV formulations, respectively. The ex vivo human nasal permeability study revealed that FAV diffusion from FAV-ASP8 was higher than from FAV-NIO9 or initial FAV. Furthermore, the in vivo animal study showed that FAV-ASP8 had a higher BBB penetration compared to FAV-NIO9 and pure FAV. The in vitro–in vivo correlation study showed good correlation between the in vitro and the in vivo pharmacokinetic data.
Conclusion: FAV-ASP8 for nose-to-brain delivery system could be a promising formulation to improve FAV bioavailability compared to FAV-NIO9.
Keywords: aspasomes, niosomes, favipiravir, nose-to-brain delivery, ex vivo nasal permeability, in vivo nasal permeability
Graphical Abstract:

Introduction
Neurotropic viruses including RNA viruses, such as poliovirus, Zika virus, influenza A and B, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), mumps virus, and measles virus, are able to access the brain and infect the central nervous system (CNS), resulting in meningitis, myelitis, encephalitis, or meningoencephalitis.1–3 Neurotropic viruses can access CNS by crossing the blood–brain barrier (BBB) or blood-cerebrospinal fluid barrier (BCSFB), nerve terminals in the nasal olfactory epithelium, or the pseudounipolar sensory neurons of the peripheral nervous system (PNS).3–5 This viral infection of the CNS leads to alteration or degeneration of neuronal cell function, which causes several neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis.5–7 Therefore, it has become urgent to search for potential drugs for the treatment of viral CNS infections. Considering these facts, intranasal delivery can be a proper choice to deliver the drug directly to the brain via the systemic pathway, olfactory and trigeminal nerve pathways.8–10 Several possible candidates exist for the intranasal delivery of virucidal drugs and agents, such as favipiravir (FAV).11,12
FAV (FAV, 6-fluoro-3-hydroxypyrazine-2-carboxamide, Figure 1), as an antiviral prodrug, was approved in Japan in 2014 for the treatment of new-onset or recurrent pandemic influenza as well as for experimental drug for Ebola virus infections. FAV is first converted by hypoxanthine-guanine phosphoribosyl transferase (HGPRT) to ribose 5′-monophosphate (T-705-RMP) and then metabolized to the triphosphate form (T-705RTP) by cellular kinases.13,14 The compound selectively inhibits viral RNA polymerase in vivo by its triphosphorylated metabolite, translating to broad-spectrum inhibition of RNA viruses.15,16
 |
Figure 1 Chemical structure of FAV and its metabolic activation.14 |
 |
Figure 2 EE% and DL% results of FAV-ASP8, FAV-ASP10 and FAV-NIO9. Results are expressed as means ± SD (n = 3), ***p-value < 0.001, ****p-value < 0.0001. |
 |
Figure 3 BBB-PAMPA results for FAV-ASP8, FAV-ASP10 and FAV-NIO9 in comparison with pure FAV. (A): permeability results, (B): flux results. Results are expressed as means ± SD (n = 6), **p-value < 0.01, and ***p-value < 0.001, ****p-value <0.0001. |
 |
Figure 4 Rapid equilibrium dialysis (RED) of the optimal formulations in comparison with pure FAV. Results are expressed as means ± SD (n = 6). |
 |
Figure 5 Cumulative in vitro release profile of the optimal formulations compared to pure FAV. Results are expressed as means ± SD (n = 3). |
 |
Figure 6 Ex vivo permeability study of FAV-ASP8 and FAV-NIO9 in comparison with pure FAV. Results are expressed as means ± SD (n = 3). |
 |
Figure 7 Plasma concentration–time profile after nasal administration of FAV-ASP8, FAV-NIO9 and pure FAV in rats. Results are expressed as means ± SD (n = 4), (ns) means non-significant. |
 |
Figure 8 The concentration of FAV in cerebrospinal fluid (CSF) after 1 hour of nasal administration. Results are expressed as means ± SD (n = 4), (ns) means non-significant, *p-value < 0.05, ***p-value < 0.001. |
 |
Figure 9 IVIVC graphs for the comparison of AUC 0-t values between the in vitro release and the in vivo PK data. Where (A): IVIVC for pure FAV, (B): IVIVC for FAV-ASP8, and (C): IVIVC for FAV-NIO9. |
 |
Figure 10 IVIVC graphs for the comparison of AUC 0-t values between the in vitro permeation and the in vivo PK data. (A): IVIVC for pure FAV, (B): IVIVC for FAV-ASP8, and (C): IVIVC for FAV-NIO9. |
FAV is available in an oral solid dosage form (Avigan® 200 mg),17 with a recommended dosage of 1600 mg twice daily on day 1, followed by 600 mg twice daily on days 2 to 5 for the treatment of influenza infection,18 which leads to patient incompliance, and results in toxicity and limited oral bioavailability (due to its low water solubility, FAV is a Biopharmaceutics Classification System (BCS) class II drug).19 FAV has a short half-life, leading to rapid renal clearance in its hydroxylated form (inactive metabolite T-705M1) after being metabolized by aldehyde oxidase/xanthine oxidase enzymes (present in the liver, lungs, small and large intestine, kidney, prostate, and adrenal glands).20 Thus, a novel formulation and alternative route of administration are critical to improve bioavailability and therapeutic efficacy while minimizing the adverse effects of FAV (that include mild to moderate diarrhoea, increase of blood uric acid and transaminase level, and decrease in the neutrophil counts).21,22
Intranasal delivery of FAV means further advantages, including improved systemic bioavailability due to increased drug solubility, absorption and nasal permeability through a highly vascularized and less acidic environment, avoiding hepatic first-pass metabolism in comparison to the gastrointestinal tract.23,24 FAV shows low brain penetrance because it has a low log P value (0.72), the critical factor for CNS penetration, and low lipophilicity, which decreases brain uptake.25 Therefore, intranasal delivery can be utilized to deliver FAV directly to the brain, where it could be activated by the HGPRT enzyme within the infected cells by neurotropic viruses, as HGPRT is also be found in the CNS. Moreover, the administration of lower doses may be therapeutically effective with fewer systemic side effects.
Lipid nanoparticles, including liposomes and niosomes, are considered a suitable antiviral drug carrier to enhance the BBB penetrating, protect the drug against enzymatic degradation, and improve its bioavailability, hence, prevent viral spread.26–28 Niosomes (NIOs), a newer generation of liposomes, rise from the self-assembly of non-ionic surfactants of the alkyl or dialkyl polyglycerol ether class and cholesterol (CH) with subsequent hydration in aqueous media.29,30 Recently, amphiphilic materials, such as ascorbyl palmitate (AP), have been used to prepare bilayer vesicles called aspasomes (ascorbyl palmitate-based nanocarriers; ASPs), a newer generation of NIOs, to enhance the stability and biological activity of NIOs.31 ASPs contain AP in combination with CH and an anionic or nonionic surfactant in various molar ratios. AP is an ester form of ascorbic acid, which is amphiphilic by nature and more stable in liquid form than ascorbic acid.31–35
These colloidal vesicles have several advantages, like the development of an effective drug delivery system to achieve a maximum effective concentration and the formation of vesicles that are able to entrap both hydrophilic and hydrophobic drug molecules with higher stability compared to liposomes. Moreover, modification of the nanoparticulate composition or surface can adjust the affinity for the target site and/or the drug release rate.
According to our knowledge, several FAV-nanocarriers for intranasal administration have been developed such as favipiravir-pyridinecarboxamide cocrystal nasal powder formulation,36 crystalline favipiravir Sodium Salt liquid formulation,37 favipiravir loaded PLGA nanoparticles,38 and favipiravir loaded mucoadhesive chitosan–alginate nanoparticles.39 Moreover, ASPs have been used for transdermal delivery, such as Acyclovir loaded aspasomal gel,40 Quercetin loaded aspasomal gel,41 Idebenone/Naproxen co-loaded aspasomes,42 and based on the literature, there are no available reported ASP formulations for intranasal administration. Therefore, FAV-loaded aspasome formulations (FAV-ASPs) and FAV-loaded niosome formulations (FAV-NIOs) seem to be a novel approach.
In this study, our aim was to develop and compare novel FAV-ASPs and FAV-NIOs intended for intranasal administration in order to improve the poor solubility of FAV, as a potential treatment for viral infections of CNS. A further aim was the characterization of nasal applicability of optimized FAV-ASP and FAV-NIO formulations.
Materials and Methods
Chemicals and Solvents
FAV was provided by Egis Pharmaceuticals Plc. (Budapest, Hungary) with a purity of 99.6% w/w (according to supplier certificate of analysis). Methanol 99.99% v/v (HPLC grade), ortho-phosphoric acid 85% v/v (HPLC grade) and anhydrous disodium hydrogen phosphate were purchased from Molar Chemicals Kft. (Budapest, Hungary). Acetonitrile 99.8% v/v (HPLC grade) was purchased from PromoChem (Wesel, Germany). Ascorbyl acid-6-palmitate (AP), sorbitan monostearate (Span® 60), chloroform, ethanol 96% v/v, dimethyl sulfoxide (DMSO) and dodecane were purchased from Merck KGaA (Darmstadt, Germany). Porcine brain polar lipid extract, dicetyl phosphate (DCP) and cholesterol (CH) were purchased from Sigma Aldrich Co. Ltd. (Budapest, Hungary). For redispersion of formulations, pH 7.4 Dulbecco’s phosphate-buffered saline (DPBS) acquired from Capricorn Scientific GmbH (Ebsdorfergrund, Germany) was applied. As nasal dissolution medium, Simulated Nasal Electrolyte Solution (SNES) was freshly prepared, containing 8.77 g sodium chloride (NaCl), 2.98 g potassium chloride (KCl), and 0.59 g anhydrous calcium chloride (CaCl2) in 1000 mL of deionized water at pH 5.6. These chemicals were acquired from Sigma-Aldrich Co. Ltd. (Budapest, Hungary). In all experiments, purified water was filtered using the Millipore Milli-Q® (Merck Ltd., Budapest, Hungary) Gradient Water Purification System.
Quantitative Analysis by RP-HPLC-DAD
FAV concentration in the experiments was analyzed using an Agilent 1260 hPLC (Agilent Technologies, San Diego, CA, USA) and Zorbax® SB-CN C18 column (5 μm, 250 mm × 4.6 mm, 100 Å). As mobile phase acetonitrile–disodium hydrogen phosphate anhydrous buffer (pH 3.1, 20 mm) in a ratio of 10:90 (v/v) was used, and FAV was eluted at an isocratic flow rate of 1.0 mL/min up to 10 min at 30°C. The injection volume was 10 µL. Chromatograms were detected at 323 nm using a UV-VIS diode-array detector.
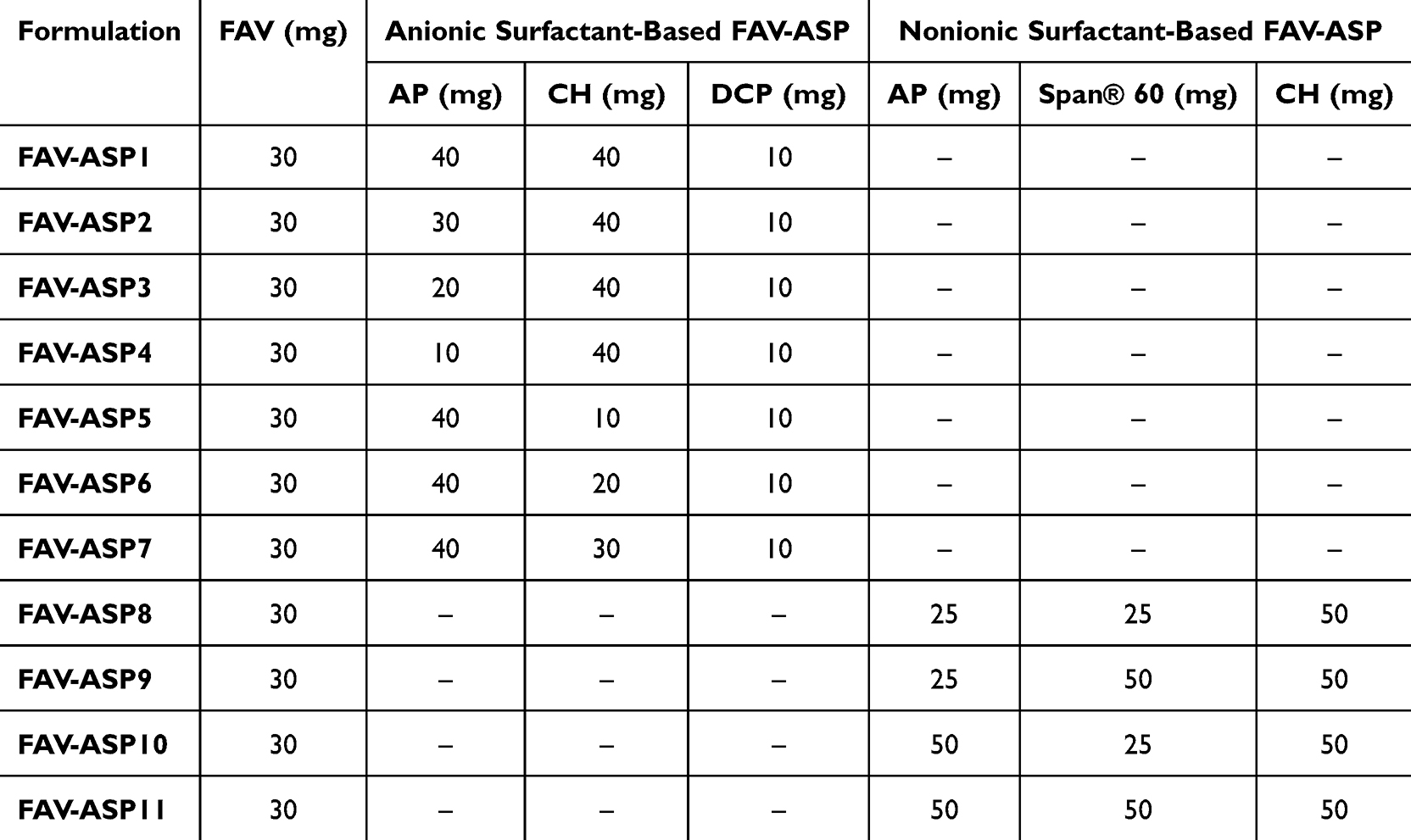 |
Table 1 Composition of FAV-Loaded Aspasome Formulations (FAV-ASP) |
 |
Table 2 Composition of FAV-Loaded Niosome Formulations (FAV-NIO) |
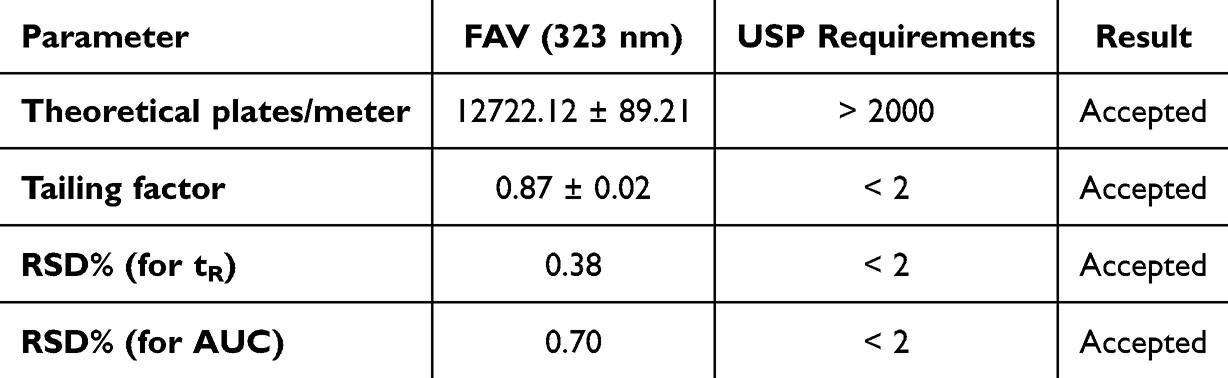 |
Table 3 Results of System Suitability (n = 6) |
Preparation of FAV-ASP Formulations
A modified film hydration method was used for the preparation of FAV-ASPs31,34 (Table 1). For anionic surfactant-based FAV-ASPs, various ratios of lipid mixture (AP:CH:DCP) (w/w) were dissolved in 10 mL of chloroform, whereas for nonionic surfactant-based FAV-ASPs, different ratio of AP/Span® 60 with a constant amount of CH (50 mg) were dissolved in 10 mL of chloroform. A fixed amount of FAV (30 mg) was dissolved in 5 mL of methanol. Then, the FAV solution was mixed with each lipid solution in a round bottom flask, and the organic solvent was evaporated at 60 °C and 633 mbar pressure using a Büchi R-210 rotary vacuum evaporator (Flawil, Switzerland), and the rotation was set at 100 rpm for 1 h, until the appearance of a thin film on the wall of the flask. Thereafter, the thin lipid film was hydrated with 15 mL of pH 7.4 phosphate-buffered saline (PBS) for 1 h.
Preparation of FAV-NIO Formulations
FAV-NIOs were formulated by a modified ethanol injection method.43 Various amounts of Span® 60 and CH (as shown in Table 2) were dissolved in 2 mL ethanol using ultrasonication (for 10 min at 30°C). A fixed amount of FAV (30 mg) was dissolved in the ethanolic solution, followed by sonication until the formation of a clear solution. Then, it was injected rapidly in 15 mL of distilled water (previously heated to 60°C) under constant stirring, followed by probe sonication (0.5 cycles for 2 min at ultrasonic power input 80%). Then, it was stirred for 1 h at room temperature, followed by a complete evaporation of ethanol at 60°C and 633 mbar pressure using a rotary vacuum evaporator, and the rotation was set at 100 rpm for 1 h. To reach the final volume (15 mL) of niosomal dispersion, distilled water was added.
Lyophilization
For freeze-drying a Scanvac, CoolSafe 100–9 Pro type apparatus (LaboGeneApS, Lynge, Denmark) was applied. Formulations were divided in 1.5 mL portions, and 5% w/v of mannitol was added as a cryoprotectant. Freeze-drying was performed at −40°C for 12 h at reduced pressure (0.013 mbar) controlled by Scanlaf CTS16a02 software. After that, a secondary drying at 25°C was applied for 3 hours. Freeze-dried samples were stored in the refrigerator at 4°C until further analysis. The samples were redispersed in adequate medium before each test.
Vesicle Size Analysis, Polydispersity Index and Zeta Potential Determination
The mean hydrodynamic diameter of vesicles (Z-average), polydispersity index (PDI) and zeta potential (ZP) for FAV-ASP and FAV-NIO formulations were investigated using a Malvern NanoZS equipment (Malvern Instrument). Samples were redispersed in distilled water and suitably diluted (1:10) before assessments. All measurements were carried out in triplicate, and results are presented as means ± SD.
Determination of Drug Content, Drug Encapsulation Efficiency (EE%) and Drug Loading (DL%)
For drug content determination 1–1 mL of formulations were diluted with 4 mL of methanol using ultrasonication bath for 10 min. Then, the suspensions were filtered using 0.45 µm syringe filters and analyzed with HPLC to determine FAV concentration. Encapsulation efficiency (EE%), ie the percentage amount of FAV that has been loaded in the vesicles, was determined by using the dialysis technique in order to select the proper formulations with the highest EE% for further studies.44 The formulations were redispersed in distilled water, and then 3 mL of each sample was transferred into a dialysis bag (12 kDa MWCO, Sigma-Aldrich, St. Louis, USA) immersed in 100 mL of purified water and constantly stirred for 30 min. The concentration of free FAV that diffused from the dialysis bag to the purified water was determined by using HPLC, and EE% and DL% were calculated as follows:45


Residual Solvent Determination
To determine the residual organic solvents (ethanol, methanol and chloroform) in the freeze-dried formulations, gas chromatography-mass spectroscopy (GC-MS) measurements were performed using a Shimadzu GCMS-QP2010 SE (Shimadzu Europa GmbH, Duisburg, Germany) with a ZBWax-Plus column (length: 30 m, diameter: 0.25 mm) and helium carrier gas.
In vitro Permeability Measurements
A blood–brain barrier-specific parallel artificial membrane permeability assay (BBB-PAMPA) was used to investigate FAV permeability (cm/s) of initial API solution and in the selected formulations (according to the EE% results) in order to find the optimal one.46 10 mm FAV solution was prepared in dimethyl sulfoxide (DMSO), which was further diluted with DPBS solution (pH 7.4) to obtain reference solution with 100 µM concentration. The donor plate (Multiscreen™-IP, with pore size 0.45 μm; Millipore, Merck Ltd., Budapest, Hungary) was preliminary coated with 5 μL of lipid solution (24 mg porcine brain polar lipid extract dissolved in 840 μL hexane and 360 μL dodecane). Then, the donor plate was inserted to the acceptor plate (Multiscreen Acceptor Plate, Millipore, Merck Ltd., Budapest, Hungary), which contained 300 μL of DPBS solution (pH 7.4). 150–150 μL of reference FAV solution, as well as of the redispersed freeze-dried formulations with DPBS (with 2 mg/mL FAV nominal concentration) were transferred on the lipid membrane of the donor plate. The plates were incubated at 37°C for 4 h (Heidolph Titramax 1000, Heidolph Instruments, Schwabach, Germany). After that the PAMPA plates were separated and the FAV concentrations both in the donor and acceptor chambers were determined using HPLC. The effective permeability and membrane retention of drugs were calculated using the following equation (3):

where Pe means the effective permeability coefficient (cm/s), VA is the volume of the acceptor well (0.3 cm3), A indicates the surface area of one well (0.24 cm2), t is the incubation time (s), τSS is the time to reach the steady state (s), CA(t) is the concentration of the compound in the acceptor phase at time point t (mol/cm3), and S is the solubility of FAV in the donor phase. The flux of the samples was calculated as follows (4):

Six parallel measurements were performed, and data are presented as means ± SD.
Rapid Equilibrium Dialysis Measurement (RED)
RED method (Thermo Scientific™, Waltham, MA, USA) was applied for the determination of the time-dependent drug release profiles of FAV and the selected optimal formulations (according to PAMPA results) after nasal absorption. For the measurement, the reference was prepared by suspending 2 mg of FAV in 1 mL DPBS solution (pH 7.4) using an Eppendorf MixMate (Thermo Scientific™, Waltham, MA, USA) vortex mixer for 30 s. RED inserts (8K MWCO) were fitted into the reusable Teflon base plate, and then 150–150 μL of reference FAV solution, as well as of the lyophilized formulations redispersed in DPBS (with 2 mg/mL FAV nominal concentration) were pipetted into the donor chambers. Then, 300 μL of DPBS was pipetted to the acceptor chambers, and the device was covered with a sealing tape to avoid evaporation and incubated at 37°C for 4 h. Aliquots from the acceptor chambers were withdrawn at predetermined time points and replaced with fresh DPBS.46 FAV concentrations were determined using HPLC. Six parallel measurements were conducted, and data were presented as means ± SD.
In vitro Drug Release Under Nasal Conditions
In vitro drug release study under nasal conditions is an important test during the development of intranasal formulations to determine the absorption through the nasal cavity. Using the modified paddle method (Hanson SR8 Plus, Teledyne Hanson Research, Chatsworth, CA, USA) drug release profile for reference FAV suspension, and the optimal freeze-dried formulations redispersed in SNES (with 2 mg/mL FAV nominal concentration47) were determined at 32°C at 50 rpm paddle rotation speed in 100 mL SNES medium for 60 min. 1 mL of reference suspension and formulations were loaded in pre-treated dialysis bags (Spectra/Por® Dialysis Membrane with 12–14 kDa MWCO, Spectrum Laboratories Inc., Rancho Dominguez, CA, USA) sealed at both ends. Quantification of aliquots was performed by HPLC.
Evaluation of in vitro Drug Release Data
Drug release profiles were compared with model-independent approaches based on calculating the area under the curve (AUC), dissolution efficiency (DE), and mean dissolution time (MDT). In addition, the model-dependent kinetic analysis (zero order, first order, Higuchi, and Korsmeyer-Peppas kinetic profiles) was fit to evaluated. Microsoft Excel DDsolver® add-in software was used for the mathematical evaluation of the release kinetics, and the fitting of each model by comparing the correlation coefficient (R2), rate constant (K), Akaike Information Criterion (AIC), and Model Selection Criterion (MSC).48–51
Storage Stability
The optimal formulations were stored at 4°C and the storage stability study was conducted for 4 weeks and analyzed in terms of Z-average, PDI, ZP to evaluate the physical stability and in terms of drug content to evaluate the chemical stability every week (as described previously).
Droplet Size Distribution Measurement
Laser diffraction method was used to evaluate the droplet size distribution of the optimal formulations.52 The evaluation was performed using a Malvern Spraytec® system (Malvern Instruments Ltd., Malvern, UK), equipped with a 300 mm lens capable of analysing droplet sizes in a range of 0.1–900 μm (Dv50: 0.5–600 μm). The tip of the nasal spray device was aligned and positioned at 45° from the horizontal plane. Measurements were conducted at room temperature. The formulations were redispersed in distilled water and placed in a nasal spray container. Each nasal spray was manually actuated three times and discharged into waste, allowing the device to function optimally. The data were analyzed using the Spraytec® software v4.00 (Malvern Panalytical Ltd., Malvern, UK), with volume diameter and described as 10% (Dv10), 50% (Dv50), and 90% (Dv90) of the cumulative volume distribution. The results were presented as means ± SD.
Ex vivo Nasal Permeability Study on Human Nasal Mucosa
The ex vivo transmucosal permeability of the optimized formulation (based on BBB-PAMPA, nasal dissolution, and RED results) and reference FAV suspension (with 2 mg/mL nominal FAV concentration) was studied in a modified Side-Bi-Side® type horizontal diffusion apparatus under artificial nasal conditions.53 Human nasal mucosa were collected during routine nasal and sinus surgeries (septoplasty, Functional Endoscopic Sinus Surgery (FESS)) under general or local anesthesia. The surgical field was infiltrated with locally administered 1% lidocaine-adrenalin injection, and the mucosa was excised with a raspatorium or Cottle elevator. Excised nasal mucosa was stored in physiological saline until further investigation. All investigations were conducted freshly within 30 min after the removal of the tissue. The experiments have been performed in line with the principles of the Declaration of Helsinki under approval of University of Szeged’s institutional ethics committee (ETT-TUKEB: IV/3880-1/2021/EKU). The participants were briefed on the study procedures, and written informed consent was obtained from all subjects prior to conducting the procedure. Nasal mucosa was cut with a surgical scalpel into uniform segments with a diameter of 6 mm and inserted between donor and acceptor phases to provide an appropriate surface for permeability study.54 To the donor Phase 8 mL of SNES was added, whereas to the acceptor Phase 9 mL of DPBS solution (pH 7.4) was pipetted. The temperature of both chambers was thermostated at 32 ± 0.5°C using a heating circulator (ThermoHaake C 10-P5, Sigma–Aldrich Co. Ltd., Budapest, Hungary). For the measurement, both FAV reference suspension and selected freeze-dried formulation were redispersed in 1 mL SNES (with 2 mg/mL FAV nominal concentration) and added to the donor compartment. Both compartments were continuously stirred at 300 rpm using magnetic stirrers. Aliquots were withdrawn (100 µL) from the acceptor phase at 5, 10, 15, 30, and 60 min and replaced with fresh DPBS. FAV concentration was determined using HPLC. The steady-state flux (Jss), permeability coefficient (Kp), and enhancement ratio (ER) were calculated as follows:55



where Jss is the steady-state flux (µg/cm2/h), mt is the quantity of FAV permeated through the nasal mucosa, A is the permeability surface of nasal mucosa (0.785 cm2), t is the duration of the investigation (h), Kp is the permeability coefficient (cm/h) and Cd is the drug concentration in the donor phase (µg/cm3).
The theoretical steady-state plasma concentration (Css) of the drug can be used for estimation of the concentration of drug that could be reached in the blood after nasal administration, was calculated as follows:56

where Jss is the steady-state flux, A is the surface area of nasal mucosa used for the permeation study (cm2), and Clp is the plasmatic clearance (human Clp value for FAV is 5.11 L/h).57
In vivo Study
Animals and Samples Collection
The animals were kept at room temperature (approximately 23°C) and light was adjusted to alternate darkness and light for 12 hours. The rats were fed with normal rodent chow and tap water ad libitum. All animals received humane care, in compliance with the “Principles of Laboratory Animal Care” according to the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals, formulated by the National Academy of Sciences, and published by the National Institute of Health (NIH Publication No. 86–23, revised 1985). Healthy male Sprague-Dawley rats (339 ± 39 g, mean ± SD) were anaesthetized with an i.p. injection of ketamine (50 mg/kg) and xylazine (10 mg/kg). The femoral vein was cannulated for plasma collection.58 A total of 50 µL FAV (2 µg/µL) or the corresponding formulation of FAV was administered nasally. Animals were divided into three groups (FAV, FAV-ASP8, and FAV-NIO9), and 0.5 mL of blood samples were taken at 0, 5, 10, 15, 30 and 60 min post-FAV administration, then cerebrospinal fluid samples were collected at the end of the experiments. After 45 min, the clot was removed by centrifuging at 1000 × g for 10 min. The resulting supernatant (non-hemolytic serum) was extracted with 2 volumes of acetonitrile. Accordingly, acetonitrile is suitable for precipitating and removing high-abundance proteins from the serum and eliminate the intra-molecular protein interactions. The precipitate was removed by centrifuging at 1000 × g for 10 min. The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of Institutional Animal Care and Use Committee of the University of Debrecen, Debrecen, Hungary. (Approval number: 2/2021/DEMÁB)
Sample Preparation
Real or spiked (standard) plasma or cerebrospinal fluid samples were precipitated with acetonitrile (1:2) and centrifuged at 1000 × g for 10 min. The supernatants were collected, and the samples were dried under a gentle stream of N2 at 60°C using a Turbovap LV concentrator. The samples were re-dissolved in 50 µL pyridine, and then 50 µL N,O-bis(trimethylsilyl)tri- fluoroacetamide (BSTFA) with 1% of trimethylchlorosilane (TMCS) was added into the tubes. The solutions were transferred into 1.5 mL vials sealed, and derivatization was carried out at 80°C for 30 min at 500 rpm using an Eppendorf Thermomixer C device.59 Finally, 1 µL of the reaction mixture was injected into GC-MS equipment.
Gas Chromatograph-Mass Spectrometric (GC-MS) Analysis
Derivatized FAV was measured by GC-MS using a Shimadzu GCMS-QP2010 (Shimadzu, Kyoto, Japan) equipment. The GC was equipped with an SLB-5 MS capillary column (Supelco, Bellefonte, PA, USA) (30 m × 0.25 mm i.d.; 0.25 µm film thickness). Operating conditions were as follows: carrier gas: He, flow rate 32 cm/s; column temperature program: 1 min at 60°C, 60–250°C at 40°C/min and finally 7 min at 250°C. The temperature of the injection port and the interface were 250°C and 270°C, respectively. 1 µL of the samples was injected into the GC-MS using an AOC-20i autosampler; the split ratio was 1:10. The MS was equipped with an electron ionization (EI) ion source, and the operating conditions were as follows: ionization energy 70 eV, ion source temperature 200°C, solvent cut time 4 min. The measurements were carried out in selected ion monitoring (SIM) mode. The registered ions were 301 m/z, 286 m/z and 270 m/z, which were selected based on the EI spectrum measured in SCAN mode.
Evaluation of Pharmacokinetic (PK) Parameters
The PK parameters of pure FAV, FAV-ASP8, and FAV-NIO9 were determined in the plasma and the CSF following a single intranasal dose to evaluate the brain targeting. PK Solver 2.0 software was used to determine pharmacokinetic parameters by non-compartmental analysis.60 The maximum concentrations (Cmax) and times taken to reach these concentrations (Tmax) were determined from the mean concentration-time profiles. The elimination rate constant (ke) was determined by plotting log-linear concentration versus time. The elimination half-life (t1/2) was calculated using equation (9):61

The area under the plasma concentration versus time curve (AUC0-t) was calculated using the linear trapezoidal method from time 0 to time t (the last time point to withdraw blood samples), then the AUC from 0 to infinity, AUC0–∞ (µg hour/mL) was calculated using the following equation:61,62

where Ct (µg/mL) is the last measured concentration at time t.
The clearance (CL), the apparent volume of distribution (Vd), the mean residence time (MRT), and the relative bioavailability (F%) were calculated using the following equations:46,63–66




where AUMC is the first moment of the concentration-time integral.
Evaluation of in vitro-in vivo Correlations (IVIVC)
A point-to-point IVIVC for pure FAV, FAV-ASP8, and FAV-NIO9 was mathematically examined based on the in vitro data (release and permeation) and the in vivo pharmacokinetics data (based on FDA guidance67–69). The relationship between in vitro values of AUC0-t (µg×min/mL) and in vivo values of AUC0-t (µg×min/mL) were evaluated using linear regression. R2 values have been calculated for each graph.
Statistical Analysis
All experimental results are expressed as the mean ± SD. For statistical analysis, GraphPad Prism version 10.12 software (GraphPad Software, San Diego, CA) was used. A one-way analysis of variance (ANOVA) and Tukey’s post-hoc test were performed to compare the groups. The significance level was set at p-value < 0.05, where (ns) means non-significant, *p-value < 0.05, **p-value < 0.01, and ***p-value < 0.001, ****p-value < 0.0001.
Results and Discussion
Quantitative Analysis by RP-HPLC-DAD
The optimized method showed a good retention time (4.95 ± 0.01 min), symmetrical peak shape (0.87 ± 0.02) and the required number of theoretical plates (12722.12 ± 89.21) as per United States Pharmacopoeia (USP) and International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q2 (R1) guidelines.70,71 The correlation coefficient (R2) was 0.9996, which suggests an excellent correlation and good linearity for the method. System suitability is related to the reproducibility and capability of the method for routine analysis. The optimized method showed good system suitability (Table 3).
Preparation of FAV-ASP and FAV-NIO Formulations
Anionic surfactant-based FAV-ASP consisted of AP, CH and DCP. AP has a hydrophilic–lipophilic balance (HLB) value of about 8.4, which is suitable for vesicle formation.72 However, these AP vesicles are not stable. In contrast, AP may form stable ASPs when combined with CH and the negatively charged lipid DCP.42,73,74 CH is necessary to improve physical stability as it provides rigidity to the bilayer membrane. DCP is used as a negative charge inducer to create vesicles with stronger interaction with the mucosal surface, achieving improved delivery. It also forms electrostatic repulsion between the vesicles, inhibiting aggregations. In addition, the double hydrocarbon chains in DCP results in tighter packing of the bilayer membrane, increasing the encapsulation efficiency in ASP formulations.75,76
Nonionic surfactant-based FAV-ASPs were formed by combining AP, CH and nonionic surfactants (Span® 60). All Span types have the same head group but a different alkyl chain, and increasing the alkyl chain length leads to a higher encapsulation efficiency. In this study, Span® 60 (HLB = 4.7) was used as a vesicle stabilizer due to its long chain length (C18) and high transition temperature (53−55°C).34 The temperature of the rotary evaporator was 60°C, above the gel-to-liquid phase transition temperature of Span® 60.
The NIO vesicles were formed by the combination of CH and Span® 60. The impact of CH and Span® 60 on the formation of the vesicles was described previously. According to the literature, Span® 60 can form vesicles-NIOs only in the presence of a suitable amount of cholesterol.77,78
 |
Table 4 The Critical Parameters for the Selection of the Optimal Formulations and the Justification |
 |
Table 5 Z-Average, PDI and ZP of Selected Formulations. Results are Expressed as Means ± SD (n = 3) |
 |
Table 6 The Summarized Results of the Optimal Formulations |
 |
Table 7 Comparison of Release Profiles Using Model-Independent Approaches. Results are Expressed as Means ± SD (n = 3) |
 |
Table 8 Droplet Size Distribution of the Optimal Formulations. Results are Presented as Means ± SD (n = 3) |
 |
Table 9 The Biopharmaceutical Parameters of the Formulations are Compared to the Initial FAV. Results are Expressed as Means ± SD (n = 3) |
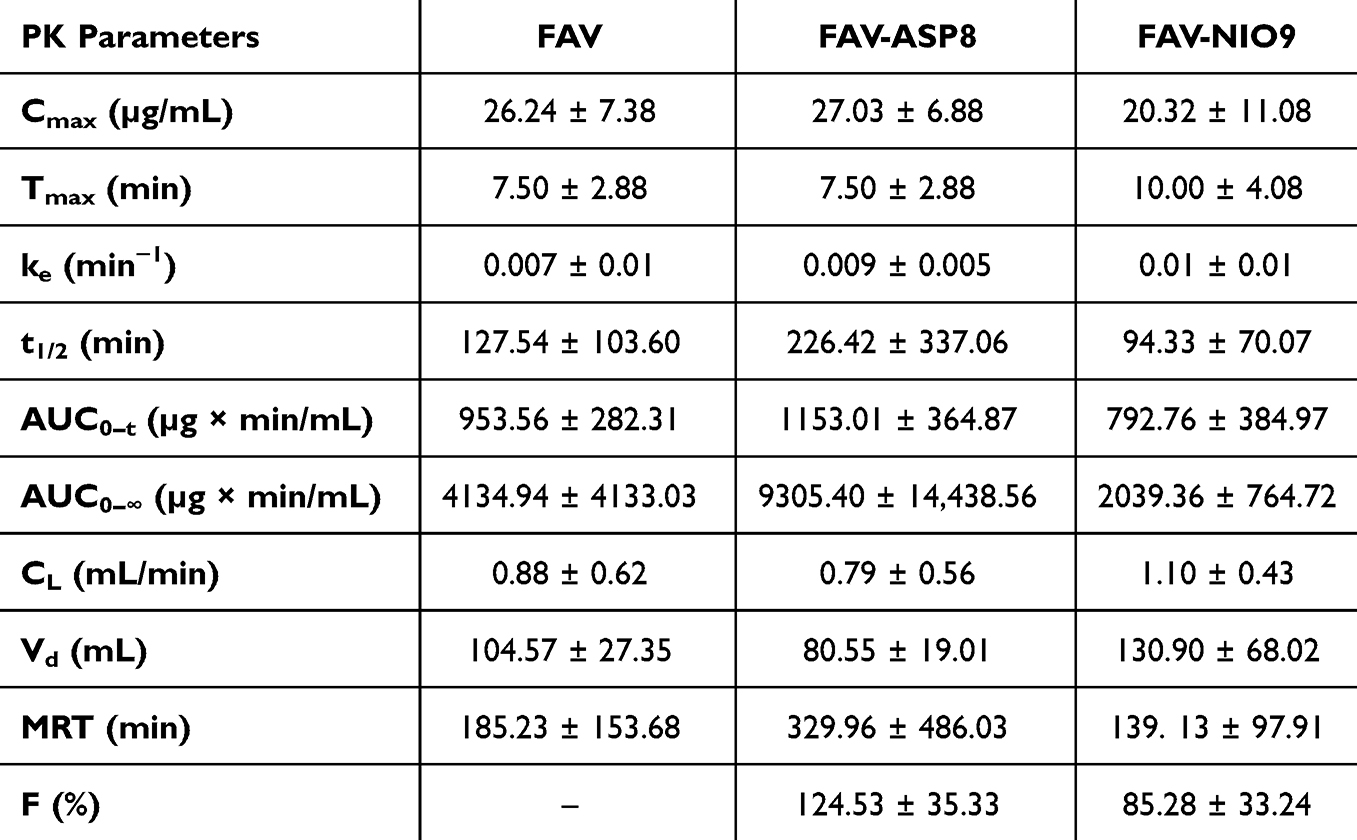 |
Table 10 Pharmacokinetic (PK) Parameters of Pure FAV, FAV-ASP8, and FAV-NIO9 Obtained After Non-Compartmental Analysis Using PK Solver Software. Results are Expressed as Means ± SD (n = 4) |
The optimal formulations were selected based on the critical parameters of the drug carrier for the nose-to-brain delivery system, as summarized in Table 4. Firstly, we selected the formulations within the acceptable range of Z-average, PDI and ZP. Then, we selected those with the highest EE% and DL%. After that, depending on PAMPA results, we selected those with the highest permeability and flux values to evaluate the release rate, permeation through the human nasal mucosa and in vivo permeability. Finally, we selected the promising formulation based on the in vivo pharmacokinetics results.
Vesicle Size (Z-Average), Polydispersity Index (PDI) and Zeta Potential (ZP) Analysis
Table 5 shows the results of Z-average, PDI and ZP for the selected formulations (Z-average, PDI and ZP results of all formulations are presented in Supplementary Table 1). The results for FAV-ASPs showed that when the amount of AP was decreased, the vesicle size of the ASPs significantly increased (p-value < 0.0001). Furthermore, the vesicle size significantly increased with increasing CH amount (p-value < 0.0001). This result was similar to the findings of a previous study by Babar et al (2025), where they developed Acyclovir loaded aspasomal gel for transdermal delivery system. Their results revealed that the amount of AP and CH had a positive impact on Z-average.40 CH intercalates in the bilayer with its polar head, and due to its hydrophobic properties, it occupies the interior portion of lipid bilayers and can fill the gaps. The nonionic surfactant-based FAV-ASP and FAV-NIO formulations showed that with the increase in Span® 60 amount, the Z-average significantly increased (p-value < 0.0001). This result was also in accord with a previous study by Mathure et al (2018). They developed Buspirone loaded niosomes for nose-to-brain delivery system. Their results revealed that Z-average increased by increasing Span® 60 amount from 20 to 40 mg.87 Soni et al (2024) explained the effect of Span® 60 on Z-average by the high interfacial activity and a critical packing parameter between 0.5 and 1, where the lower value means tight packed vesicles, thus a smaller Z-average, while the further increase in Span® 60 amount led to an increase in Z-average due to increase CH amount to fill the gaps and counteract the effect of Span® 60 leading to higher rigidity of the Methotrexate loaded niosomes.88
Anionic surfactant-based FAV-ASPs exhibited bigger vesicles, which could be attributed to the influence of DCP that had a negative charge, leading to an increase in the interbilayer distance and the surface area of the vesicles. Similar results obtained by Varshosaz et al (2003) for Insulin loaded niosomes.89
Moreover, FAV-ASP formulations had a broad size distribution (PDI > 0.3). However, PDI values < 0.5 were reported to be acceptable.81 Whereas FAV-NIO formulations showed a significantly smaller particle size compared with FAV-ASP formulations (p-value < 0.0001), and a narrow size distribution (PDI < 0.3). The results of the prepared formulations showed an acceptably homogenous size distribution; this distribution may depend on the synthesis of mixed multilamellar and large unilamellar vesicles during the hydration stage, as reported in previous studies.42,72
ZP is a predictive indicator of the intensity of repulsive and attractive forces between vesicles, which allows the evaluation of the vesicle stability. The formulations had negative values of ZP, which maintain electric repulsion. These negative values depended on the chemical properties of AP and CH, also because of the application of negatively charged lipids (anionic surfactant-based FAV-ASP formulations). These results agreed with data previously reported by Gopinath et al.31 According to these results, the produced FAV-ASPs and FAV-NIOs were physically stable, preventing vesicle aggregation, and they have a longer residence time inside the brain without being effluxed.90
Z-average, PDI and ZP are critical parameters which could impact the release, transport through direct or indirect pathways, biodistribution and stability.91,92 As reported in the literature, nanoparticles with a Z-average of 150–300 nm were successfully taken up into the brain.79 Accordingly, we selected the formulations with a Z-average < 300 nm for further investigations.
Determination of Drug Content, Encapsulation Efficiency (EE%) and Drug Loading (DL%)
The evaluation of the selected formulations (with a Z-average < 300 nm) indicating that FAV was successfully encapsulated into the vesicles with drug content ranged from 1.60 ± 0.36 mg to 1.85 ± 0.60 mg, and EE% values ranging between 41.01 ± 1.07% and 55.33 ± 0.41%. Drug content of the selected formulations did not alter significantly with the compositions of the formulations (p-value > 0.05). The EE% of FAV-NIOs was varied, and it did not alter significantly with changing of CH amount (p-value > 0.05), and the same result was obtained by Gopinath et al,31 while increasing the ratio of Span® 60 to CH increased EE% (p-value < 0.01). This outcome might be explained by an increase in the rigidity of the NIO membrane as CH amount increased to fill the gaps and counteract the effect of Span® 60, leading to higher rigidity; thus, the membrane fluidity decreased.93 El-Sayed et al (2017) studied the effect of CH amount on EE% of Flurbiprofen loaded niosomes, and the results revealed a significant increment in EE% upon increasing CH amount.94 The results of FAV-ASPs demonstrated that nonionic surfactant-based FAV-ASPs had a significantly higher EE% compared to anionic surfactant-based FAV-ASPs (p-value < 0.0001), which could be related to the impact of using AP and Span® 60. Moreover, EE% increased significantly by increasing the amount of Span® 60 (p-value < 0.0001), or by increasing the amount of AP (p-value < 0.01), or by increasing both AP and Span® 60 amounts (p-value < 0.0001).
Most nanoparticle systems have relatively low drug loading (<10 w%). It is critical to increase drug loading in order to improve the therapeutic effect and minimize adverse effects.95 The nano-carrier system can be considered efficient if EE% > 50% and DL% > 5%.86 The results showed that using ASP and NIO formulations enhanced DL % of FAV (as shown in Figure 2). FAV-ASP8, FAV-ASP10 and FAV-NIO9 showed acceptable EE % (>50%), high DL % (>5%) and drug content. Accordingly, we selected these formulations for further investigation. The results of drug content, EE % and DL % of the selected formulations are presented in Supplementary Table 2.
Residual Solvent Determination
Residual solvents are classified into four classes based on the toxicity level and the degree to which they can be considered as an environmental hazard. Both chloroform and methanol belong to Class II solvents; thus, their residual concentration should be less than 60 ppm and 3000 ppm, respectively, while ethanol belongs to Class III solvents (the accepted residual limit is 5000 ppm) in the daily dose of final product as a requirement of the International Council of Harmonisation (ICH) Q3C (R5) guideline for residual solvents. The results of GC measurement showed that the residual organic solvent contents were under LOD (less than 1 ppm for chloroform, methanol and ethanol), which supports that they were totally evaporated from the ASP and NIO vesicles during the film hydration method, ethanol injection method and freeze-drying.
In vitro Permeability Measurements
PAMPA is a cost-effective, relative, and reproducible technique for rapidly predicting passive transcellular transport,96,97 which is one of the absorption routes of lipophilic molecules through the nasal mucosa and BBB,98,99 so it is unable to give a complete description of the permeability via the BBB. However, PAMPA would be useful for choosing the most promising formulation, hence reducing in vitro and in vivo testing.100
The BBB-PAMPA results showed that the formulations (FAV-ASP8, FAV-ASP10 and FAV-NIO9) had significantly higher permeability than pure FAV (Figure 3A). The addition of the surfactant agent to the formulations enhanced the solubility of FAV and increased its membrane permeability through BBB.87 The effect of Span® 60 could also be observed in the flux values in the PAMPA study (Figure 3B). Based on these results, we selected FAV-ASP8 and FAV-NIO9 as optimal formulations due to the high permeability and flux values through the porcine brain polar lipid extract, high EE% and DL%. The results of the optimal formulations were summarized in Table 6.
Rapid Equilibrium Dialysis Measurement (RED)
RED assay was used to investigate the in vitro drug release of FAV from ASPs and NIOs vesicles at blood circulation conditions (DPBS, pH = 7.4). The incubation time of 4 h was sufficient for test formulations to reach equilibrium. The result showed that the dissolution rate of the optimal formulations was increased by approximately 3.5-fold compared to pure FAV (p-value < 0.0001), which could be related to the nanosized particles and the effect of AP and Span® 60. As shown in Figure 4, the amount released of FAV increased rapidly during the first hour, and the equilibrium state of the formulations started after 2 h. This result could be related to the release of desorption FAV from the surface of the vesicles, followed by diffusion of FAV through the bilayers. Similar result obtained by Taymouri et al (2016).101
In vitro Drug Release Under Nasal Conditions
Figure 5 presents the cumulative in vitro release of FAV from the optimal formulations in SNES. The study indicated that all formulations had a significantly higher release rate than FAV suspension. FAV-ASP8 released 77.31 ± 1.96% of the loaded drug after 60 min (p-value < 0.001), whereas FAV-NIO9 released 57.01 ± 1.22% (p-value < 0.05), in comparison with pure FAV (46.31 ± 1.83%). AP is a more stable amphiphilic derivative of ascorbic acid. It reduces the surface tension, especially when integrated into a phospholipid monolayer.102 Therefore, the integration of AP with CH and Span® 60 could effect on the fluidity of the vesicle membrane resulting in higher release rate compared to NIOs. Moreover, AP is pH neutral, so the hydrolysis of AP at pH 5.6 is slow, which could impact FAV release by diffusion without affecting the vesicle membrane integrity. In addition, increasing Span® 60:CH ratio may improve the integrity of NIOs membrane and reduce the FAV leakage, resulting in less drug release from the vesicles compared to ASPs. The result indicated that the dialysis bag did not restrict the diffusion of FAV. Furthermore, the use of ASPs and NIOs as carriers enhanced the encapsulation of FAV and increased the release rate due to the small particle size of the carrier, which increased the surface area and its hydrophilic properties.
In vitro Drug Release Data Evaluation
The release kinetics of the optimal formulations were statically compared with pure FAV using DDsolver®. The results showed that FAV-ASP8 had the highest AUC and DE values (Table 7). The AUC and DE values of all the optimal formulations were significantly higher than the AUC value of pure FAV. Moreover, the MDT values of FAV-ASP8 were significantly lower than those of pure FAV, which means a faster release rate in comparison to pure FAV and FAV-NIO9, which could increase the absorption through the nasal cavity.103,104
The model with the highest R2, the smallest AIC, and the highest MSC values is the best kinetics model.50,51,105 The release profiles of the optimal formulations were fitted Korsmeyer-Peppas kinetic model. The release exponent “n” values of FAV-ASP8 and FAV-NIO9 were 0.44 ± 0.01 and 0.38 ± 0.01, respectively, which refers to a quasi-Fickian diffusion (n < 0.45)50,106 (Supplementary Table 3).
Storage Stability
The stability test is important for the evaluation of the ability of the vesicles to resist environmental conditions, such as temperature, and for the protection of the drug from degradation, thus determining the recommended storage conditions and the retest period for the drug. The stability results for the optimal formulations showed that there was a decrease in Z-average value after 4 weeks (p-value < 0.0001) in case of FAV-ASP8, which can be explained by the change of mean intensity distribution of the size (Z-average is calculated by the intensity of the particle),107 and the effect of AP as antioxidant which prevents the oxidative degradation of lipid bilayer. However, FAV-NIO9 showed a significant increase in Z-average value (p-value < 0.0001), presumably because of the aggregation of NIOs over time due to attractive forces (Van der Waals forces) or electrostatic interactions (repulsive forces). Moreover, the evaluation of PDI for FAV-ASP8 and FAV-NIO9 showed a non-significant increase, and both formulations maintained acceptably homogenous size distribution. The evaluation of ZP over time showed that FAV-ASP8 had a non-significant change in ZP, while FAV-NIO9 exhibited a decrease in ZP (non-significant), indicating a higher possibility to aggregate over time.108 Furthermore, the six hydroxyl groups of mannitol can form hydrogen bonds with FAV (using the hydroxyl and carboxamide groups), which could impact the crystallization of mannitol and form a partially amorphous state. The amorphous state is less chemical stability and more hygroscopic.109,110 Therefore, hydrolysis or oxidative degradation could occur over time. FAV-NIO9 showed a higher reduction of FAV concentration compared to FAV-ASP8. Thus, we can conclude that FAV-ASP8 had a higher stability than FAV-NIO9 (Supplementary Figures 1–4).
Droplet Size Distribution Measurement
Targeting the CNS requires particle deposition in the posterior region of the nasal cavity (to reach the olfactory and trigeminal nerves).111 The deposition depends on many factors, including the interaction of the spray device with the nose (the angle of insertion and the plume angle),112 nasal cavity (the mucociliary clearance mechanism and the human variabilities),113 and the formulation characteristics (droplet size, viscosity, elasticity).114
US FDA and EMA recommended the droplet size distribution measurement as an indicator of in vitro bioavailability for the liquid nasal formulation.113,114 The nasal spray is recommended to be placed 3 cm below the receiving lens,113 with an application angle of 45°. A study by Jüptner et al (2025) demonstrated a more posterior deposition profile for 45° compared to 60° due to increased deposition in the turbinate region, and the overall nasal deposition was excellent at ~98%, with minimal loss from the nose.111
The recommended droplet size ranges between 20 to around 200 µm.112 Droplets with Dv50 < 10 µm can be inhaled through the nasopharynx and reach the lungs,115 or deposited in the anterior region of the nasal cavity which enhances formulation clearance from the nasal cavity.113
The results demonstrated that FAV-ASP8 and FAV-NIO9 were suitable for nasal administration (Dv50 values were 174.80 ± 12.08 and 89.05 ± 7.33 µm, respectively), which indicates a more posterior deposition, thus, increasing the absorption through the nasal cavity.
FAV-ASP8 showed a significantly higher Dv50 values compared to FAV-NIO9 (p-value < 0.01), which could be attributed to the effect of AP and Span® 60. Span® 60, as a non-ionic surfactant, reduced the interfacial tension and facilitated the formation of smaller droplets (FAV-NIO9),114 while the incorporation of AP could counteract the effect of Span® 60 leading to larger droplets (FAV-ASP8). Moreover, FAV-ASP8 and FAV-NIO9 showed small Span values (no significant differences), indicating a narrow width of droplet size distribution116 (Table 8).
Ex vivo Nasal Diffusion Study on Human Nasal Mucosa
The transmucosal permeability measurement of the optimal formulations in comparison with pure FAV was performed using human nasal mucosa. The study showed that both FAV-ASP8 and FAV-NIO9 had a higher amount of FAV diffused through the human nasal mucosa to the acceptor chamber within 60 min (1311.74 ± 41.70 µg/cm2 and 402.94 ± 22.32 µg/cm2, respectively) compared to pure FAV (128.17 ± 13.64 µg/cm2, p-value < 0.0001), which means an enhancement in the absorption and permeation through the nasal mucosa (Figure 6). These results could be attributed to the nanosized particles (particle sizes < 500 nm can pass through the aqueous, non-viscous holes of the mucin network, resulting in higher mucosal penetration and absorption into the mucosal cells), negative ZP value (avoid slow penetration by limiting adherence to the negatively charged membrane), in addition to the effect of vesicles components.117 Furthermore, FAV suspension has high fluidity and negligible mucosal adherence.38 As a result of the shorter contact time with the nasal mucosa, FAV suspension has a lower permeability.
Moreover, FAV-ASP8 showed a significantly higher permeability compared to FAV-NIO9 (p-value < 0.0001), which could be attributed to the surfactant-like property of AP (like sorbitan derivatives), Its lipophilic properties which enhance the tissue penetration, and its deformability which allowed ASPs to bypass through the tight junctions of the nasal cavity. D’Avanzo et al (2022) evaluated the permeation profile of Idebenone/Naproxen co‐loaded aspasomes by human stratum corneum and viable epidermis (SCE) membrane, and the results demonstrated enhancement in the permeability due to the impact of AP.42
Table 9 summarizes the biopharmaceutical parameters, including permeation and prediction parameters. FAV-ASP8 had higher steady-state flux (Jss), permeability coefficient (Kp) and theoretical plasma concentration at the steady-state in humans compared to FAV-NIO9 and pure FAV.
In vivo Study
Pharmacokinetic Parameters
FAV has a low solubility in water (8.7 mg/mL) and a low CNS permeability, which could be attributed to the low passive permeability due to its three H-bonding donors.25 The capacity of FAV-ASP8 and FAV-NIO9 to deliver FAV to the CNS via the intranasal route was evaluated using Sprague–Dawley rats.
The average plasma drug concentration–time curves after a single nasal dose of pure FAV, FAV-ASP8 and FAV-NIO9 are demonstrated in Figure 7. FAV was detected and reached a plasma concentration peak (Cmax) of 26.24 ± 7.382, 27.03 ± 6.88, and 20.32 ± 11.08 µg/mL at 7.5 ± 2.88, 7.5 ± 2.88, and 10 ± 4.08 min (Tmax) after the administration of pure FAV, FAV-ASP8 and FAV-NIO9, respectively. These results indicate that the nasal mucosa effectively and rapidly transports FAV into the systemic circulation. In addition, FAV-ASP8 showed the highest t1/2 value (226.42 ± 337.06 min) compared to pure FAV and FAV-NIO9. This could be related to the use of AP to prepare the drug carrier, which may reduce the elimination rate and thus extend the duration in the systemic circulation (CL value was 0.79 ± 0.56 mL/min). The relative bioavailability (F%) values of FAV-ASP8 and FAV-NIO9 were 124.53 ± 35.33% and 85.28 ± 33.24%, respectively (Table 10).
Due to restrictions in CNS tissue access, CSF drug concentrations are used as an alternative for CNS tissue delivery, with the hypothesis that there is a direct, though not absolute, link between CSF and CNS tissue penetration.118 Furthermore, the olfactory nerves in the nasal cavity provide additional link with the CSF in the subarachnoid space through the interstitial fluid surrounding the olfactory nerve bundle.119,120 In our study, FAV was detected in the CSF samples (Figure 8). The evaluation of CSF samples showed a significant elevation of FAV concentration into CSF after 1 hour of nasal administration of FAV-ASP8 compared to FAV suspension (*p-value < 0.05) and FAV-NIO9 (***p-value < 0.001), while there was no significant increase in the case of FAV-NIO9 compared to FAV suspension. These results can be attributed to an increase in the extent of drug absorption through the nasal mucosa (olfactory nerves) due to the physical parameter, the lipophilicity nature of the vesicles and the presence of a permeation enhancer (Span® 60), which improves the membrane penetration. Furthermore, AP can overcome biological barriers, enter the brain121 and resist hydrolysis,122 thereby preventing the degradation of FAV. According to the chemical structure of FAV, amide hydrolysis and oxidation are the potential major degradation pathways.123
Abou-Taleb et al (2018) developed Nefopam loaded niosomes for intranasal administration using CH:Span® 80 (1:1.85 molar ratio). Their study illustrated a 4.77-fold increase in bioavailability compared with oral solution of drug.61 Moreover, Ammar et al (2017) developed Diltiazem loaded niosomes using CH:Span® 60 (1:1 molar ratio), which showed the highest EE% and release rate. The in vivo study on male Wistar rats showed an increase in MRT, t1/2 and AUC with a decrease in Ke.124
Due to the lipophilic nature of AP, it can easy bypass phospholipid bilayers of membranes, especially neuronal ones and BBB.121,125,126 AP has been used in nasal formulations as a cross-linker, as reported by J. Varshosaz et al (2004). They developed insulin chitosan microspheres by emulsification-cross linking process using ascorbic acid or ascorbyl palmitate as a cross-linker. The formulation consists of 400 mg of chitosan and 70 mg of AP showed a 67% reduction of blood glucose compared to IV route.127
Evaluation of in vitro–in vivo Correlations (IVIVC)
IVIVC is a useful tool to predict the in vivo performance of FAV based on its in vitro data. Figure 9 demonstrates the IVIVC graphs for the comparison of AUC0-t values between the in vitro release and the in vivo PK data. The correlation coefficient (R2) values were 0.971, 0.982, and 0.903 for pure FAV, FAV-ASP8, and FAV-NIO9, respectively. Furthermore, R2 values obtained from IVIVC graphs for the comparison of AUC0-t values between the in vitro permeation and the in vivo PK data (Figure 10) showed a good point-to-point correlation (0.977, 0.992, and 0.839 for pure FAV, FAV-ASP8, and FAV-NIO9, respectively). This result indicates that the use of in vitro release and permeation data to establish the IVIVC could be useful in predicting the in vivo properties of the formulations.
Conclusion
In the present study, ASP and NIO formulations have been developed as promising intranasal delivery systems for FAV to enhance its bioavailability. The optimized formulation FAV-ASP8 (consisting of FAV:AP:Span® 60:CH in 30:25:25:50 w/w ratio) can encapsulate FAV inside ASP vesicles with an encapsulation efficiency of 55.33 ± 0.41% drug loading of 12.79 ± 0.22%. The formulation showed a small particle size (292.06 ± 2.10 nm), a narrow PDI value (0.36 ± 0.03) and negative values of ZP (−74.73 ± 3.28 mV), indicating a high physical stability for FAV-ASP8. The other optimized formulation, FAV-NIO9 contained FAV, Span® 60 and CH in 30:30:40 w/w ratio with a Z-average value of 167.13 ± 1.60 nm, a narrow PDI value (0.07 ± 0.01), adequate ZP (−27.1 ± 1.24 mV), acceptable encapsulation efficiency (51.30 ± 0.69%) and advantageous drug loading (15.42 ± 0.38%).
FAV-ASP8 and FAV-NIO9 formulations enhanced the solubility of FAV, elevated its membrane permeability through BBB, and increased its release under both nasal (pH = 5.6) and blood circulation conditions (pH = 7.4), with Dv50 values ranged between 20 and 200 µm. The ex vivo study showed that FAV-ASP8 had a higher amount of FAV diffused through the human nasal mucosa than FAV-NIO9 and pure FAV. The in vivo study indicated that there was no significant difference in plasma concentrations for the optimal formulations in comparison to pure FAV. However, only FAV-ASP8 had higher CSF concentrations than FAV-NIO9 and pure FAV after a single intranasal dose. IVIVC showed a good point-to-point correlation between the in vitro and the in vivo PK data.
These encouraging results suggest that ASPs could be promising intranasal delivery systems for hydrophobic drugs to enhance solubility, avoid first-pass metabolism, and decrease systemic adverse effects.
Data Sharing Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The publication was funded by The University of Szeged Open Access Fund (FundRef, Grant No. 7468). This work was supported by Project no. TKP2021-EGA-32 implemented with support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vazquez C, Jurado KA. Neurotropic RNA Virus Modulation of Immune Responses within the Central Nervous System. International Journal of Molecular Sciences. 2022;23(7):4018. doi:10.3390/ijms23074018
2. Salinas S, Funk KE. Editorial: cellular and: molecular Mechanisms of Neurotropic Viral Infection. Front Cell Neurosci. 2024;18:1365755. doi:10.3389/fncel.2024.1365755
3. Klein RS, Garber C, Funk KE, et al. Neuroinflammation During RNA Viral Infections. Annu Rev Immunol. 2019;37(1):73–95. doi:10.1146/annurev-immunol-042718-041417
4. Yachou Y, El Idrissi A, Belapasov V, Ait Benali S. Neuroinvasion, Neurotropic, and Neuroinflammatory Events of SARS-CoV-2: understanding the Neurological Manifestations in COVID-19 Patients. Neurol Sci. 2020;41(10):2657. doi:10.1007/S10072-020-04575-3
5. Sun M, Manson ML, Guo T, De Lange ECM. CNS Viral Infections-What to Consider for Improving Drug Treatment: a Plea for Using Mathematical Modeling Approaches. CNS Drugs. 2024;38:349–373. doi:10.1007/s40263-024-01082-3
6. Wongchitrat P, Chanmee T, Govitrapong P. Molecular Mechanisms Associated with Neurodegeneration of Neurotropic Viral Infection. Mol Neurobiol. 2024;61(5):2881–2903. doi:10.1007/s12035-023-03761-6
7. Zhao YJ, Xu KF, Shu FX, Zhang F. Neurotropic Virus Infection and Neurodegenerative Diseases: potential Roles of Autophagy Pathway. CNS Neurosci Ther. 2024;30:6. doi:10.1111/CNS.14548
8. Rinaldi F, Hanieh P, Chan L, et al. Chitosan Glutamate-Coated Niosomes: a Proposal for Nose-to-Brain Delivery. Pharmaceutics. 2018;10(2):38. doi:10.3390/pharmaceutics10020038
9. Djupesland PG, Messina JC, Mahmoud RA. The Nasal Approach to Delivering Treatment for Brain Diseases: an Anatomic, Physiologic, and Delivery Technology Overview. Ther Deliv. 2014;5(6):709–733. doi:10.4155/TDE.14.41
10. Martins PP, Smyth HDC, Cui Z. Strategies to Facilitate or Block Nose-to-Brain Drug Delivery. Int J Pharm. 2019;570:118635. doi:10.1016/J.IJPHARM.2019.118635
11. Higgins TS, Wu AW, Illing EA, et al. Intranasal Antiviral Drug Delivery and Coronavirus Disease 2019 (COVID-19): a State of the Art Review. Otolaryngol Head Neck Surg. 2020;163(4):682–694. doi:10.1177/0194599820933170
12. Delshadi R, Bahrami A, McClements DJ, Moore MD, Williams L. Development of Nanoparticle-Delivery Systems for Antiviral Agents: a Review. J Control Release. 2021;331:30–44. doi:10.1016/J.JCONREL.2021.01.017
13. Konstantinova ID, Andronova VL, Fateev IV, Esipov RS. Favipiravir and Its Structural Analogs: antiviral Activity and Synthesis Methods. Acta Naturae. 2022;14(2):16–38. doi:10.32607/actanaturae.11652
14. Al-Horani RA, Kar S. Potential Anti-SARS-CoV-2 Therapeutics That Target the Post-Entry Stages of the Viral Life Cycle: a Comprehensive Review. Viruses. 2020;12:10. doi:10.3390/V12101092
15. Shiraki K, Daikoku T. Favipiravir, an Anti-Influenza Drug against Life-Threatening RNA Virus Infections. Pharmacol Ther. 2020;209:107512. doi:10.1016/j.pharmthera.2020.107512
16. Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a Broad Spectrum Inhibitor of Viral RNA Polymerase. Proceedings Japan Academy Ser B: Physical and Biological Sciences. 2017;93(7):449–463. doi:10.2183/pjab.93.027
17. Borbone N, Piccialli G, Roviello GN, Oliviero G. Nucleoside Analogs and Nucleoside Precursors as Drugs in the Fight against SARS-CoV-2 and Other Coronaviruses. Molecules. 2021;26(4):986. doi:10.3390/molecules26040986
18. Rattanaumpawan P, Jirajariyavej S, Lerdlamyong K, Palavutitotai N, Saiyarin J. Real-World Effectiveness and Optimal Dosage of Favipiravir for Treatment of COVID-19: results from a Multicenter Observational Study in Thailand. Antibiotics. 2022;11(6):805. doi:10.3390/ANTIBIOTICS11060805
19. Sajadian SA, Ardestani NS, Esfandiari N, Askarizadeh M, Jouyban A. Solubility of Favipiravir (as an Anti-COVID-19) in Supercritical Carbon Dioxide: an Experimental Analysis and Thermodynamic Modeling. J Supercrit Fluids. 2022;183:105539. doi:10.1016/J.SUPFLU.2022.105539
20. Hayden FG, Lenk RP, Epstein C, Kang LL. Oral Favipiravir Exposure and Pharmacodynamic Effects in Adult Outpatients With Acute Influenza. J Infect Dis. 2024;230(2):e395–e404. doi:10.1093/INFDIS/JIAD409
21. Du Y, Chen X. Favipiravir: pharmacokinetics and Concerns About Clinical Trials for 2019‐nCoV Infection. Clin Pharmacol Ther. 2020;108(2):242–247. doi:10.1002/cpt.1844
22. Almutairi AO, El-Readi MZ, Althubiti M, et al. Liver Injury in Favipiravir-Treated COVID-19 Patients: retrospective Single-Center Cohort Study. Trop Med Infect Dis. 2023;8(2). doi:10.3390/TROPICALMED8020129
23. Pires A, Fortuna A, Alves G, Falcão A. Intranasal Drug Delivery: how, Why and What For? J Pharm Pharm Sci. 2009;12(3):288. doi:10.18433/J3NC79
24. Erdő F, Bors LA, Farkas D, Bajza Á, Gizurarson S. Evaluation of Intranasal Delivery Route of Drug Administration for Brain Targeting. Brain Res Bull. 2018;143:155–170. doi:10.1016/j.brainresbull.2018.10.009
25. Rong J, Zhao C, Xia X, et al. Evaluation of [18F]Favipiravir in Rodents and Nonhuman Primates (NHP) with Positron Emission Tomography. Pharmaceuticals. 2023;16(4):524. doi:10.3390/ph16040524
26. Khan MS, Baskoy SA, Yang C, et al. Lipid-Based Colloidal Nanoparticles for Applications in Targeted Vaccine Delivery. Nanoscale Adv. 2023;5(7):1853. doi:10.1039/D2NA00795A
27. Xinchen Y, Jing T, Jiaoqiong G. Lipid-Based Nanoparticles via Nose-to-Brain Delivery: a Mini Review. Front Cell Dev Biol. 2023;11:1214450. doi:10.3389/FCELL.2023.1214450/FULL
28. Battaglia L, Panciani PP, Muntoni E, et al. Lipid Nanoparticles for Intranasal Administration: application to Nose-to-Brain Delivery. Expert Opin Drug Deliv. 2018;15(4):369–378. doi:10.1080/17425247.2018.1429401
29. Rajera R, Nagpal K, Singh SK, Mishra DN. Niosomes: a Controlled and Novel Drug Delivery System. Biol Pharm Bull. 2011;34(7):945–953. doi:10.1248/bpb.34.945
30. Marianecci C, Di Marzio L, Rinaldi F, et al. Niosomes from 80s to Present: the State of the Art. Adv Colloid Interface Sci. 2014;205:187–206. doi:10.1016/j.cis.2013.11.018
31. Gopinath D, Ravi D, Rao BR, Apte SS, Renuka D, Rambhau D. Ascorbyl Palmitate Vesicles (Aspasomes): formation, Characterization and Applications. Int J Pharm. 2004;271(1–2):95–113. doi:10.1016/j.ijpharm.2003.10.032
32. Moribe K, Limwikrant W, Higashi K, Yamamoto K. Drug Nanoparticle Formulation Using Ascorbic Acid Derivatives. J Drug Deliv. 2011;2011:1–9. doi:10.1155/2011/138929
33. Palma S, Manzo R, Lo Nostro P, Allemandi D. Nanostructures from Alkyl Vitamin C Derivatives (ASCn): properties and Potential Platform for Drug Delivery. Int J Pharm. 2007;345(1–2):26–34. doi:10.1016/J.IJPHARM.2007.09.014
34. Khalil RM, Abdelbary A, Arini SKE, Basha M, El-Hashemy HA, Farouk F. Development of Tizanidine Loaded Aspasomes as Transdermal Delivery System: ex-vivo and in-vivo Evaluation. J Liposome Res. 2021;31(1):19–29. doi:10.1080/08982104.2019.1684940
35. Han S. Structure of Ascorbyl Palmitate Bilayers (Aspasomes) from Molecular Dynamics Simulation. Bull Korean Chem Soc. 2018;39(7):887–890. doi:10.1002/BKCS.11475
36. Wong SN, Li S, Low KH, et al. Development of Favipiravir Dry Powders for Intranasal Delivery: an Integrated Cocrystal and Particle Engineering Approach via Spray Freeze Drying. Int J Pharm. 2024;653:123896. doi:10.1016/J.IJPHARM.2024.123896
37. Darne P, Vidhate S, Shintre S, et al. Advancements in Antiviral Therapy: favipiravir Sodium in Nasal Formulation. AAPS Pharm Sci Tech. 2024;25(8):1–15. doi:10.1208/S12249-024-02986-5/TABLES/4
38. Gattani V, Dawre S. Development of Favipiravir Loaded PLGA Nanoparticles Entrapped in In-Situ Gel for Treatment of Covid-19 via Nasal Route. J Drug Deliv Sci Technol. 2023;79:104082. doi:10.1016/j.jddst.2022.104082
39. Alcantara KP, Nalinratana N, Chutiwitoonchai N, et al. Enhanced Nasal Deposition and Anti-Coronavirus Effect of Favipiravir-Loaded Mucoadhesive Chitosan–Alginate Nanoparticles. Pharmaceutics. 2022;14(12):2680. doi:10.3390/PHARMACEUTICS14122680/S1
40. Vimalakar Babar A, Prasanna Sutar K, Vijay Usulkar S. S121-S131. Formulation and Evaluation of Acyclovir Loaded Aspasomal Gel for Effective Management of Herpes. Indian J Pharm Educ Res. 2025;59(1s). doi:10.5530/ijper.20254263
41. Kar M, Saquib M, Jain DK. Formulation Development and Evaluation of Aspasomes Containing Skin Whitening Agent. Manipal J Pharma Sci. 2020. 6(1):47–53.
42. d’Avanzo N, Cristiano MC, Di Marzio L, et al. Multidrug Idebenone/Naproxen Co-Loaded Aspasomes for Significant in vivo Anti-Inflammatory Activity. ChemMedChem. 2022;17(9):e202200067. doi:10.1002/CMDC.202200067
43. Sita VG, Jadhav D, Vavia P. Niosomes for Nose-to-Brain Delivery of Bromocriptine: formulation Development, Efficacy Evaluation and Toxicity Profiling. J Drug Deliv Sci Technol. 2020;58:101791. doi:10.1016/J.JDDST.2020.101791
44. Tavano L, Muzzalupo R, Trombino S, Cassano R, Pingitore A, Picci N. Effect of Formulations Variables on the in vitro Percutaneous Permeation of Sodium Diclofenac from New Vesicular Systems Obtained from Pluronic Triblock Copolymers. Colloids Surf B Biointerfaces. 2010;79(1):227–234. doi:10.1016/j.colsurfb.2010.03.055
45. Peng W, Jiang XY, Zhu Y, et al. Oral Delivery of Capsaicin Using MPEG-PCL Nanoparticles. Acta Pharmacol Sin. 2015;36(1):139–148. doi:10.1038/APS.2014.113
46. Katona G, Balogh GT, Dargó G, et al. Development of Meloxicam-Human Serum Albumin Nanoparticles for Nose-to-Brain Delivery via Application of a Quality by Design Approach. Pharmaceutics. 2020;12(2):97. doi:10.3390/pharmaceutics12020097
47. Nigam K, Kaur A, Tyagi A, et al. Nose-to-Brain Delivery of Lamotrigine-Loaded PLGA Nanoparticles. Drug Deliv Transl Res. 2019;9(5):879–890. doi:10.1007/s13346-019-00622-5
48. Zhang Y, Huo M, Zhou J, et al. DDSolver: an Add-In Program for Modeling and Comparison of Drug Dissolution Profiles. AAPS J. 2010;12(3):263. doi:10.1208/S12248-010-9185-1
49. OZ Ameer. In vitro Pharmaco-Equivalence Analysis of Diclofenac Potassium Oral Film-Coated Tablet Relative to Marketed Generics. JPPRes. 2023;11:585. doi:10.56499/jppres23.1641_11.4.585
50. Abdul Rasool BK, Mohammed AA, Salem YY. The Optimization of a Dimenhydrinate Transdermal Patch Formulation Based on the Quantitative Analysis of in vitro Release Data by DDSolver through Skin Penetration Studies. Sci Pharm. 2021;89(3):33. doi:10.3390/SCIPHARM89030033/S1
51. Pascoal ADSMR, da Silva PM, Coelho Pinheiro MN. Drug Dissolution Profiles from Polymeric Matrices: data versus Numerical Solution of the Diffusion Problem and Kinetic Models. Int J Heat Mass Transf. 2015;61:118–127. doi:10.1016/J.ICHEATMASSTRANSFER.2014.12.011
52. Salamah M, Budai-Sz M, Ucs M, et al. Development and Characterization of In Situ Gelling Nasal Cilostazol Spanlastics. Gels. 2025;11(2):82. doi:10.3390/GELS11020082
53. Katona G, Sabir F, Sipos B, et al. Development of Lomustine and N-Propyl Gallate Co-Encapsulated Liposomes for Targeting Glioblastoma Multiforme via Intranasal Administration. Pharmaceutics. 2022;14(3):631. doi:10.3390/pharmaceutics14030631
54. Sipos B, Bella Z, Gróf I, et al. Soluplus® Promotes Efficient Transport of Meloxicam to the Central Nervous System via Nasal Administration. Int J Pharm. 2023;632:122594. doi:10.1016/j.ijpharm.2023.122594
55. Abdullah G, Abdulkarim M, Salman I, et al. In vitro Permeation and in vivo Anti-Inflammatory and Analgesic Properties of Nanoscaled Emulsions Containing Ibuprofen for Topical Delivery. IJN. 2011;2011:387. doi:10.2147/IJN.S14667
56. Espinoza LC, Silva-Abreu M, Clares B, et al. Formulation Strategies to Improve Nose-to-Brain Delivery of Donepezil. Pharmaceutics. 2019;11(2):64. doi:10.3390/pharmaceutics11020064
57. Irie K, Nakagawa A, Fujita H, et al. Population Pharmacokinetics of Favipiravir in Patients with COVID‐19. CPT Pharmacom Syst Pharma. 2021;10(10):1161–1170. doi:10.1002/psp4.12685
58. Jespersen B, Knupp L, Northcott CA. Femoral Arterial and Venous Catheterization for Blood Sampling, Drug Administration and Conscious Blood Pressure and Heart Rate Measurements. JoVE. 2012;(59):3496. doi:10.3791/3496
59. Jain R, Jain B, Kabir A, Bajaj A, Ch R, Sharma S. Fabric Phase Sorptive Extraction-Gas Chromatography-Mass Spectrometry for the Determination of Favipiravir in Biological and Forensic Samples. Adv Sample Preparation. 2023;6:100058. doi:10.1016/j.sampre.2023.100058
60. Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an Add-in Program for Pharmacokinetic and Pharmacodynamic Data Analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. doi:10.1016/J.CMPB.2010.01.007
61. Abou-Taleb HA, Khallaf RA, Abdel-Aleem JA. Intranasal Niosomes of Nefopam with Improved Bioavailability: preparation, Optimization, and in-vivo Evaluation. Drug Des Devel Ther. 2018;12:3501. doi:10.2147/DDDT.S177746
62. Ahmed TA. Preparation of Finasteride Capsules-Loaded Drug Nanoparticles: formulation, Optimization, in vitro, and Pharmacokinetic Evaluation. Int J Nanomed. 2016;11:515–527. doi:10.2147/IJN.S98080
63. Ibrahim MM, Basalious EB, El-Nabarawi MA, Makhlouf AIA, Sayyed ME, Ibrahim IT. Nose to Brain Delivery of Mirtazapine via Lipid Nanocapsules: preparation, Statistical Optimization, Radiolabeling, in vivo Biodistribution and Pharmacokinetic Study. Drug Deliv Transl Res. 2024;14(9):2539–2557. doi:10.1007/S13346-024-01528-7
64. Rompicherla SKL, Arumugam K, Bojja SL, Kumar N, Rao CM. Pharmacokinetic and Pharmacodynamic Evaluation of Nasal Liposome and Nanoparticle Based Rivastigmine Formulations in Acute and Chronic Models of Alzheimer’s Disease. Naunyn Schmiedebergs Arch Pharmacol. 2021;394(8):1737–1755. doi:10.1007/S00210-021-02096-0/FIGURES/13
65. Bhandari R, Kaur IP. Pharmacokinetics, Tissue Distribution and Relative Bioavailability of Isoniazid-Solid Lipid Nanoparticles. Int J Pharm. 2013;441(1–2):202–212. doi:10.1016/J.IJPHARM.2012.11.042
66. Kohajda Z, Virág L, Hornyik T, et al. In vivo and Cellular Antiarrhythmic and Cardiac Electrophysiological Effects of Desethylamiodarone in Dog Cardiac Preparations. Br J Pharmacol. 2022;179(13):3382–3402. doi:10.1111/BPH.15812
67. Akel H, Ismail R, Katona G, Sabir F, Ambrus R, Csóka I. A Comparison Study of Lipid and Polymeric Nanoparticles in the Nasal Delivery of Meloxicam: formulation, Characterization, and in vitro Evaluation. Int J Pharm. 2021;604:120724. doi:10.1016/J.IJPHARM.2021.120724
68. D’Souza S, Faraj JA, Giovagnoli S, DeLuca PP. IVIVC from Long Acting Olanzapine Microspheres. Int J Biomater. 2014;2014:1–11. doi:10.1155/2014/407065
69. Sipos B, Szabó-Révész P, Csóka I, et al. Quality by Design Based Formulation Study of Meloxicam-Loaded Polymeric Micelles for Intranasal Administration. Pharmaceutics. 2020;12(8):1–29. doi:10.3390/PHARMACEUTICS12080697
70. Brill Nijhoff. International Conference On Harmonisation Of Technical Requirements For Registration Of Pharmaceuticals For Human Use. In: Tietje C, Brouder A, editors. Handbook of Transnational Economic Governance Regimes. Brill Nijhoff. 2010:1041–1053. doi:10.1163/ej.9789004163300.i-1081.897
71. Shabir GA. Validation of High-Performance Liquid Chromatography Methods for Pharmaceutical Analysis Understanding the Differences and Similarities between Validation Requirements of the US Food and Drug Administration, the US Pharmacopeia and the International Conference on Harmonization. J Chromatogr A. 2003;2023:1.
72. Aboul-Einien MH, Kandil SM, Abdou EM, Diab HM, Zaki MSE. Ascorbic Acid Derivative-Loaded Modified Aspasomes: formulation, in vitro, ex vivo and Clinical Evaluation for Melasma Treatment. J Liposome Res. 2020;30(1):54–67. doi:10.1080/08982104.2019.1585448
73. Moribe K, Maruyama S, Inoue Y, et al. Ascorbyl Dipalmitate/PEG-Lipid Nanoparticles as a Novel Carrier for Hydrophobic Drugs. Int J Pharm. 2010;387(1–2):236–243. doi:10.1016/j.ijpharm.2009.12.007
74. Honary S, Zahir F. Effect of Process Factors on the Properties of Doxycycline Nanovesicles. Trop J Pharm Res. 2012;11(2):169–175. doi:10.4314/TJPR.V11I2.1
75. Amer SS, Nasr M, Abdel-Aziz RTA, et al. Cosm-Nutraceutical Nanovesicles for Acne Treatment: physicochemical Characterization and Exploratory Clinical Experimentation. Int J Pharm. 2020;577:119092. doi:10.1016/j.ijpharm.2020.119092
76. Zaid Alkilani A, Abu-Zour H, Alshishani A, Abu-Huwaij R, Basheer HA, Abo-Zour H. Formulation and Evaluation of Niosomal Alendronate Sodium Encapsulated in Polymeric Microneedles: in vitro Studies, Stability Study and Cytotoxicity Study. Nanomaterials. 2022;12(20):3570. doi:10.3390/NANO12203570
77. Saraswathi TS, Mothilal M. Development of Rivastigmine Loaded Self Assembled Nanostructures of Nonionic Surfactants for Brain Delivery. Int J Appl Pharm. 2021;13:205–215. doi:10.22159/ijap.2021v13i5.42664
78. Manosroi A, Wongtrakul P, Manosroi J, et al. Characterization of Vesicles Prepared with Various Non-Ionic Surfactants Mixed with Cholesterol. Colloids Surf B Biointerfaces. 2003;30(1–2):129–138. doi:10.1016/S0927-7765(03)00080-8
79. Wohlfart S, Gelperina S, Kreuter J. Transport of Drugs across the Blood–Brain Barrier by Nanoparticles. J Control Release. 2012;161(2):264–273. doi:10.1016/j.jconrel.2011.08.017
80. SahiN A, Tonbul H, Çapan Y, Seko I. Brain-Targeted Nanoparticles to Overcome the Blood-Brain Barrier. jpt. 2020;1(1):26–40. doi:10.37662/jpt.2020.4
81. Amasya G, Badilli U, Aksu B, Tarimci N. Quality by Design Case Study 1: design of 5-Fluorouracil Loaded Lipid Nanoparticles by the W/O/W Double Emulsion — solvent Evaporation Method. Eur J Pharm Sci. 2016;84:92–102. doi:10.1016/j.ejps.2016.01.003
82. Dutta L, Mukherjee B, Chakraborty T, et al. Lipid-Based Nanocarrier Efficiently Delivers Highly Water Soluble Drug across the Blood–Brain Barrier into Brain. Drug Deliv. 2018;25(1):504–516. doi:10.1080/10717544.2018.1435749
83. Eid HM, Naguib IA, Alsantali RI, Alsalahat I, Hegazy AM. Novel Chitosan-Coated Niosomal Formulation for Improved Management of Bacterial Conjunctivitis: a Highly Permeable and Efficient Ocular Nanocarrier for Azithromycin. J Pharm Sci. 2021;110(8):3027–3036. doi:10.1016/J.XPHS.2021.04.020
84. Zolghadri S, Asad AG, Farzi F, et al. Span 60/Cholesterol Niosomal Formulation as a Suitable Vehicle for Gallic Acid Delivery with Potent in vitro Antibacterial, Antimelanoma, and Anti-Tyrosinase Activity. Pharmaceuticals. 2023;16(12):1680. doi:10.3390/ph16121680
85. Neves AR, Queiroz JF, Reis S. Brain-Targeted Delivery of Resveratrol Using Solid Lipid Nanoparticles Functionalized with Apolipoprotein E. J Nanobiotechnol. 2016;14(1):27. doi:10.1186/s12951-016-0177-x
86. Korchia L, Bouilhac C, Aubert A, Robin -J-J, Lapinte V. Light-Switchable Nanoparticles Based on Amphiphilic Diblock, Triblock and Heterograft Polyoxazoline. RSC Adv. 2017;7(68):42690–42698. doi:10.1039/C7RA07094B
87. Mathure D, Madan R. Formulation and Evaluation of Niosomal in Situ Nasal Gel of a Serotonin Receptor Agonist, Buspirone Hydrochloride for the Brain Delivery via Intranasal Route. PNT. 2018;6(1):69–78. doi:10.2174/2211738506666180130105919
88. Soni S, Baghel K, Soni ML, Kashaw SK, Soni V. Size-Dependent Effects of Niosomes on the Penetration of Methotrexate in Skin Layers. Future J Pharm Sci. 2024;10(1):1–18. doi:10.1186/S43094-024-00624-2
89. Varshosaz J, Pardakhty A, Hajhashemi VI, Najafabadi AR. Development and Physical Characterization of Sorbitan Monoester Niosomes for Insulin Oral Delivery. Drug Delivery. 2003;10(4):251–262. doi:10.1080/DRD_10_4_251
90. Ammar HO, Ghorab MM, Mahmoud AA, Higazy IM. Lamotrigine Loaded Poly-ɛ-(d,l-Lactide-Co-Caprolactone) Nanoparticles as Brain Delivery System. Eur J Pharm Sci. 2018;115:77–87. doi:10.1016/J.EJPS.2018.01.028
91. Hoseini B, Jaafari MR, Golabpour A, Momtazi-Borojeni AA, Karimi M, Eslami S. Application of Ensemble Machine Learning Approach to Assess the Factors Affecting Size and Polydispersity Index of Liposomal Nanoparticles. Sci Rep. 2023;13(1):18012. doi:10.1038/s41598-023-43689-4
92. Danaei M, Dehghankhold M, Ataei S, et al. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics. 2018;10(2):57. doi:10.3390/pharmaceutics10020057
93. Khallaf RA, Aboud HM, Sayed OM. Surface Modified Niosomes of Olanzapine for Brain Targeting via Nasal Route; Preparation, Optimization, and in vivo Evaluation. J Liposome Res. 2020;30(2):163–173. doi:10.1080/08982104.2019.1610435
94. El-Sayed MM, Hussein AK, Sarhan HA, Mansour HF. Flurbiprofen-Loaded Niosomes-in-Gel System Improves the Ocular Bioavailability of Flurbiprofen in the Aqueous Humor. Drug Dev Ind Pharm. 2017;43(6):902–910. doi:10.1080/03639045.2016.1272120
95. Yang G, Liu Y, Wang H, et al. Bioinspired Core–Shell Nanoparticles for Hydrophobic Drug Delivery. Angew Chem Int Ed. 2019;58(40):14357–14364. doi:10.1002/anie.201908357
96. Passeleu-Le Bourdonnec C, Carrupt P-A, Scherrmann JM, Martel S. Methodologies to Assess Drug Permeation Through the Blood–Brain Barrier for Pharmaceutical Research. Pharm Res. 2013;30(11):2729–2756. doi:10.1007/s11095-013-1119-z
97. Józsa L, Nemes D, Pető Á, et al. Recent Options and Techniques to Assess Improved Bioavailability: in vitro and ex vivo Methods. Pharmaceutics. 2023;15(4):1146. doi:10.3390/PHARMACEUTICS15041146
98. Ozsoy Y, Gungor S, Cevher E. Nasal Delivery of High Molecular Weight Drugs. Molecules. 2009;14(9):3754–3779. doi:10.3390/molecules14093754
99. Wong AD, Ye M, Levy AF, Rothstein JD, Bergles DE, Searson PC. The Blood-Brain Barrier: an Engineering Perspective. Front Neuroeng. 2013;6. doi:10.3389/fneng.2013.00007
100. Henriques P, Bicker J, Silva S, Doktorovová S, Fortuna A. Nasal-PAMPA: a Novel Non-Cell-Based High Throughput Screening Assay for Prediction of Nasal Drug Permeability. Int J Pharm. 2023;643:123252. doi:10.1016/j.ijpharm.2023.123252
101. Taymouri S, Varshosaz J. Effect of Different Types of Surfactants on the Physical Properties and Stability of Carvedilol Nano-Niosomes. Adv Biomed Res. 2016;5(1):48. doi:10.4103/2277-9175.178781
102. Imran M, Titilayo B, Adil M, et al. Ascorbyl Palmitate: a Comprehensive Review on Its Characteristics, Synthesis, Encapsulation and Applications. Process Biochem. 2024;142:68–80. doi:10.1016/J.PROCBIO.2024.04.015
103. Mekasha YT, Chali BU, Feissa AB, Godena GH, Hassen HK, Wega SS. Quality Evaluation of the Azithromycin Tablets Commonly Marketed in Adama, and Modjo Towns, Oromia Regional State, Ethiopia. PLoS One. 2023;18(3):e0282156. doi:10.1371/JOURNAL.PONE.0282156
104. Mazumder R, Mahanti B, Majumdar S, Pal R, Chowdhury AD. Response Surface Method for Optimization of Prepared Satranidazole Powder Layered Pellets. Future J Pharm Sci. 2021;7(1):1–11. doi:10.1186/S43094-021-00337-W
105. Öztürk AA, Yenilmez E, Yazan Y. Dexketoprofen Trometamol-Loaded Eudragit ® RL 100 Nanoparticle Formulation, Characterization and Release Kinetics. Acta Pharma Sci. 2019;57:1. doi:10.23893/1307-2080.APS.05705
106. Ekenna IC, Abali SO. Comparison of the Use of Kinetic Model Plots and DD Solver Software to Evaluate the Drug Release from Griseofulvin Tablets. J Drug Delivery Ther. 2022;12(2–S):5–13. doi:10.22270/JDDT.V12I2-S.5402
107. Basiri L, Rajabzadeh G, Bostan A. Physicochemical Properties and Release Behavior of Span 60/Tween 60 Niosomes as Vehicle for α-Tocopherol Delivery. LWT. 2017;84:471–478. doi:10.1016/j.lwt.2017.06.009
108. Elfadl AA, Boughdady M, Meshali M. New PeceolTM/SpanTM 60 Niosomes Coated with Chitosan for Candesartan Cilexetil: perspective Increase in Absolute Bioavailability in Rats. Int J Nanomed. 2021;16:5581–5601. doi:10.2147/IJN.S324171
109. Shi Q, Li F, Yeh S, Wang Y, Xin J. Physical Stability of Amorphous Pharmaceutical Solids: nucleation, Crystal Growth, Phase Separation and Effects of the Polymers. Int J Pharm. 2020;590:378–5173. doi:10.1016/j.ijpharm.2020.119925
110. Chen Q, Ji Y. Thermodynamic Mechanism of Physical Stability of Amorphous Pharmaceutical Formulations. Ind Eng Chem Res. 2023;62(3):1596–1605. doi:10.1021/ACS.IECR.2C02953/ASSET/IMAGES/LARGE/IE2C02953_0012.JPEG
111. Jüptner A, Scherließ R. Investigation of Powder Properties and Application Aspects Impacting Nasal Deposition of Spray-Dried Powders in a Nasal Cast. Eur J Pharm Biopharm. 2025;2025:114666. doi:10.1016/j.ejpb.2025.114666
112. Doub WH, Suman JM, Copley M, Goodey AP, Hosseini S, Mitchell JP. Laboratory Performance Testing of Aqueous Nasal Inhalation Products for Droplet/Particle Size Distribution: an Assessment from the International Pharmaceutical Aerosol Consortium on Regulation and Science (IPAC-RS). AAPS Pharm Sci Tech. 2023;24(7):1–13. doi:10.1208/S12249-023-02665-X/TABLES/4
113. Pina Costa C, Nižić Nodilo L, Silva R, et al. In Situ Hydrogel Containing Diazepam-Loaded Nanostructured Lipid Carriers (DZP-NLC) for Nose-to-Brain Delivery: development, Characterization and Deposition Studies in a 3D-Printed Human Nasal Cavity Model. Int J Pharm. 2023;644:123345. doi:10.1016/J.IJPHARM.2023.123345
114. Dayal P, Shaik MS, Singh M. Evaluation of Different Parameters That Affect Droplet‐size Distribution from Nasal Sprays Using the Malvern Spraytec®. J Pharm Sci. 2004;93(7):1725–1742. doi:10.1002/jps.20090
115. Casula E, Manconi M, Vázquez J, et al. Design of a Nasal Spray Based on Cardiospermum Halicacabum Extract Loaded in Phospholipid Vesicles Enriched with Gelatin or Chondroitin Sulfate. Molecules. 2021;26(21):6670. doi:10.3390/molecules26216670
116. Patterlini V, Guareschi F, D’Angelo D, et al. Clinically Relevant Characterization and Comparison of Ryaltris and Other Anti-Allergic Nasal Sprays. Pharmaceutics. 2024;16(8). doi:10.3390/PHARMACEUTICS16080989/S1
117. Abourehab MAS, Khames A, Genedy S, et al. Sesame Oil-Based Nanostructured Lipid Carriers of Nicergoline, Intranasal Delivery System for Brain Targeting of Synergistic Cerebrovascular Protection. Pharmaceutics. 2021;13(4):581. doi:10.3390/pharmaceutics13040581
118. League-Pascual JC, Lester-McCully CM, Shandilya S, et al. Plasma and Cerebrospinal Fluid Pharmacokinetics of Select Chemotherapeutic Agents Following Intranasal Delivery in a Non-Human Primate Model. J Neurooncol. 2017;132(3):401–407. doi:10.1007/s11060-017-2388-x
119. Huang Q, Chen X, Yu S, Gong G, Shu H. Research Progress in Brain-Targeted Nasal Drug Delivery. Front Aging Neurosci. 2023;15:1341295. doi:10.3389/FNAGI.2023.1341295/BIBTEX
120. Formica ML, Real DA, Picchio ML, Catlin E, Donnelly RF, Paredes AJ. On a Highway to the Brain: a Review on Nose-to-Brain Drug Delivery Using Nanoparticles. Appl Mater Today. 2022;29:101631. doi:10.1016/J.APMT.2022.101631
121. Pokorski M, Marczak M, Dymecka A, Suchocki P. Ascorbyl Palmitate as a Carrier of Ascorbate into Neural Tissues. J Biomed Sci. 2003;10(2):193–198. doi:10.1007/BF02256054
122. Pokorski M, Marczak M. Stability of Ascorbyl Palmitate Molecule in the Rat. J Physiol Pharmacol. 2005;56:197.
123. Tuesuwan B, Mueannoom W, Jamnongtanachot P, et al. Basis to Aid Crisis: favipiravir Oral Solution for Hospital Compounding During COVID-19 Drug Shortage. J Pharm Sci. 2023;112(2):610–617. doi:10.1016/j.xphs.2022.10.026
124. Ammar HO, Haider M, Ibrahim M, El Hoffy NM. In vitro and in vivo Investigation for Optimization of Niosomal Ability for Sustainment and Bioavailability Enhancement of Diltiazem after Nasal Administration. Drug Deliv. 2017;24(1):414–421. doi:10.1080/10717544.2016.1259371
125. Mane SD, Thoh M, Sharma D, Sandur SK, Naidu KA. Ascorbyl Stearate Promotes Apoptosis Through Intrinsic Mitochondrial Pathway in HeLa Cancer Cells. Anticancer Res. 2016;36(12):6409–6417. doi:10.21873/ANTICANRES.11238
126. Zhang L, Li G, Lin B, He H, Zhou R, Jiang W. Ascorbyl Palmitate Ameliorates Inflammatory Diseases by Inhibition of NLRP3 Inflammasome. Int Immunopharmacol. 2024;131. doi:10.1016/J.INTIMP.2024.111915
127. Varshosaz J, Sadrai H, Alinagari R. Nasal Delivery of Insulin Using Chitosan Microspheres. J Microencapsul. 2004;21(7):761–774. doi:10.1080/02652040400015403
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.