")
Back to Journals » Journal of Inflammation Research » Volume 17
Prevention and Treatment Strategies for Alzheimer’s Disease: Focusing on Microglia and Astrocytes in Neuroinflammation
Authors Zhang S , Gao Z, Feng L , Li M
Received 19 June 2024
Accepted for publication 2 October 2024
Published 13 October 2024 Volume 2024:17 Pages 7235—7259
DOI https://doi.org/10.2147/JIR.S483412
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Shenghao Zhang,1,* Zhejianyi Gao,2,* Lina Feng,1,3 Mingquan Li1
1Department of Neurology, The Third Affiliated Hospital of Changchun University of Chinese Medicine, Changchun, Jilin Province, 130021, People’s Republic of China; 2Department of Orthopaedics, Fushun Hospital of Chinese Medicine, Fushun, Liaoning Province, 113008, People’s Republic of China; 3Shandong Key Laboratory of TCM Multi-Targets Intervention and Disease Control, The Second Affiliated Hospital of Shandong First Medical University, Taian, Shandong Province, 271000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lina Feng; Mingquan Li, Department of Neurology, the Third Affiliated Hospital of Changchun University of Chinese Medicine, Changchun, Jilin, 130117, People’s Republic of China, Tel +86-15543120222 ; +86-15948000577, Email [email protected]; [email protected]
Abstract: Alzheimer’s disease (AD) is a fatal neurodegenerative disease characterized by its insidious onset and progressive development, making it the most common form of dementia. Despite its prevalence, the exact causes and mechanisms responsible for AD remain unclear. Recent studies have highlighted that inflammation in the central nervous system (CNS) plays a crucial role in both the initiation and progression of AD. Neuroinflammation, an immune response within the CNS triggered by glial cells in response to various stimuli, such as nerve injury, infection, toxins, or autoimmune reactions, has emerged as a significant factor alongside amyloid deposition and neurofibrillary tangles (NFTs) commonly associated with AD. This article aims to provide an overview of the most recent research regarding the involvement of neuroinflammation in AD, with a particular focus on elucidating the specific mechanisms involving microglia and astrocytes. By exploring these intricate processes, a new theoretical framework can be established to further probe the impact of neuroinflammation on the development and progression of AD. Through a deeper understanding of these underlying mechanisms, potential targets for therapeutic interventions and novel treatment strategies can be identified in the ongoing battle against AD.
Keywords: alzheimer’s disease, neuroinflammation, microglia, astrocyte, review
Introduction
Alzheimer’s disease (AD), also known as senile dementia, is a progressive neurodegenerative disease that impacts the CNS in the elderly population. Presently, approximately 10 million individuals in China are living with AD, and estimates suggest that this figure will exceed 40 million by 2050. AD is characterized by cognitive decline, alterations in behaviour, and a decreased capacity to perform daily activities.1 The main pathological hallmarks include cortical atrophy, neuronal loss in memory regions, and the accumulation of β-amyloid protein (Aβ) and NFTs.2 In recent years, the number of AD cases has increased; however, the exact causes of this disease and its underlying mechanisms remain incompletely elucidated. The “β-amyloid cascade theory” and the “Tau theory” are widely recognized for elucidating the progression of AD. Nevertheless, the continuous disappointment with therapeutic approaches targeting Aβ has raised scepticism regarding the dominance of the “Aβ cascade theory”. Additionally, as research advances, neuroinflammation, disturbed calcium homeostasis, malfunctioning mitochondria, and defective autophagy pathways have emerged as closely linked to the initiation of AD.3 Increased levels of neuroinflammatory markers have been detected in the cerebral cortex and cerebrospinal fluid (CSF) of AD patients, indicating persistent inflammatory responses within the brain. Through the identification of AD risk genes related to innate immune responses and insights from pertinent autopsies, neuroinflammation significantly contributes to AD development.4,5
Different phenotypes of microglia play different roles at different stages of AD. While microglia can phagocytose Aβ during the pathological progression of AD,6 excessive activation of microglia and persistent inflammatory responses can worsen the course of the disease.7 Peripheral blood immune cells, such as monocytes, macrophages, neutrophils, and natural killer cells, also contribute to the development of AD. Notably, memory CD8+ T cells are present in the brain and CSF of AD patients, and their increased abundance correlates with cognitive decline.8 These cells tend to aggregate in Aβ-rich regions, indicating a potential role for adaptive immunity in the progression of AD. A 2020 study in Nature highlighted the role of interferon-inducible transmembrane protein 3 (IFITM3) in enhancing γ-secretase activity, which promotes the formation of Aβ. This study provides direct evidence of the involvement of the immune response in Aβ generation.9 T cells, including their subpopulations, have been shown to breach the blood‒brain barrier (BBB) in AD patients, contributing to neuroinflammation. Additionally, the neurodegeneration associated with the Tau protein impacts the pathological manifestations and cognitive decline in AD patients.10 This complex interaction between immune cells, inflammatory responses, and pathological proteins underscores the intricate mechanisms involved in the progression of AD. To date, seven drugs have been approved by the Food and Drug Administration (FDA) to treat AD. These medications include three acetylcholinesterase inhibitors (galantamine, donepezil, and cabalatin), the N-methyl-D-aspartate receptor antagonist (NMDAR-A) known as memantine, and the combination of memantine and donepezil. Furthermore, two monoclonal antibodies targeting Aβ, aducanumab and lecanemab, were approved in January 2021 and 2023, respectively. Notably, certain treatments may improve AD symptoms, yet they are not capable of reversing the disease. The effectiveness of other treatment options has not been verified in clinical trials and could worsen cognitive function in individuals with AD. The cause of AD is still not fully understood, highlighting the necessity for the development of medications that can either alleviate or cure the symptoms of AD. Neuroinflammation has been identified as a crucial process in AD and could be targeted for new treatment strategies. This paper examines recent research on neuroinflammation, with a specific focus on microglia and astrocytes in the context of AD pathogenesis and the goal of presenting novel treatment approaches for AD (Figure 1).
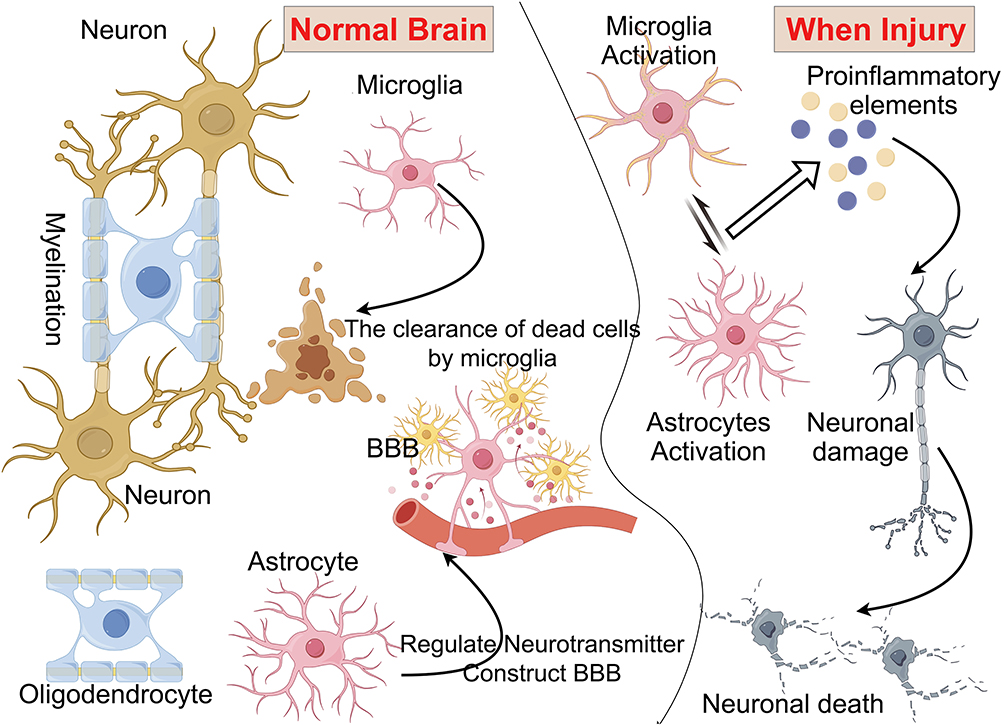 |
Figure 1 The function of microglia and astrocytes in the cerebral nervous system. (Brain homeostasis relies on the interactions between various cell types, particularly microglia and astrocytes, which play crucial roles in numerous neuronal functions. In the adult brain, these cells are involved in neuromodulation, surveillance, synaptic plasticity, learning and memory, as well as maintaining the integrity of BBB. They also contribute to metabolic coupling, ion buffering, neurotransmitter homeostasis, production of neuroactive factors such as ATP and TNF-α, and the normal functioning of circuits that regulate neuronal synchronization and synapses. Additionally, microglia and astrocytes can act as local immune cells, participating in inflammatory responses). Some cartoon components were from www.figdraw.com for model drawing. |
Current Clinical Research Status of Anti-Neuroinflammation Treatments for AD
The emergence of anti-Aβ monoclonal antibodies has generated considerable interest in the treatment of AD focused on Aβ. Treatment strategies target the reduction of Aβ production (via BACE-1 inhibitors and gamma-secretase inhibitors), hinder the aggregation of Aβ, and enhance its clearance11 (by promoting Aβ metabolism or employing active/passive immunity strategies against Aβ).12 Since Aβ accumulation leads to permanent damage to the nervous system before clinical symptoms manifest, the use of active Aβ immunotherapy is more appropriate for early intervention rather than for managing high-risk AD patients.13 Monoclonal antibodies targeting Aβ in passive immunization strategies can interact with various forms of Aβ. Several monoclonal antibodies, such as Crenezumab, Donanemab, and Gantenerumab, have been developed to address different forms of Aβ, including those for oligomers, while others like bapineuzumab and solanezumab are intended for monomers. Moreover, antibodies such as eculizumab and lecanemab focus on soluble fibrils, with aducanumab specifically aimed at Aβ fibrils and soluble oligomers. In June 2021, the US FDA granted marketing approval for aducanumab, setting a precedent for the authorization of additional disease-modifying therapies driven by biomarker responses.14 Following this, the Phase III Clarity-AD trial of lecanemab reported in January 2023 a 27% decrease in cognitive decline and other functional impairments over a period of 18 months, thereby achieving its primary endpoint.15 Additionally, in May 2023, Lilly announced favorable results from the phase III TRAILBLAZER-ALZ2 trial of donanemab, indicating a notable slowdown in cognitive decline among individuals with early symptomatic AD.16 These medications have shown promise in experimental phases, underscoring the essential function of the immune response in AD and its potential in shaping future therapeutic options.
Many of the therapies presently undergoing clinical trials aimed at enhancing Tau protein clearance focus on immunotherapy. These trials remain at the initial phases of clinical research. Anti-Tau protein vaccines, employing active immunity approaches, seek to activate the patient’s immune system to generate antibodies against the harmful Tau protein. This objective is usually accomplished through the use of synthetic peptides that imitate pathological Tau protein epitopes as antigens.17 For example, AADvac1 has showcased remarkable immunogenicity, with 98.2% of participants generating specific anti-Tau protein antibodies. Evidence indicates that it can significantly reduce brain atrophy in AD patients, enhance cognitive function, and display favorable safety and tolerability profiles.18,19 Gosuranemab (BIIB092) showed promising safety and tolerability during Phase I clinical trials while also revealing potential for disease modification. Nonetheless, the Phase II clinical trial was halted due to insufficient efficacy (NCT03352557).20 Phase II clinical trials are currently evaluating the safety and efficacy of Tilavonemab (ABBV-8E12, NCT02880956) and Zagotenemab (LY3303560, NCT03518073) in individuals with early AD, although results are still awaited for publication.21 Semorinemab (RO7105705) demonstrated favorable safety in phase II clinical trials, but did not exhibit significant efficacy in patients experiencing prodromal to mild AD.22 Despite the potential of these antibodies, their overall effectiveness remains uncertain. Active immunity strategies targeting the N-terminus of Aβ42, including CAD106, ACC-001 with Aβ1-7, and ACI-24 with Aβ1-15 epitopes, can produce Aβ-specific antibodies without the activation of T-cells, yet there is a risk of inducing aseptic meningitis.23,24 Both bapineuzumab and solanezumab encountered challenges in phase III clinical trials in 2012 and 2016, respectively.25
Aducanumab is recognized as the first monoclonal antibody approved by the FDA for the treatment of AD.26 This medication has shown effectiveness in decreasing Aβ accumulation in the cerebral cortex of patients with AD, with its effects dependent on both the dosage and treatment duration. It demonstrates promise in reducing cognitive decline in individuals experiencing mild AD or mild cognitive impairment (MCI). However, elevated dosage levels may result in amyloid-related imaging abnormalities (ARIA), which can include edema and effusion.26 On January 6, 2023, lecanemab-irmb received accelerated approval for marketing in the United States, making it the second Aβ monoclonal antibody to gain global marketing authorization after Aducanumab. This antibody also poses risks for ARIA that can involve edema, effusion, and bleeding.27 High doses of Crenezumab have shown encouraging clinical outcomes for patients with mild AD.28,29 Nonetheless, in phase III clinical studies, the higher doses of Crenezumab have not yet proven to exhibit significant benefits in those with prodromal AD.30 A phase II clinical trial revealed that Donanemab significantly lowered levels of plasma pTau-217 and glial fibrillary acidic protein (GFAP) in patients with early-stage AD, resulting in enhancements in cognitive function and daily activity ratings, although ARIA with edema and effusion was noted as the main adverse effect.31 Consequently, additional research is warranted to elucidate the clinical advantages of the immune response in AD.
Microglia and ADβ
Microglia in AD
The CNS is primarily composed of microglia, astrocytes, and oligodendrocytes. Microglia, the most abundant mononuclear transitional phagocytes in brain tissue, play a crucial role in the immune response and defence mechanisms of the brain.32,33 In their resting state, microglia undergo immune surveillance via their branched structure. However, when activated due to disease or injury, they transform into an amoeboid shape and serve as inflammatory cells within the CNS. This activation involves a transition from a resting state to an activated state of classically activated (M1) microglia, where proinflammatory factors and toxic substances are released to eliminate pathogens. Clinical studies have shown that overactivation of M1 microglia can lead to neuronal dysfunction, injury, and degeneration, particularly in conditions such as AD. Alternatively activated (M2) microglia function as phagocytes, clearing harmful substances such as bacteria, dead cells, and aggregated proteins that can affect the CNS. In addition to their phagocytic role, M2 microglia also secrete soluble factors such as chemoattractants, cytokines, and neurotrophins. These substances contribute to the immune response, repair damaged brain tissue, and provide neuroprotection in the CNS. By understanding the roles of both M1 and M2 microglia in the immune response and maintenance of brain health, researchers and clinicians can better target interventions for conditions that involve neuroinflammation and neurodegeneration.
In recent years, numerous genome-wide association studies (GWASs) conducted on clinical samples from AD patients have shown that more than half of the confirmed genetic variations in AD-related genes are associated with innate immune function and microglial function. Specifically, genes such as triggering receptor expressed on myeloid cells-2 (TREM2), CD33, apolipoprotein E4 (APOE4), complement component receptor 1 (CR1), and human leukocyte antigen-DR isotype beta-1 (HLA-DRB1) have been implicated. Studies have indicated that surface receptors on microglia, including TREM2 and Toll-like receptors (TLRs), become active under pathological conditions such as AD and bind to Aβ and APOE prior to migrating to the injury site.34,35 In October 2020, researchers discovered a new gene, FAM171A2, with the ability to control progranulin (PGRN) production in endothelial cells lining blood vessels. PGRN, a secreted glycoprotein, participates in crucial biological processes, such as neural development, regeneration, neuroinflammation, and autophagy. Moreover, irregularities in PGRN regulation are associated with neurodegenerative diseases such as AD, Parkinson’s disease, and frontotemporal dementia.36 Furthermore, some scientists hypothesize that microglia can detect Aβ plaques during the initial stages of AD progression, encircling the plaques to form a physical barrier, which could hinder further spread of neurotoxicity. These findings suggest that neuroinflammation mediated by microglia plays a pivotal role in the development of AD and is potentially influenced by the wide-ranging diversity and functional variability of microglia that are worsened by AD.
Trem2
The TREM2 gene encodes a protein that acts as an innate immune cell receptor and is primarily located on the surface of myeloid cells, with predominant expression on microglia in the brain.37 It is also localized in the intracellular golgi apparatus and plays a crucial role in regulating the energy metabolism and phenotype of microglia. Currently, TREM2 is being explored as a potential therapeutic target for AD. Research has linked the TREM2 gene variant R47H to AD, and its impact on late-onset AD is comparable to that of APOEε4. In vitro, the common variant of TREM2 has been shown to interact with anionic lipids, whereas the R47H mutation inhibits this binding ability. In one study, scientists developed transgenic mice expressing a common human variant or R47H TREM2 while lacking endogenous TREM2 in the 5XFAD AD model.38 The results indicated that only the common variant transgene could restore Aβ-induced microgliosis and microglial activation, suggesting that R47H impairs TREM2 function in vivo.38 TREM2 is a single-channel transmembrane receptor of the immunoglobulin superfamily. It consists of an immunoglobulin-containing extracellular domain, a transmembrane domain, and a short cytoplasmic tail region. This receptor binds to immunoglobulin domain-binding pathogen-associated molecular pattern (PAMP) molecules, damage-associated molecular pattern (DAMP) molecules, cell debris, lipids, and apolipoproteins. When cleaved by the ADAM family enzymes ADAM10 and ADAM17, a secreted variant of the TREM2 extracellular domain, sTREM2, is released. This cleavage-mediated activation stops the TREM2 signalling cascade, thereby playing a crucial role in microglial survival and proinflammatory signalling.
The transmembrane domain transmits intracellular signals by binding to the ligand transmembrane immune signalling adaptor (TYROBP or DAP12), also known as the 12 kDa DNAX-activating protein. DAP12 is a signal transduction adaptor protein expressed in cells involved in the innate immune response and contains an immune receptor tyrosine-based activation motif (ITAM). When TREM2 is activated through extracellular ligand binding, it subsequently activates DAP12, initiating a cascade of intracellular signalling events. Lysine on TREM2 interacts with aspartic acid on DAP12 via electrostatic interactions. Upon ligand binding to TREM2, the tyrosine residue on DAP12 becomes phosphorylated, leading to the recruitment of spleen tyrosine kinase (Syk), triggering a series of tyrosine phosphorylation events and activating downstream mediators such as phospholipase Cγ2 (PLCγ2), phosphatidylinositol 3-kinase (PI3K), mammalian target of rapamycin (mTOR), and mitogen-activated protein kinase (MAPK),39 This signalling ultimately results in cell activation (Figure 2). Functional studies have shown that the absence of TREM2 impairs proper microglial responses to damage and cues that typically induce cell movement. In ex vivo brain slice assays, the lack of TREM2 reduces the migration distance of microglia.40 According to in vivo experiments, microglia lacking TREM2 exhibit decreased migration towards injected apoptotic neurons. Furthermore, the extension of microglial processes towards areas of laser-induced focal CNS injury in the somatosensory cortex is delayed.40 These findings suggest that alterations in TREM2 at the gene level impact microglial function.
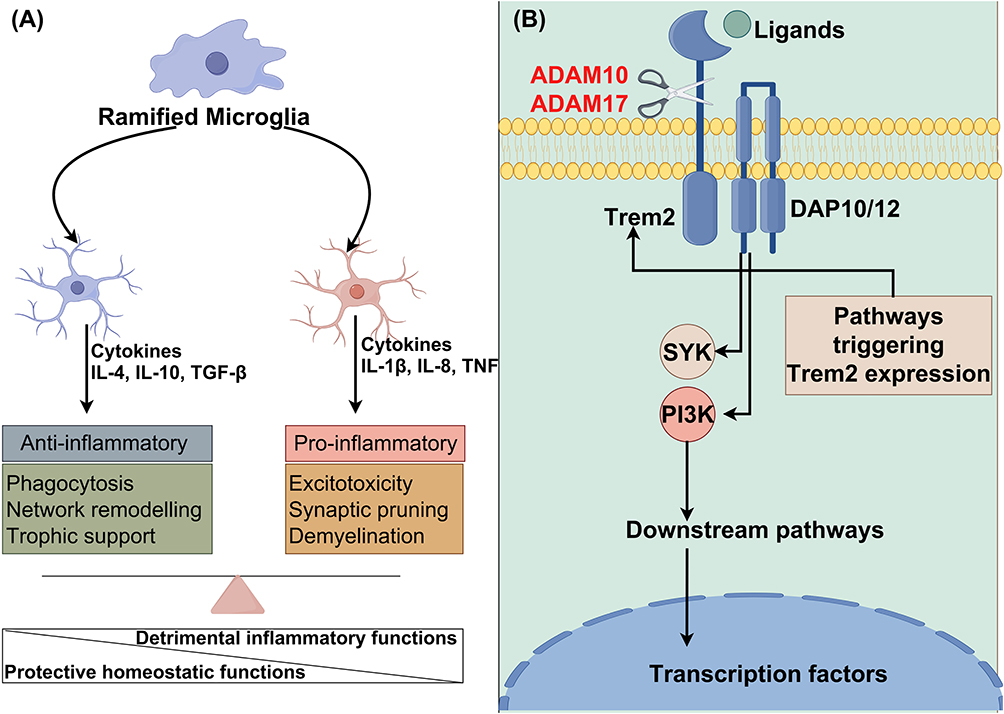 |
Figure 2 A schematic diagram illustrating the interaction between neuroinflammation and microglial activation, as well as a separate schematic diagram depicting the TREM2 signaling pathway. (A): The interaction between neuroinflammation and microglial activation; (B) TREM2 signaling pathway. TREM downstream signaling regulation is contingent upon the activity status of DAP10 and DAP12. Following the binding of TREM2 to its ligand, the co-receptor molecule undergoes phosphorylation and subsequently recruits intracellular signal transduction molecules (DAP12 associates with Syk, while DAP10 associates with PI3K); Syk: spleen tyrosine kinase; PI3K: phosphatidylinositol 3-kinase). Some cartoon components were from www.figdraw.com for model drawing. |
TREM2 in AD
In the pathogenesis of AD, the accumulation of Aβ in the brain is often accompanied by gliosis and lipid deposition, leading to the continuous progression of neuroinflammation. Microglia play a crucial role in recognizing Aβ through receptors such as TREM2, TLRs, and CR1, initiating a signalling cascade that prompts microglia to migrate to the lesion site.41–44 Subsequently, soluble Aβ and tau proteins are engulfed by microglia through phagocytosis and other mechanisms. Upon fusion with lysosomes, the vesicles and their contents are degraded, reducing Aβ levels, slowing senile plaque formation, preventing NFT production, and potentially delaying the onset of AD.45 Senile plaques are abnormal neural patches resulting from Aβ deposition in the brain, with Aβ 39–42 being a protein fragment derived from β-amyloid precursor protein (APP) cleavage by β-secretase and γ-secretase. Aβ exists in monomeric, oligomeric, and fibrillar forms, with oligomeric Aβ primarily contributing to cognitive decline and neurodegeneration in AD. Aβ oligomers can bind to various receptors and modulate nerve cell function through distinct signalling pathways. Research findings indicate that microglia abundant in TREM2 play a critical role in surrounding and compacting early amyloid fibrils and plaques. In mice lacking TREM2 or DAP12, as well as in individuals with the R47H mutation in the TREM2 gene, microglia exhibited a significantly diminished capacity to encase amyloid deposits. Consequently, an increase in the formation of more diffuse plaques containing elongated and branched amyloid fibrils was observed, leading to increased exposure of neighbouring neurites. This change was accompanied by increased neuritic tau hyperphosphorylation and axonal degeneration around amyloid deposits.46 The extracellular domain of TREM2 is responsive to the accumulation of Aβ in AD, as it can bind to various forms of Aβ, showing the highest affinity for Aβ oligomers.47,48 When TREM2 is knocked out, the proliferation and activation of microglia near the lesion area are impaired, preventing microglia from fulfilling their normal protective role in the brain in terms of both quantity and effectiveness. This inhibitory effect exacerbates the damage caused by the disease. In mouse models of AD, microglia lacking TREM2 are unable to multiply and form clusters around the Aβ plaques associated with the disease.38 Additionally, TREM2-deficient N9 microglial cell lines, macrophages, and primary microglia all show significantly decreased uptake of antibody-bound Aβ, resulting in reduced clearance of amyloid plaques.49
A decrease in APOE and APOC1 expression was observed in TREM2-null human microglia. The differentially expressed genes were enriched in pathways related to “calcium signalling pathway regulation”, “ERK1/ERK2 cascade”, and “cell migration”. Upon activation of the TREM2 pathway, the downregulated genes in TREM2-null microglia were significantly upregulated, leading to a shift in enriched pathways to “positive regulation of leukocyte chemotaxis”, “cellular response to tumour necrosis factor”, and ‘positive regulation of ERK1 and ERK2 cascades’.50 TREM2 plays a role in maintaining cellular energy metabolism and biosynthesis in microglia, impacting the pathological process of AD.51 Previous studies in TREM2 knockout 5XFAD mice revealed an increase in autophagic vesicles in microglia, which was mitigated by cyclocreatine supplementation to enhance energy storage, indicating an energy metabolism disorder in TREM2 knockout mice.51 Additionally, TREM2 enhances microglial survival through the Wnt/β-catenin signalling pathway, suggesting that strategies targeting the TREM2/β-catenin signalling pathway could hold promise for AD treatment. Furthermore, research has identified lipoprotein particles such as LDL and apolipoproteins such as CLU/APOJ and APOE as ligands of TREM2,49 with disease-associated mutations such as those in AD hindering or reducing the binding of these ligands by TREM2. Aβ binds to lipoproteins, which are then efficiently taken up by microglia in a manner dependent on TREM2.49 Microglia without Trem2 showed reduced internalization of LDL and CLU. Moreover, macrophages from individuals with a TREM2 AD variant exhibited decreased absorption of Aβ–lipoprotein complexes.49 These results indicate a link between three genetic risk factors for AD and provide insights into a possible mechanism by which mutant TREM2 increases the risk of AD.
Microglia play a crucial role in controlling synaptic remodelling within the CNS. Activation of the classical complement pathway facilitates microglia-driven synaptic pruning both in developmental stages and during disease progression.52 Studies using conditional knockout mice have revealed that the specific deletion of SIRPα in microglia leads to a reduction in synaptic density.52 In human tissue, a decrease in microglial SIRPα expression correlates with the progression of AD.52 CD47, which shields synapses from excessive pruning during development, indicates the involvement of microglial SIRPα, a receptor for CD47, in the process of synaptic remodelling.52 Furthermore, the absence of microglial SIRPα results in increased synapse loss due to microglial engulfment, ultimately leading to increased cognitive impairment.52 A study revealed that the extracellular domain of TREM2 hinders the activation of the classical complement cascade by binding strongly to complement component 1q (C1q), the initiator of the classical complement pathway.53 In postmortem brain tissues from AD patients, TREM2 and C1q form protein complexes. The number of these complexes is inversely related to the deposition level of complement protein C3 but is positively associated with the level of synaptin.53 Researchers utilized a TREM2-null mouse model of AD and found that in the early stages of the disease, Trem2 gene deletion did not impact the deposition of AD pathological proteins or the morphology or number of microglia.53 However, complement activation in the brain was significantly increased, leading to increased microglial phagocytosis of synapses and exacerbated synaptic loss, suggesting that TREM2 plays a role in complement-mediated synaptic loss in AD pathology.53 sTREM2 can traverse the BBB and is elevated in the CSF of patients with various neurological diseases, including AD, Parkinson’s disease with PGRN mutations, and natural ageing in cognitively normal individuals. The level of sTREM2 reflects microglial activation and neuronal degeneration, serving as a useful marker for detecting AD.54 Furthermore, TREM2-deficient microglia do not exhibit vesicular phagocytosis of APOE.50
Studies have shown that sTREM2 can decrease the apoptosis of microglia through the activation of the AKT-GSK3β-β-catenin signalling pathway and enhance the survival of microglia via the PI3K/Akt signalling pathway. An infusion of the sTREM2-Fc protein into the hippocampus of normal mice and TREM2 knockout mice upregulates the expression of inflammatory factors, alters the morphology of microglia, and boosts the immune response. These findings indicate that enhancing the sTREM2 signalling pathway could serve as a potential treatment for AD.55 Furthermore, the direct administration of the recombinant sTREM2 protein or the overexpression of sTREM2 in 5xFAD mice increases the proliferation and phagocytic activity of microglia surrounding Aβ plaques, leading to accelerated clearance of Aβ plaques.55 A study examining CSF sTREM2 levels within the amyloid/tau/neurodegeneration (A/T/N) classification framework revealed that elevated CSF sTREM2 levels in early disease stages are associated with increased levels of biomarkers of tau pathology and neurodegeneration. Conversely, lower CSF sTREM2 levels are linked to Aβ deposition in the absence of tau deposition and neurodegeneration. Additionally, research has indicated that microglial phagocytosis may not always be beneficial, particularly in specific clinical stages. In individuals with pathological changes characteristic of AD but minimal or no clinical symptoms, increased microglial phagocytosis has been shown to decrease Aβ levels and slow the production of senile plaques (SP) and NFTs.45 However, due to a reduced number of lysosomes, the phagocytic capacity of microglia diminishes after the engulfment of Aβ and tau.56 Following phagocytosis, inflammatory vesicles generated by cell metabolism, including NLRP3 and cytokines such as TNF-α and IL-1, trigger microglial polarization towards a proinflammatory state. This shift leads to the release of proinflammatory cytokines, exacerbating inflammation, inhibiting Aβ phagocytosis, and furthering Tau protein pathology.57,58
Microglia Autophagy in AD
The pathway involving autophagy and lysosomes plays a crucial role in cellular catabolism, facilitating the removal and recycling of damaged proteins, protein aggregates, and organelles.59 Beyond its well-known function in maintaining protein homeostasis and ensuring quality control, recent studies suggest that autophagy serves as a significant regulator of inflammatory responses. Evidence of the interaction between autophagy and inflammation has been recorded in conditions associated with both organ and systemic inflammation, including inflammatory bowel diseases,60 type 2 diabetes, heart disorders, and cystic fibrosis.61 Additionally, numerous autophagy-related genes are connected to the progression of autoimmune diseases.62 In neurodegenerative diseases, microglial autophagy is both impaired and downregulated.63 The transition from the M1 to the M2 phenotype can protect the body from excessive inflammatory injury, with autophagy being one of the mechanisms that influence macrophage polarization.64 Research demonstrated that microglial autophagy facilitates the transformation of microglia from a proinflammatory state to a beneficial anti-inflammatory state, while also inhibiting NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome-mediated inflammatory responses, thereby acting as a negative regulator.65
Dysfunctional autophagy has been linked to the development of AD. Previous research indicated that various stages of the autophagy-lysosomal pathway are disrupted in neurons affected by the disease. Choi et al66 demonstrate that autophagy is activated in microglia, especially in disease-associated microglia around Aβ. The inhibition of autophagic processes in microglia leads to their detachment from Aβ, a reduction in disease-associated microglia, and exacerbation of the neuropathology observed in AD mice. Mechanistically, a deficiency in autophagy fosters the emergence of senescence-associated microglia, as indicated by decreased cell proliferation, elevated levels of Cdkn1a/p21Cip1, dystrophic cellular shapes, and the presence of a senescence-associated secretory phenotype.66 Qin et al67 demonstrated that cognitive function was enhanced in the 5XFAD/NLRP3-KO mouse model through the reduction of the pro-inflammatory response of microglia, coupled with the preservation of their phagocytic and clearance capabilities concerning deposited Aβ plaques. Ubiquitinated NLRP3 can be recognized by p62 and subsequently degraded via the autophagy-lysosome pathway, which mitigates the pro-inflammatory activity and pyroptosis of microglia. Additionally, the expression of autophagy pathway-related proteins, such as LC3B/A and p62, was elevated in the in vitro AD model.67 Elevated risk of developing AD is associated with hypomorphic variants of TREM2, a surface receptor essential for microglial responses to neurodegeneration, including proliferation, survival, clustering, and phagocytosis. Ulland et al51 demonstrated that microglia from AD patients carrying TREM2 risk variants, as well as TREM2-deficient mice exhibiting AD-like pathology, possess an abundance of autophagic vesicles. Similarly, TREM2-deficient macrophages under conditions of growth-factor limitation or endoplasmic reticulum (ER) stress also display numerous autophagic vesicles. Recent studies indicate that autophagy in microglia plays a protective role for neurons against neurodegeneration. Postnatal neurogenesis, defined as the generation of new neurons from adult neural stem cells (NSCs), is impaired in patients with AD as well as in AD animal models. However, the extent to which microglial autophagy influences adult NSCs and neurogenesis in these AD animal models remains to be investigated.68 To confirm the roles of autophagy in microglia during postnatal hippocampal neurogenesis, Tang et al generated additional conditional knockout (cKO) mice to delete the autophagy-essential genes Rb1cc1 or Atg14 specifically in microglia. However, these mice did not exhibit defects in neurogenesis within the context of a female AD mouse model. In contrast, the use of a CSF1R antagonist to deplete ATG5-deficient microglia resulted in the restoration of neurogenesis in the hippocampus of 5×FAD mice.68
These studies illustrate the protective role of microglial autophagy in maintaining the homeostasis of amyloid plaques and preventing cellular senescence. The removal of senescent microglia emerges as a promising therapeutic strategy.
Astrocytes and AD
The CNS is primarily composed of two types of cells: neurons and glial cells. Astrocytes are the most abundant, widely distributed, and largest glial cells in the CNS. They play a crucial role in maintaining CNS homeostasis by participating in neuronal signal transduction, providing structural and metabolic support to neurons, regulating the formation of neuronal synapses, and forming a three-component synaptic structure with presynaptic and postsynaptic neurons. Additionally, astrocytes regulate the transmission of synaptic neurotransmitters, release neurotrophic factors to protect neurons, maintain the BBB, and clear Aβ.69 Astrocytes typically serve as the primary regulators of inflammatory responses in the brain. However, in pathological situations, astrocytes undergo a transformation marked by functional, morphological, and molecular changes, becoming reactive astrocytes (RAs). This transformation allows them to respond to neuroinflammation and contributes to the progression of neurodegenerative diseases such as AD.70 For instance, astrocytes in close proximity to amyloid plaques become activated and produce RAs in response to prolonged exposure to these plaques, which are composed of Aβ and Tau proteins. This activation can in turn lead to an increase in Aβ production, ultimately contributing to neuronal death.71 Recent studies have indicated that RAs play a crucial role in amyloid plaques, with a correlation between cognitive decline and increased RA levels.72 Furthermore, emerging research suggests that targeting RAs could be a novel approach for AD treatment. For instance, studies of AD animal models have shown that specific modulation of activated RAs can ameliorate neuropathological changes in AD mice.73 Moreover, RAs have been identified as a potential drug target for AD treatment. By inhibiting RA-induced energy metabolism and oxidative stress in the brains of AD mice through drug interventions, neuroinflammation is mitigated, Aβ deposition is reduced, memory function is enhanced, and AD progression is delayed.74,75
Astrocytes
Astrocytes are the most abundant glial cells in the CNS, with various functions: (1) providing mechanical support and nutrition by filling spaces between neurons and blood vessels, forming bridges between neurons and capillaries, transporting nutrients, and releasing neurotrophic factors;76 (2) serving as barriers and isolating signals from different afferent fibres, protecting neighbouring neurons, and forming the BBB;77 (3) guiding migrating neurons to their final locations; (4) aiding in repair and proliferation after hypoxia, injury, or disease; (5) participating in immune responses by presenting antigens to T lymphocytes; (6) maintaining stable extracellular K+ concentrations by transferring K+ into cells and dispersing it to other glial cells;78 and (7) the metabolism of specific neurotransmitters and active substances by taking up glutamate and γ-aminobutyric acid released by neurons, converting them into glutamine, and transferring them back to neurons.79 Communication between neurons and glial cells is essential for modulating synaptic connection properties in the brain. Astrocytes play a crucial and complex role in synapse development, maintenance, and plasticity, and neurons reciprocally influence astrocyte physiology. Research has shown that Eph receptor tyrosine kinases and ephrins are important for contact-dependent communication between neurons and glia at synapses. Binding of these cell surface proteins initiates bidirectional signalling pathways that regulate the structural and physiological characteristics of both neurons and astrocytes.80
Abnormal Calcium Signalling in AD
Ca2+ acts as a crucial second messenger in eukaryotic cells, playing a role in regulating various cellular functions such as cell proliferation, differentiation, survival, gene expression, and neuronal synaptic activity. It triggers multiple intracellular signals and impacts neuronal function and cellular information transmission.81 Recent studies have shown imbalanced calcium homeostasis in the brains of AD patients and APP/PS1 mice, leading to a significant increase in intracellular Ca2+ levels.82 Elevated Ca2+ in the axoplasm binds to calmodulin (CaM), forming a Ca2+-CaM complex, which activates Ca2+-CaM-dependent protein kinase II (Ca2+-CaM K II) and facilitates neurotransmitter release at synapses.83 RAs, a type of glial cell, lack action potentials and are nonexcitable, yet their membrane potential responds to extracellular K+ concentrations and can be stimulated by changes in intracellular Ca2+ and Na+ levels in neurons.84 Calcium stores within neurons are typically referred to as endoplasmic reticulum (ER) calcium stores. In the normal resting state, the intracellular Ca2+ concentration in neurons is approximately 100 nmol/L. Upon neuronal stimulation, the intracellular Ca2+ concentration rapidly increases to 1–3 µmol/L. This increase in the calcium signal is primarily due to extracellular calcium influx (1~2 mmol/L) and calcium released from the ER (100~800 µmol/L).81 The transient receptor potential A1 (TRPA1) channel is a nonselective cation channel expressed in the terminals of sensory nerves that is highly permeable to Ca2+ and widely distributed in both nerve and nonnerve cells. Activation of TRPA1 channels can occur in response to endogenous inflammatory mediators (such as bradykinin and trypsin) and exogenous noxious stimuli (such as cold, heat, and irritating compounds). An experiment has shown that astrocytes contribute to early Aβ oligomer toxicity by displaying both global and local Ca2+ hyperactivity involving TRPA1 channels, and these cells are associated with neuronal hyperactivity.85
The inositol 1,4,5-trisphosphate (IP3) second messenger system is a widely recognized signal transduction pathway in cells. IP3, a primary form of phosphatidylinositol, is a small, water-soluble molecule resulting from the enzymatic hydrolysis of triphosphoinositide. Under physiological conditions, bioactive substances such as neurotransmitters and peptides bind to G protein-coupled receptors on the cell surface, activating phospholipase C on the plasma membrane to generate IP3 from 4,5-diphosphate phosphatidylinositol. Upon diffusion into the cytoplasm, IP3 triggers IP3 receptors in nonmitochondrial Ca2+ storage sites such as the ER or sarcoplasmic reticulum, leading to the release of Ca2+ from intracellular stores. This rapid increase in the cytosolic Ca2+ concentration initiates the calcium signalling pathway, subsequently eliciting various biological responses, such as cell growth, metabolism, and ion channel activation. Studies have shown that Aβ can influence IP3 receptors in the ER, resulting in the release of Ca2+ from the ER. Shilling et al86 conducted experiments using mice lacking IP3R1 crossed with PS1M146V knock-in (PS1KIN) mice and revealed improved long-term potentiation in the hippocampal CA1-CA3 region in 5-week-old and 3-month-old mice. Through techniques such as patch clamp, calcium imaging, and two-photon imaging, they confirmed that IP3R-mediated calcium release was at least three times higher in the brain slices of PS mutant mice than in those of control mice.87 Further research showed that a 50% decrease in IP3R1 expression can restore abnormal calcium signalling in neurons and reduce amyloid deposition in the brains of PS mutant AD model mice. Moreover, behavioural tests such as the open field test and novel object recognition test have revealed that memory deficits associated with the hippocampus and amygdala can be ameliorated by reducing the expression of IP3Rs.86 APP, a precursor protein of Aβ and a transmembrane protein, is normally cleaved by α-secretase and γ-secretase to generate soluble amyloid precursor protein α (sAPPα) and C83. This process leads to a decrease in intracellular Ca2+ levels.88 PS, an aspartyl protease consisting of the PS1 and PS2 forms, is synthesized in the ER and serves as the catalytic core of γ-secretase.89 γ-Secretase, an enzyme involved in the production of Aβ from amyloid precursor protein, plays a crucial role in the cleavage, transport, and processing of APP.90 Mutation of the PS gene can lead to an increase in the production and clearance of Aβ, resulting in the formation of excessive Aβ products and an increase in the Ca2+ concentration in neurons.91 Aβ can activate AS to RAs, which in turn induce BACE1 to produce more Aβ. Additionally, RAs can enhance the activities of neuronal BACE1 and γ-secretase, further promoting Aβ production.92 Aβ not only directly affects the cell membrane by forming calcium osmotic channels and causing calcium influx but also increases calcium influx through N-methyl-D-aspartate receptors (NMDARs) and voltage-gated calcium channels (VGCCs).93,94 The concentration of Ca2+ outside normal neurons is significantly higher than that inside normal neurons. Neurons can precisely regulate calcium signals by opening VGCCs on the cell membrane.95 The VGCC is a calcium channel with high selectivity and affinity for calcium signalling. Various ligand-gated calcium channels are present on the cell membrane, with ionotropic glutamate receptors (iGluRs) being unique to neurons. iGluRs are categorized into several subtypes, including NMDARs.96 In the resting state, the NMDAR channel is obstructed by extracellular Mg2+. Upon stimulation by glutamate released from the presynaptic membrane, extracellular Na+ and Ca2+ flow into the cell, causing partial depolarization of the cell membrane and removal of Mg2+ from the NMDAR channel, leading to its opening and the subsequent entry of Ca2+ into the cell. This influx of Ca2+ results in calcium overload in neurons, which can trigger a cascade of downstream pro-death signalling events, such as calpain activation, ROS generation, and mitochondrial damage, ultimately culminating in cellular necrosis or apoptosis. In AD, abnormal calcium signalling can also contribute to reactive hyperplasia and neuroinflammation in AS, leading to abnormal neuronal calcium signaling.97
Brain Energy Metabolism in AD
During nervous system development, astrocytes play a crucial role in guiding neurons to migrate to specific locations and form synaptic connections with other cells. Astrocytes are primarily responsible for utilizing glucose in the brain, accounting for 25% of the body’s total glucose consumption. By acting as the main reservoir of glycogen in the brain, astrocytes can breakdown glycogen into glucose to provide energy for neurons when blood glucose levels from the blood–brain barrier are insufficient to meet the demands of highly active neurons, thus maintaining energy metabolism in brain tissue.98 Enhanced synaptic activity triggers the astrocyte–neuron lactate shuttle (ANLS), where lactate is transferred from astrocytes to neurons. Essentially, glutamate released by neuronal synapses is absorbed by astrocytes through anaerobic glycolysis and glucose uptake pathways. Subsequently, astrocytes convert glucose through glycolysis into lactate, which is then utilized by neurons as an energy source.99 Research has shown that astrocytes in AD patients exhibit reduced lactate secretion and compromised neuronal support function.100 Increasing lactate production through a neuroprotective and therapeutic agent in the brains of AD patients has been found to alleviate Aβ-induced neuronal toxicity.101–103 The transport of lactate between neurons and astrocytes relies on monocarboxylate transporters (MCTs), a family of 14 members. MCT1-4 facilitate the passive transport of monocarboxylic acids and protons, including lactate, pyruvate, and ketone bodies, across cell membranes. Specifically, MCT2 is located on neuronal cell membranes, while MCT1 and MCT4 are located on astrocyte cell membranes, with MCT4 being cell-specific.104 Within the brain, MCTs transfer lactate generated by astrocytes to neurons for use as an oxidative fuel. Dysfunction of MCTs is linked to CNS pathologies such as neurodegeneration, cognitive deficits and metabolic disorders.105 Sustained astrocyte glycolysis maintains extracellular lactate levels to fulfil neuronal energy requirements. Under conditions of sufficient glucose, the MCT2 blocker α-cyano-4-hydroxycinnamic acid disrupts energy metabolism by inhibiting MCT2-mediated lactate uptake in neurons. This interference results in reduced synaptic evoked potentials in the hippocampal CA3 region and altered concentrations of K+, Na+, and Ca2+, indicating the contribution of the ANLS to ion homeostasis, synaptic signalling, and energy metabolism.106 The neuroenergetic hypothesis proposes that AD stems from early brain glucose hypometabolism, with astrocytes playing a crucial role.
Tarczyluk et al107 reported that Aβ1-42 can reduce energy metabolism in neurons and astrocyte networks in the brain, affecting various compounds such as glucose, lactate, pyruvate, glutamate, and glutathione. The authors suggested that this reduction may be linked to a decrease in the tricarboxylic acid cycle and ANLS substrates. Disruption of the ANLS can result in oxidative stress and neuroinflammation, disrupting astrocyte–neuron interactions. This interaction is crucial for physiological brain processes, including the tricarboxylic acid cycle and neurotransmitter balance. Additionally, damaged astrocytes can stimulate Aβ production, exacerbating disease progression. Nilsen et al108 utilized13 C nuclear magnetic resonance spectroscopy and13 C-labelled precursors to reveal that AD pathogenesis involves alterations in brain metabolic pathways, leading to energy and neurotransmitter imbalances, reduced mitochondrial metabolism in astrocytes and neurons, and impaired glutamate–glutamine cycling.
Glutamate Metabolism in AD
Astrocytes play a crucial role in glutamatergic synapses as the third component of the synapse, forming what is known as the “tripartite synapse” with presynaptic and postsynaptic nerve terminals and perisynaptic stellate cell terminal processes (PAPs). Glutamate, an excitatory amino acid neurotransmitter primarily found in the CNS, is utilized by numerous nerve synapses for neuromodulation.109 Glutamate is synthesized in the brain through four main pathways: the α-ketoglutarate pathway, GABA pathway, ornithine pathway, and glutamine pathway. Among these, only glutamic acid, produced through the hydrolysis of glutamine by glutaminase, serves as a neurotransmitter. Glutamate, a nonessential amino acid that cannot cross the BBB, is therefore produced within the brain. The synthesis of glutamate/glutamine involves two pathways: the de novo synthesis pathway and the glutamate/glutamine cycle. De novo synthesis may occur through the TCA cycle. In mammals, oxaloacetate (OAA) can be supplemented by pyruvate carboxylate catalysed by pyruvate carboxylase (PC), an enzyme expressed only in astrocytes and not in neurons. Therefore, glutamatergic neurons depend on astrocytes to replenish the glutamate pool for transmission. As glutamate itself is excitotoxic, it is converted to glutamine by glutamine synthetase (GS) before being transported by astrocytes to neurons. Glutaminase 1 (GLS1), also known as PAG (phosphate-activated glutaminase), is expressed in glutamatergic neurons and converts glutamine back into glutamate.
Glutamate is crucial for maintaining neuronal excitability and normal synaptic activity. The levels of glutamate in the brain are primarily regulated by the astrocyte–neuron glutamate–glutamine cycle.110 Astrocytes convert glutamate to glutamine through the action of GS. This glutamine is then transported to neurons by sodium-coupled neutral amino acid transporters (SNATs). Neuronal mitochondria deaminate glutamine back to glutamate using GLS1. The recycled glutamate is then loaded into synaptic vesicles by vesicular glutamate transporters (VGLUTs) in preparation for the next transmission cycle. Research indicates that decreased levels of GS in astrocytes in AD lead to an excitotoxic effect of glutamate, contributing to the pathogenesis of AD. The imbalance of glutamate/glutamine between astrocytes and neurons is thought to be a key factor in AD development.111 Glutamate transporters play a crucial role in terminating excitatory signals, recycling excitatory amino acids, and preventing neurotoxicity by selective transport mechanisms.112 Upon release, glutamate binds to glutamate receptors on the postsynaptic membrane. These receptors include ion channel types (AMPAR/GluA, KainateR/GluK, and NMDAR/GluN receptors) and G-protein coupled metabotropic glutamate receptors (mGluRs). Ionotropic receptors facilitate cation influx, while metabotropic receptors primarily act through intracellular second messengers. In the pathogenesis of AD, Aβ directly binds to iGluRs, leading to calcium influx, cell death, and Tau phosphorylation. Research has highlighted the significance of both ionotropic and mGluRs in AD pathogenesis, with increasing focus on NMDARs.113 Glutamate release from astrocytes is facilitated by Ca2+ activation of Bestrophin-1 (BEST1), a chloride channel activated by intracellular Ca2+. The released glutamate then binds to NMDARs at neuronal synapses.114 NMDARs play crucial roles in excitatory synaptic transmission, synaptic plasticity, and excitotoxicity in the nervous system. Studies suggest that hyperactivation of NMDARs can lead to synaptic dysfunction in early AD stages, contributing to various nervous system lesions, including AD.115 Overactivation of glutamate receptors results in excitotoxicity, known as glutamate excitatory toxicity. Astrocytes promptly eliminate glutamate through excitatory amino acid transporters (EAATs), particularly EAAT1 and EAAT2, to prevent glutamate excitotoxicity due to excessive receptor activation.112 A small fraction of glutamate can be removed by neurons via EAAT3. Research has shown that EAAT1 is expressed only in astrocytes when cultured alone, but both EAAT1 and EAAT2 are expressed in astrocytes and neurons when cocultured. This finding suggested that neuron-derived factors may influence the expression of the EAAT2 protein and mRNA.116,117 EAAT2, which is primarily expressed in astrocytes and is responsible for nearly 90% of glutamate uptake, plays a crucial role in terminating glutamate-mediated neurotransmission. This activity helps maintain the appropriate concentration of synaptic glutamate and limits the excitotoxic effects of glutamate on the CNS. Selective upregulation of EAAT2 expression could have therapeutic effects on diseases caused by excess glutamic acid.118 Studies have indicated that EAAT2 levels are temporarily reduced in AD models, and increasing EAAT2 expression through pharmacological approaches leads to a decrease in pathological Tau accumulation and the recovery of synaptic proteins.119
Synaptic Plasticity in AD
One of the pathological changes observed in AD is synapse loss. Current research suggests that cognitive and memory deficits in AD may be attributed to Aβ targeting the tripartite synapse, resulting in synaptic loss and reduced plasticity (Figure 3).120,121 In the cerebral cortex and hippocampus, synaptic dysfunction, decline, and loss of synaptic plasticity are prominent features of AD.121 Aβ can accumulate between neurons, disrupting normal synaptic signal transmission and ultimately leading to chronic dysfunction throughout the brain. Both in vivo and in vitro experimental results have confirmed that Aβ oligomers can lead to synaptic atrophy and disintegration122 and can cause abnormal changes in brain excitability.123 A cross-sectional study based on autopsy specimens revealed that the loss of synapses occurred before the onset of clinical symptoms of AD.124 These findings suggest that synaptic dysfunction plays a crucial role in the progression of AD, highlighting the importance of early detection and intervention in synaptic dysfunction for the early diagnosis and treatment of AD. Synapses serve as functional connections between neurons or other cell types and are categorized into chemical synapses and electrical synapses. Electrical synapses transmit signals through a current via gap junctions, while chemical synapses release neurotransmitters for communication.125 The process of classical synaptic transmission involves depolarization of the presynaptic membrane, opening of VGCCs, release of neurotransmitters into the synaptic cleft, and their interactions with specific receptors on the postsynaptic membrane.126 This process can lead to depolarization or hyperpolarization of the postsynaptic membrane.127 Synaptic plasticity refers to the ability of the nervous system to adapt to continuous external stimuli, resulting in long-lasting changes in synaptic morphology and function. It is fundamental to the development of the nervous system, as well as to learning and memory.128 Synaptic plasticity encompasses both structural and functional changes in synapses.
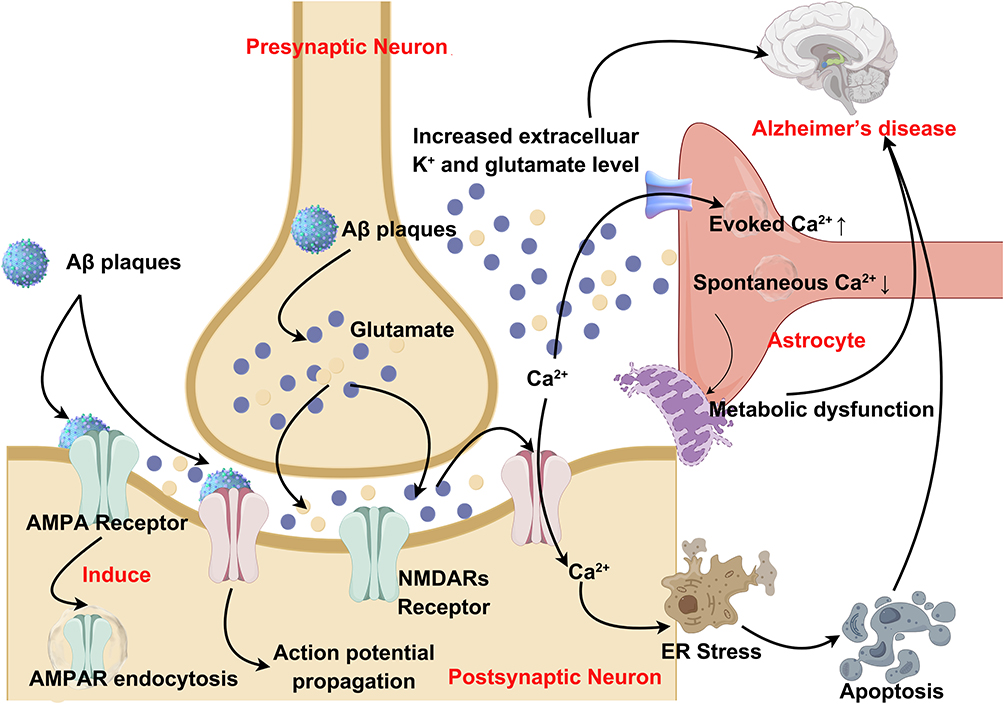 |
Figure 3 A schematic representation of the tripartite synapse and elucidates its mechanism of action in AD. (The tripartite synapse is composed of presynaptic and postsynaptic nerve terminals along with perisynaptic stellate cell terminal processes (PAP). Astrocytes play a crucial role in this system by uptake of glutamate from the synaptic cleft to prevent glutamate excitotoxicity, and also by providing glutamine as a precursor for glutamate). Some cartoon components were from www.figdraw.com for model drawing. |
Synaptic structural plasticity encompasses changes in physical characteristics such as the structure of the presynaptic membrane, synaptic cleft, and postsynaptic membrane, as well as factors such as the number of synapses, connection strength, thickness of the postsynaptic density (PSD), length of synaptic activation areas, synaptic curvature, number of synaptic vesicles, and dendritic spines. These changes are influenced by external factors and can be categorized into presynaptic plasticity and postsynaptic plasticity.125–127 Presynaptic plasticity involves alterations in the presynaptic membrane, including synaptic vesicles122 and neurotransmitters.123 Rab3/Rab27, small-molecule G proteins, play a role in vesicle transport to synaptic activation zones,124 while dysfunction in presynaptic plasticity can impact transmitter release.129 SNAP-25, a target protein at the presynaptic membrane, aids in the localization of synaptic vesicles. Studies have shown significant synaptic dysfunction in various dementia groups, with associations between cognitive decline rates and decreased levels of Rab3 in individuals with dementia with Lewy bodies (DLB) and SNAP25 in patients with AD. Synaptic proteins have high sensitivity and specificity in differentiating dementia patients from controls.130 Postsynaptic plasticity involves changes in the postsynaptic membrane, with changes in the concentration of cytoskeletal and signalling protein molecules in the PSD.131 The structural plasticity of the PSD is considered fundamental for enhancing synaptic efficacy.132
One study utilized innovative Simoa assays to assess CSF levels of candidate synaptic biomarkers, including PSD-95/DLG4, SNAP-25, and neurogranin (Ng). CSF samples from two cohorts (n=178 and n=156) were selected from stored samples collected during diagnostic lumbar punctures, encompassing individuals with Aβ-positive AD, non-AD neurodegenerative disorders, other neurological conditions, and healthy controls (HCs). The results showed that AD patients had higher concentrations of all three synaptic markers than individuals with non-AD neurodegenerative diseases, individuals with other neurological disorders, and HCs.133 Research has shown that oxysterols in brains with mild and severe AD can induce distinct morphological changes in mouse primary astrocytes, along with the upregulation of reactive astrocyte markers such as lipocalin-2 (Lcn2). Moreover, a significant decrease in PSD95 levels was observed in neurons cocultured with astrocytes treated with oxysterols, suggesting that astrocyte-released substances can impact neurons. Specifically, Lcn2 plays a crucial role in synaptic function by influencing neurite morphology and reducing dendritic spine density.134
Cytokines in AD
Astrocytes play an important role in AD-related amyloid deposition, neuronal loss and synaptic damage.135,136 Research indicates that astrocytes exhibit dual responses to AD, depending on the specific stimulatory factors involved. Factors such as transforming growth factor-β, interferon-γ, glycoprotein 130, signal transducer and activator of transcription 3, brain-derived neurotrophic factor, and oestrogen promote a protective phenotype in astrocytes. Conversely, factors such as IL-17, sphingolipid, neurotrophin receptor, suppressor of cytokine signalling 3, nuclear factor kappa B (NF-кB), chemokines, and vascular endothelial growth factor trigger a destructive phenotype. The production of LacCer by activated astrocytes facilitates the recruitment of interferon regulatory factor 1 (IRF-1) and NF-kB to the promoter regions of Ccl2, Csf2, and Nos2, resulting in cytokine production and microglial activation.137 In vivo studies reveal that astrocytes respond to a variety of factors, forming a complex network of responses with varying effects on the progression of AD.138,139 Astrocytes can exhibit either proinflammatory or anti-inflammatory behaviour by modifying their phenotypes and releasing inflammatory cytokines, particularly in response to inflammatory mediators released during microglial activation. Both astrocytes and microglia contribute to the regulation of inflammation in the CNS through the secretion of various cytokines and inflammatory mediators.140
When activated by lipopolysaccharide (LPS), microglia induce a neurotoxic phenotype in RAs. The secretion of IL-1a, TNF-a, and C1q by microglia triggers a transcriptional response in astrocytes, leading to the production of unidentified neurotoxic factors, decreased phagocytic activity, and reduced expression of neurotrophins.141 Neuroinflammatory stimulation can activate astrocytes to undergo oxidative stress in the brain affected by AD. Under normal conditions, astrocytes can increase the release of the antioxidant glutathione. However, the neuroinflammatory response in AD leads to oxidative damage in astrocytes, resulting in the production of large amounts of ROS that damage both astrocytes and neurons.100
Oksanen et al142 further discovered that the activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) had anti-inflammatory and antioxidant effects on astrocytes in AD. This activation led to a reduction in the secretion of Aβ and cytokines while increasing glutathione production in astrocytes. Complement 3 (C3) is a crucial complement protein necessary for activation of the complement pathway. Classical complement activation involves the cleavage of C3 into C3a and C3b, which then bind to the C3a receptor (C3aR) and CR3, respectively.143 Previous research has shown that the C3aR mRNA is expressed in the cerebral cortex, cerebellum, and spinal cord tissues, primarily in astrocytes and microglia.144 Rahpeymai et al145 confirmed that a reduction in neurogenesis was observed in mice with C3 or C3aR knockdown, as well as after the administration of C3aR antagonists (SB290157, C3aRA).146 This reduction in neurogenesis could further impact neuronal function by modulating the astrocyte–microglia axis, ultimately playing a role in the pathogenesis of AD.143
Previous research has shown a link between abnormal activation of NF-кB and AD.147 Activation of NF-кB in astrocytes is triggered by various proinflammatory stimuli, such as TNF-α, IL-1β, IL-17, ROS, myelin phagocytosis, Toll-like receptors, and other factors related to CNS inflammation.148 Studies by Lian et al149 documented that Aβ plaques can induce astrocytes to release C3 through NF-кB activation, which then interacts with C3aR in neurons, causing dendritic and synaptic changes in AD mouse models and brain tissue samples from AD patients. Treatment with a C3aRA improved cognitive deficits in AD model mice. Conversely, research by Wyss-Coray et al150 suggested that increased C3 expression may mitigate Aβ-induced neurotoxicity by either reducing Aβ production or enhancing its clearance. Among them, AD models, their pathogenesis, and potential mechanisms of neuroinflammation are presented in Table 1.
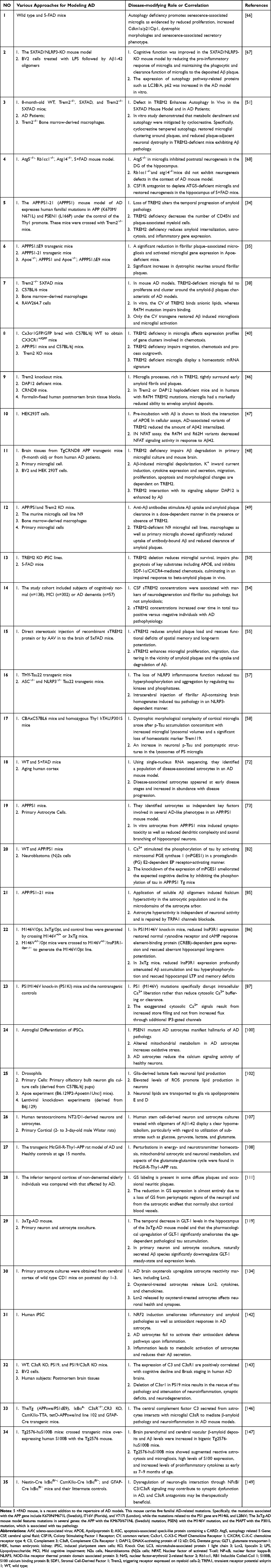 |
Table 1 Various AD Models, Their Pathogenesis, and Potential Mechanism in Neuroinflammation |
Discussion
Neuroinflammation, an immune response activated by glial cells in the CNS, typically arises in response to nerve injury, infection, toxins, or other stimuli, as well as in the context of autoimmunity. It is closely linked to the progression of neurodegenerative diseases. Recent studies have identified neuroinflammation as the third major pathological feature of AD, predominantly focusing on microglia, astrocytes, and neurons. Aberrant and sustained activation of microglia can trigger and exacerbate AD. Future investigations may aim to mitigate neuroinflammation by shifting activated M1 microglia to M2 microglia in the later stages of AD and suppressing the release of proinflammatory cytokines from microglia to identify potential therapeutic targets for AD. In the pathological mechanism of AD, astrocytes are commonly viewed as a downstream response to microglial activation. Under pathological conditions, the disruption of the astrocytic network structure, the inhibition of neurotrophic effects, and the direct neuronal damage caused by RAs underscore the significance of the cascade reaction initiated by astrocytes. Future interventions targeting the astrocytic cascade may be explored as potential therapeutic avenues for AD, such as supplementing neurotrophic factors and rebuilding the astrocytic network.
The interaction between microglia and astrocytes has garnered increasing attention from researchers due to its impacts on various aspects of neuroinflammation, neurotoxicity, neuroprotection, and nerve regeneration in AD. Microglia–astrocyte crosstalk plays a crucial role in regulating neuronal activity and brain function. Microglia typically respond more rapidly than astrocytes to pathological stimuli, influencing astrocyte activation and fate. Conversely, astrocytes can also activate microglia. This crosstalk involves the release of various molecules, such as cytokines, chemokines, complement molecules, and growth factors, as well as direct contact through surface molecules, contributing significantly to brain development, function, and homeostasis. However, dysregulated microglia–astrocyte crosstalk can exacerbate neuropathology under inflammatory or traumatic conditions. Promoting protective microglia–astrocyte crosstalk is considered a promising therapeutic approach for AD. Nevertheless, most research on microglia–astrocyte crosstalk is based on animal models and requires validation in human studies. Currently, our understanding of the molecular mechanisms underlying microglia–astrocyte crosstalk in AD is limited, necessitating further investigation to elucidate the comprehensive mechanisms and provide a solid scientific foundation for clinical AD treatment. Challenges remain in inhibiting CNS inflammation. The presence of the BBB hinders the efficient delivery of anti-inflammatory drugs to the CNS, allowing the inflammatory response and overactivation of glial cells to persist. Simply enhancing the permeability of the BBB to allow entry of anti-inflammatory drugs may lead to the uncontrolled influx of peripheral inflammatory factors into the brain, potentially exacerbating neuroinflammation.
Current research can explore targeted modifications of anti-inflammatory drugs to enhance their ability to cross the BBB and specifically target the CNS. Although this approach has shown promise, large-scale clinical validation is still needed. The identification of an effective route of administration to inhibit neuroinflammation, glial cell activation, neuronal damage, and axonal damage remains crucial in the prevention and treatment of AD.
Abbreviations
AD, Alzheimer’ s disease; ANLS, Astrocyte–neuron lactate shuttle; APOE4, Apolipoprotein E4; APP, β-amyloid precursor protein; ARIA, Amyloid-related imaging abnormalities; Aβ, β-amyloid protein; BBB, Blood‒brain barrier; BEST1, Bestrophin-1; C1q Complement component 1q; C3, Complement 3; C3aR, C3a receptor; C3aRA C3aR antagonist; CaM, Calmodulin; CNS, Central nervous system; CR1, Component receptor 1; CSF, Cerebrospinal fluid; DAMP, Damage-associated molecular pattern; DAP12, DNAX-activating protein of 12 kDa; DLB, Dementia with Lewy bodies; EAATs, Excitatory amino acid transporters; FDA, Food and Drug Administration; GFAP, Glial fibrillary acidic protein; GLS1, Glutaminase 1; GS, Glutamine synthetase; GWASs, Genome-wide association studies; HCs, Healthy controls; HLA-DRB1, Human leukocyte antigen-DR isotype beta-1; IFITM3, Interferon-inducible transmembrane protein 3; iGluRs, Ionotropic glutamate receptors; IP3, Inositol 1,4,5-trisphosphate; IRF-1, Interferon regulatory factor 1; ITAM, Immune receptor tyrosine-based activation motif; Lcn2, Lipocalin-2; LPS, Lipopolysaccharide; MAPK, Mitogen-activated protein kinase; MCI, Mild cognitive impairment; MCTs, Monocarboxylate transporters; mGluRs, Metabotropic glutamate receptors; mTOR, Mammalian target of rapamycin; NFTs, Neurofibrillary tangles; NF-кB, Nuclear factor kappa B; Ng, Neurogranin; NMDAR, N-methyl-D-aspartate receptor; NMDAR-A, N-methyl-D-aspartate receptor antagonist; NMDARs, N-methyl-D-aspartate receptors; Nrf2, Nuclear factor-erythroid 2-related factor 2; OAA, Oxaloacetate; PAMP, Pathogen-associated molecular pattern; PAPs, Perisynaptic astroglial processes; PC, Pyruvate carboxylase; PGRN, Progranulin; PI3K, Phosphatidylinositol 3-kinase; PI3K, Phosphatidylinositol 3-kinase; PLCγ2, Phospholipase Cγ2; PSD, Postsynaptic density; PSD, Postsynaptic density; RAs, Reactive astrocytes; sAPPα, Soluble amyloid precursor protein α; SNATs, Sodium-coupled neutral amino acid transporters; Syk, Spleen tyrosine kinase; TLRs, Toll-like receptors; TREM2, Triggering receptor expressed on myeloid cells-2; TREM2, Triggering receptor expressed on myeloid cells-2; TRPA1, Transient receptor potential A1; VGCCs, Voltage-gated calcium channels; VGLUTs, Vesicular glutamate transporters.
Acknowledgment
Some cartoon components were from www.figdraw.com for model drawing. The main figures were assembled in Adobe illustrator.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Jilin Province Science and Technology Development Plan (YDZJ202201ZYTS232), Natural Science Foundation of Shandong (ZR2023QH159) and Shandong Province Medical Health Science and Technology Development Plan Project (202313011384).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
1. Yan H, Feng L, Li M. The role of traditional Chinese medicine natural products in β-amyloid deposition and tau protein hyperphosphorylation in alzheimer’s disease. Drug Des Devel Ther. 2023;17:3295–3323. doi:10.2147/DDDT.S380612
2. Azarpazhooh MR, Avan A, Cipriano LE, et al. A third of community-dwelling elderly with intermediate and high level of Alzheimer’s neuropathologic changes are not demented: a meta-analysis. Ageing Res Rev. 2020;58(101002). doi:10.1016/j.arr.2019.101002
3. Nizami S, Hall-Roberts H, Warrier S, Cowley SA, Di Daniel E. Microglial inflammation and phagocytosis in alzheimer’s disease: potential therapeutic targets. Br J Pharmacol. 2019;176(18):3515–3532. doi:10.1111/bph.14618
4. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–172. doi:10.1038/s41582-020-00435-y
5. Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 2016;12(6):719–732. doi:10.1016/j.jalz.2016.02.010
6. Stuart KE, King AE, King NE, Collins JM, Vickers JC, Ziebell JM. Late-life environmental enrichment preserves short-term memory and may attenuate microglia in male app/ps1 mice. Neuroscience. 2019;408:282–292. doi:10.1016/j.neuroscience.2019.04.015
7. Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of alzheimer’s disease. Curr Opin Neurobiol. 2016;36:74–81. doi:10.1016/j.conb.2015.10.004
8. Gate D, Saligrama N, Leventhal O, et al. Clonally expanded cd8 t cells patrol the cerebrospinal fluid in alzheimer’s disease. Nature. 2020;577:399–404. doi:10.1038/s41586-019-1895-7
9. Hur JY, Frost GR, Wu X, et al. The innate immunity protein ifitm3 modulates γ-secretase in Alzheimer’s disease. Nature. 2020;586:735–740. doi:10.1038/s41586-020-2681-2
10. Chen X, Firulyova M, Manis M, et al. Microglia-mediated t cell infiltration drives neurodegeneration in tauopathy. Nature. 2023;615:
11. Wu T, Lin D, Cheng Y, et al. Amyloid cascade hypothesis for the treatment of alzheimer’s disease: progress and challenges. Aging Dis. 2022;13(6):1745–1758. doi:10.14336/AD.2022.0412
12. Valiukas Z, Ephraim R, Tangalakis K, Davidson M, Apostolopoulos V, Feehan J. Immunotherapies for alzheimer’s disease-a review. Vaccines. 2022;10(9). doi:10.3390/vaccines10091527
13. Imbimbo BP, Ippati S, Watling M, Imbimbo C. Role of monomeric amyloid-β in cognitive performance in alzheimer’s disease: insights from clinical trials with secretase inhibitors and monoclonal antibodies. Pharmacol Res. 2023;187(106631). doi:10.1016/j.phrs.2022.106631
14. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces aβ plaques in alzheimer’s disease. Nature. 2016;537(7618):50–56. doi:10.1038/nature19323
15. Dhadda S, Kanekiyo M, Li D, et al. Consistency of efficacy results across various clinical measures and statistical methods in the lecanemab Phase 2 trial of early alzheimer’s disease. Alzheimers Res Ther. 2022;14(1):182. doi:10.1186/s13195-022-01129-x
16. Alzheimer’s Disease Branch of China Association of Geriatric Health Care. Chinese association of traditional Chinese medicine, brain disease drug research specialized committee. Chinese expert consensus on integrated Chinese and Western medicine for the treatment of alzheimer’s disease. Chin J Behav Med Brain Sci. 2024;33(02):97–108.
17. Plotkin SS, Cashman NR. Passive immunotherapies targeting aβ and tau in alzheimer’s disease. Neurobiol Dis. 2020;144(105010). doi:10.1016/j.nbd.2020.105010
18. Novak P, Schmidt R, Kontsekova E, et al. Safety and immunogenicity of the tau vaccine aadvac1 in patients with alzheimer’s disease: a randomised, double-blind, placebo-controlled, Phase 1 trial. Lancet Neurol. 2017;16(2):123–134. doi:10.1016/S1474-4422(16)30331-3
19. Novak P, Schmidt R, Kontsekova E, et al. Fundamant: an interventional 72-week phase 1 follow-up study of aadvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res Ther. 2018;10(1):108. doi:10.1186/s13195-018-0436-1
20. Boxer AL, Qureshi I, Ahlijanian M, et al. Safety of the tau-directed monoclonal antibody biib092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol. 2019;18(6):549–558. doi:10.1016/S1474-4422(19)30139-5
21. West T, Hu Y, Verghese PB, et al. Preclinical and clinical development of abbv-8e12, a humanized anti-tau antibody, for treatment of alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis. 2017;4(4):236–241. doi:10.14283/jpad.2017.36
22. Teng E, Manser PT, Pickthorn K, et al. Safety and efficacy of semorinemab in individuals with prodromal to mild alzheimer disease: a randomized clinical trial. JAMA Neurol. 2022;79(8):758–767. doi:10.1001/jamaneurol.2022.1375
23. Hull M, Sadowsky C, Arai H, et al. Long-term extensions of randomized vaccination trials of acc-001 and qs-21 in mild to moderate alzheimer’s disease. Curr Alzheimer Res. 2017;14(7):696–708. doi:10.2174/1567205014666170117101537
24. Rafii MS, Sol O, Mobley WC, et al. Safety, tolerability, and immunogenicity of the aci-24 vaccine in adults with down syndrome: a phase 1b randomized clinical trial. JAMA Neurol. 2022;79(6):565–574. doi:10.1001/jamaneurol.2022.0983
25. The LN. Solanezumab: too late in mild alzheimer’s disease? Lancet Neurol. 2017;16(2):97. doi:10.1016/S1474-4422(16)30395-7
26. Fillit H, Green A. Aducanumab and the fda - where are we now? Nat Rev Neurol. 2021;17(3):129–130. doi:10.1038/s41582-020-00454-9
27. Reardon S. Fda approves Alzheimer’s drug lecanemab amid safety concerns. Nature. 2023;613:
28. Cummings JL, Cohen S, van Dyck CH, et al. Abby: a phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018;90(21):e1889–e1897. doi:10.1212/WNL.0000000000005550
29. Yang T, Dang Y, Ostaszewski B, et al. Target engagement in an alzheimer trial: crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann Neurol. 2019;86(2):215–224. doi:10.1002/ana.25513
30. Ostrowitzki S, Bittner T, Sink KM, et al. Evaluating the safety and efficacy of crenezumab vs placebo in adults with early Alzheimer disease: two Phase 3 randomized placebo-controlled trials. JAMA Neurol. 2022;79(11):1113–1121. doi:10.1001/jamaneurol.2022.2909
31. Espay AJ. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;385(7):666–667. doi:10.1056/NEJMc2109455
32. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;
33. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:
34. Jay TR, Hirsch AM, Broihier ML, et al. Disease progression-dependent effects of trem2 deficiency in a mouse model of alzheimer’s disease. J Neurosci. 2017;37(3):637–647. doi:10.1523/JNEUROSCI.2110-16.2016
35. Ulrich JD, Ulland TK, Mahan TE, et al. Apoe facilitates the microglial response to amyloid plaque pathology. J Exp Med. 2018;215(4):1047–1058. doi:10.1084/jem.20171265
36. Xu W, Han SD, Zhang C, et al. The fam171a2 gene is a key regulator of progranulin expression and modifies the risk of multiple neurodegenerative diseases. Sci Adv. 2020;6(43). doi:10.1126/sciadv.abb3063
37. Hansen DV, Hanson JE, Sheng M. Microglia in alzheimer’s disease. J Cell Biol. 2018;217(2):459–472. doi:10.1083/jcb.201709069
38. Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized trem2 mice reveal microglia-intrinsic and -extrinsic effects of r47h polymorphism. J Exp Med. 2018;215(3):745–760. doi:10.1084/jem.20171529
39. Ulrich JD, Ulland TK, Colonna M, Holtzman DM. Elucidating the role of trem2 in Alzheimer’s disease. Neuron. 2017;94(2):237–248. doi:10.1016/j.neuron.2017.02.042
40. Mazaheri F, Snaidero N, Kleinberger G, et al. Trem2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017;18(7):1186–1198. doi:10.15252/embr.201743922
41. Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi:10.1016/j.cell.2017.05.018
42. Ulland TK, Colonna M. Trem2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol. 2018;14(11):667–675. doi:10.1038/s41582-018-0072-1
43. Zhao L. Cd33 in Alzheimer’s disease - biology, pathogenesis, and therapeutics: a mini-review. Gerontology. 2019;65(4):323–331. doi:10.1159/000492596
44. Das R, Chinnathambi S. Microglial remodeling of actin network by tau oligomers, via g protein-coupled purinergic receptor, p2y12r-driven chemotaxis. Traffic. 2021;22(5):153–170. doi:10.1111/tra.12784
45. Liu RX, Huang C, Bennett DA, Li H, Wang R. The characteristics of astrocyte on aβ clearance altered in alzheimer’s disease were reversed by anti-inflammatory agent (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate. Am J Transl Res. 2016;8(10):4082–4094.
46. Yuan P, Condello C, Keene CD, et al. Trem2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;90(4):724–739. doi:10.1016/j.neuron.2016.05.003
47. Lessard CB, Malnik SL, Zhou Y, et al. High-affinity interactions and signal transduction between aβ oligomers and trem2. Embo Mol Med. 2018;10(11). doi:10.15252/emmm.201809027
48. Zhao Y, Wu X, Li X, et al. Trem2 is a receptor for β-amyloid that mediates microglial function. Neuron. 2018;97(5):1023–1031.e7. doi:10.1016/j.neuron.2018.01.031
49. Xiang X, Werner G, Bohrmann B, et al. Trem2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. Embo Mol Med. 2016;8(9):992–1004. doi:10.15252/emmm.201606370
50. McQuade A, Kang YJ, Hasselmann J, et al. Gene expression and functional deficits underlie trem2-knockout microglia responses in human models of alzheimer’s disease. Nat Commun. 2020;11(1):5370. doi:10.1038/s41467-020-19227-5
51. Ulland TK, Song WM, Huang SC, et al. Trem2 maintains microglial metabolic fitness in alzheimer’s disease. Cell. 2017;170(4):649–663.e13. doi:10.1016/j.cell.2017.07.023
52. Ding X, Wang J, Huang M, et al. Loss of microglial sirpα promotes synaptic pruning in preclinical models of neurodegeneration. Nat Commun. 2021;12(1):2030. doi:10.1038/s41467-021-22301-1
53. Zhong L, Sheng X, Wang W, et al. Trem2 receptor protects against complement-mediated synaptic loss by binding to complement c1q during neurodegeneration. Immunity. 2023;56(8):1794–1808.e8. doi:10.1016/j.immuni.2023.06.016
54. Rauchmann BS, Schneider-Axmann T, Alexopoulos P, Perneczky R. Csf soluble trem2 as a measure of immune response along the alzheimer’s disease continuum. Neurobiol Aging. 2019;
55. Zhong L, Xu Y, Zhuo R, et al. Soluble trem2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat Commun. 2019;10(1):1365. doi:10.1038/s41467-019-09118-9
56. Carosi JM, Sargeant TJ. Rapamycin and alzheimer disease: a double-edged sword? Autophagy. 2019;15(8):1460–1462. doi:10.1080/15548627.2019.1615823
57. Ising C, Venegas C, Zhang S, et al. Nlrp3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–673. doi:10.1038/s41586-019-1769-z
58. van Olst L, Verhaege D, Franssen M, et al. Microglial activation arises after aggregation of phosphorylated-tau in a neuron-specific p301s tauopathy mouse model. Neurobiol Aging. 2020:
59. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi:10.1038/nature06639
60. Shao BZ, Yao Y, Zhai JS, Zhu JH, Li JP, Wu K. The role of autophagy in inflammatory bowel disease. Front Physiol. 2021;12(621132). doi:10.3389/fphys.2021.621132
61. Wang Y, Li YB, Yin JJ, et al. Autophagy regulates inflammation following oxidative injury in diabetes. Autophagy. 2013;9(3):272–277. doi:10.4161/auto.23628
62. Yang Z, Goronzy JJ, Weyand CM. Autophagy in autoimmune disease. J Mol Med. 2015;93(7):707–717. doi:10.1007/s00109-015-1297-8
63. Li M, Sun T, Wu X, An P, Wu X, Dang H. Autophagy in the htr-8/svneo cell oxidative stress model is associated with the nlrp1 inflammasome. Oxid Med Cell Longev. 2021;2021(2353504). doi:10.1155/2021/2353504
64. Ip W, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of il-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:
65. Wu AG, Zhou XG, Qiao G, et al. Targeting microglial autophagic degradation in nlrp3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev. 2021;65(101202). doi:10.1016/j.arr.2020.101202
66. Choi I, Wang M, Yoo S, et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat Cell Biol. 2023;25(7):963–974. doi:10.1038/s41556-023-01158-0
67. Zhang D, Zhang Y, Pan J, et al. Degradation of nlrp3 by p62-dependent-autophagy improves cognitive function in alzheimer’s disease by maintaining the phagocytic function of microglia. Cns Neurosci Ther. 2023;29(10):2826–2842. doi:10.1111/cns.14219
68. Tang X, Walter E, Wohleb E, Fan Y, Wang C. Atg5 (autophagy related 5) in microglia controls hippocampal neurogenesis in alzheimer disease. Autophagy. 2024;20(4):847–862. doi:10.1080/15548627.2023.2277634
69. Li S, Selkoe DJ. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble aβ oligomers from alzheimer’s brain. J Neurochem. 2020;154(6):583–597. doi:10.1111/jnc.15007
70. Li K, Li J, Zheng J, Qin S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019;10(3):664–675. doi:10.14336/AD.2018.0720
71. Dionisio-Santos DA, Olschowka JA, O’Banion MK. Exploiting microglial and peripheral immune cell crosstalk to treat alzheimer’s disease. J Neuroinflammation. 2019;16(1):74. doi:10.1186/s12974-019-1453-0
72. Habib N, McCabe C, Medina S, et al. Disease-associated astrocytes in alzheimer’s disease and aging. Nat Neurosci. 2020;23(6):701–706. doi:10.1038/s41593-020-0624-8
73. Qiao J, Wang J, Wang H, et al. Regulation of astrocyte pathology by fluoxetine prevents the deterioration of Alzheimer phenotypes in an app/ps1 mouse model. Glia. 2016;64(2):240–254. doi:10.1002/glia.22926
74. Assefa BT, Gebre AK, Altaye BM. Reactive astrocytes as drug target in alzheimer’s disease. Biomed Res Int. 2018;2018(4160247). doi:10.1155/2018/4160247
75. Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–967. doi:10.1016/j.immuni.2017.06.006
76. Chamorro Á, Lo EH, Renú A, van Leyen K, Lyden PD. The future of neuroprotection in stroke. J Neurol Neurosurg Psychiatry. 2021;92(2):129–135. doi:10.1136/jnnp-2020-324283
77. Kellner V, Kersbergen CJ, Li S, Babola TA, Saher G, Bergles DE. Dual metabotropic glutamate receptor signaling enables coordination of astrocyte and neuron activity in developing sensory domains. Neuron. 2021;109(16):2545–2555.e7. doi:10.1016/j.neuron.2021.06.010
78. Verkhratsky A, Nedergaard M. Astroglial cradle in the life of the synapse. Philos Trans R Soc Lond B Biol Sci. 2014;369(1654):20130595. doi:10.1098/rstb.2013.0595
79. Cui Y, Yang Y, Ni Z, et al. Astroglial kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature. 2018;554:
80. Murai KK, Pasquale EB. Eph receptors and ephrins in neuron-astrocyte communication at synapses. GLIA. 2011;59(11):1567–1578. doi:10.1002/glia.21226
81. Burgoyne RD, Helassa N, McCue HV, Haynes LP. Calcium sensors in neuronal function and dysfunction. Cold Spring Harb Perspect Biol. 2019;11(5). doi:10.1101/cshperspect.a035154
82. Cao LL, Guan PP, Liang YY, Huang XS, Wang P. Calcium ions stimulate the hyperphosphorylation of tau by activating microsomal prostaglandin e synthase 1. Front Aging Neurosci. 2019;11(108). doi:10.3389/fnagi.2019.00108
83. Clapham DE. Calcium signaling. Cell. 2007;131(6):1047–1058. doi:10.1016/j.cell.2007.11.028
84. Meckelein B, Marshall DC, Conn KJ, Pietropaolo M, Van Nostrand W, Abraham CR. Identification of a novel serine protease-like molecule in human brain. Brain Res Mol Brain Res. 1998;55(2):181–197. doi:10.1016/s0169-328x(97)00366-5
85. Bosson A, Paumier A, Boisseau S, Jacquier-Sarlin M, Buisson A, Albrieux M. Trpa1 channels promote astrocytic ca(2+) hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-β peptide. Mol Neurodegener. 2017;12(1):53. doi:10.1186/s13024-017-0194-8
86. Shilling D, Müller M, Takano H, et al. Suppression of insp3 receptor-mediated ca2+ signaling alleviates mutant presenilin-linked familial alzheimer’s disease pathogenesis. J Neurosci. 2014;34(20):6910–6923. doi:10.1523/JNEUROSCI.5441-13.2014
87. Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated ip3 signaling in cortical neurons of knock-in mice expressing an alzheimer’s-linked mutation in presenilin1 results in exaggerated ca2+ signals and altered membrane excitability. J Neurosci. 2004;24(2):508–513. doi:10.1523/JNEUROSCI.4386-03.2004
88. Guan PP, Cao LL, Wang P. Elevating the levels of calcium ions exacerbate alzheimer’s disease via inducing the production and aggregation of β-amyloid protein and phosphorylated tau. Int J Mol Sci. 2021;22(11). doi:10.3390/ijms22115900
89. Annaert WG, Levesque L, Craessaerts K, et al. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol. 1999;147(2):277–294. doi:10.1083/jcb.147.2.277
90. De Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:
91. Kim S, Violette CJ, Ziff EB. Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 m146v mutant. Neurobiol Aging. 2015;36(12):3239–3246. doi:10.1016/j.neurobiolaging.2015.09.007
92. Lautrup S, Lou G, Aman Y, Nilsen H, Tao J, Fang EF. Microglial mitophagy mitigates neuroinflammation in alzheimer’s disease. Neurochem Int. 2019;129(104469). doi:10.1016/j.neuint.2019.104469
93. Zhang Z, Shen Q, Wu X, Zhang D, Xing D. Activation of pka/sirt1 signaling pathway by photobiomodulation therapy reduces aβ levels in alzheimer’s disease models. Aging Cell. 2020;19(1):e13054. doi:10.1111/acel.13054
94. Chami M, Checler F. Alterations of the endoplasmic reticulum (er) calcium signaling molecular components in alzheimer’s disease. Cells-Basel. 2020;9(12). doi:10.3390/cells9122577
95. Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium. 2018;70:87–94. doi:10.1016/j.ceca.2017.06.008
96. Scheefhals N, MacGillavry HD. Functional organization of postsynaptic glutamate receptors. Mol Cell Neurosci. 2018;91:82–94. doi:10.1016/j.mcn.2018.05.002
97. Brawek B, Garaschuk O. Network-wide dysregulation of calcium homeostasis in alzheimer’s disease. Cell Tissue Res. 2014;357(2):427–438. doi:10.1007/s00441-014-1798-8
98. Jolivet R, Allaman I, Pellerin L, Magistretti PJ, Weber B. Comment on recent modeling studies of astrocyte-neuron metabolic interactions. J Cereb Blood Flow Metab. 2010;30(12):1982–1986. doi:10.1038/jcbfm.2010.132
99. Bélanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci. 2009;11(3):281–295. doi:10.31887/DCNS.2009.11.3/mbelanger
100. Oksanen M, Petersen AJ, Naumenko N, et al. Psen1 mutant ipsc-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 2017;9(6):1885–1897. doi:10.1016/j.stemcr.2017.10.016
101. Newington JT, Harris RA, Cumming RC. Reevaluating metabolism in alzheimer’s disease from the perspective of the astrocyte-neuron lactate shuttle model. J Neurodegener Dis. 2013;2013(234572). doi:10.1155/2013/234572
102. Liu L, MacKenzie KR, Putluri N, Maletić-Savatić M, Bellen HJ. The glia-neuron lactate shuttle and elevated ros promote lipid synthesis in neurons and lipid droplet accumulation in glia via apoe/d. Cell Metab. 2017;26(5):719–737.e6. doi:10.1016/j.cmet.2017.08.024
103. Zulfiqar S, Garg P, Nieweg K. Contribution of astrocytes to metabolic dysfunction in the Alzheimer’s disease brain. Biol Chem. 2019;400(9):1113–1127. doi:10.1515/hsz-2019-0140
104. Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94(1):1–14. doi:10.1111/j.1471-4159.2005.03168.x
105. Pérez-Escuredo J, Van Hée VF, Sboarina M, et al. Monocarboxylate transporters in the brain and in cancer. Biochim Biophys Acta. 2016;1863(10):2481–2497. doi:10.1016/j.bbamcr.2016.03.013
106. Angamo EA, Rösner J, Liotta A, Kovács R, Heinemann U. A neuronal lactate uptake inhibitor slows recovery of extracellular ion concentration changes in the hippocampal ca3 region by affecting energy metabolism. J Neurophysiol. 2016;116(5):2420–2430. doi:10.1152/jn.00327.2016
107. Tarczyluk MA, Nagel DA, Rhein PH, et al. Amyloid β 1-42 induces hypometabolism in human stem cell-derived neuron and astrocyte networks. J Cereb Blood Flow Metab. 2015;35(8):1348–1357. doi:10.1038/jcbfm.2015.58
108. Nilsen LH, Witter MP, Sonnewald U. Neuronal and astrocytic metabolism in a transgenic rat model of alzheimer’s disease. J Cereb Blood Flow Metab. 2014;34(5):906–914. doi:10.1038/jcbfm.2014.37
109. Cheng XR, Zhou WX, Zhang YX. Glutamate transporters in central nervous system. Chin Bull Life Sci. 2009;21(02):246–252.
110. Tani H, Dulla CG, Farzampour Z, Taylor-Weiner A, Huguenard JR, Reimer RJ. A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron. 2014;81(4):888–900. doi:10.1016/j.neuron.2013.12.026
111. Robinson SR. Changes in the cellular distribution of glutamine synthetase in alzheimer’s disease. J Neurosci Res. 2001;66(5):972–980. doi:10.1002/jnr.10057
112. Bezzi P, Volterra A. Astrocytes: powering memory. Cell. 2011;144(5):644–645. doi:10.1016/j.cell.2011.02.027
113. Walton HS, Dodd PR. Glutamate-glutamine cycling in alzheimer’s disease. Neurochem Int. 2007;50(7–8):1052–1066. doi:10.1016/j.neuint.2006.10.007
114. Esteras N, Abramov AY. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells-Basel. 2020;9(9). doi:10.3390/cells9092135
115. Ferreiro E, Oliveira CR, Pereira C. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis. 2008;30(3):331–342. doi:10.1016/j.nbd.2008.02.003
116. Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the glt1 and glast glutamate transporters in cultured astroglia. J Neurochem. 1997;69(6):2612–2615. doi:10.1046/j.1471-4159.1997.69062612.x
117. Sonnewald U, Qu H, Aschner M. Pharmacology and toxicology of astrocyte-neuron glutamate transport and cycling. J Pharmacol Exp Ther. 2002;301(1):1–6. doi:10.1124/jpet.301.1.1
118. Hu YY, Xu J, Zhang M, Wang D, Li L, Li WB. Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J Neurochem. 2015;132(2):194–205. doi:10.1111/jnc.12958
119. Zumkehr J, Rodriguez-Ortiz CJ, Cheng D, et al. Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of alzheimer’s disease. Neurobiol Aging. 2015;36(7):2260–2271. doi:10.1016/j.neurobiolaging.2015.04.005
120. Mota SI, Ferreira IL, Rego AC. Dysfunctional synapse in alzheimer’s disease - a focus on NMDA receptors. Neuropharmacology. 2014;76:
121. Coomans EM, Schoonhoven DN, Tuncel H, et al. In vivo tau pathology is associated with synaptic loss and altered synaptic function. Alzheimers Res Ther. 2021;13(1):35. doi:10.1186/s13195-021-00772-0
122. Sáez-Orellana F, Fuentes-Fuentes MC, Godoy PA, et al. P2x receptor overexpression induced by soluble oligomers of amyloid beta peptide potentiates synaptic failure and neuronal dyshomeostasis in cellular models of alzheimer’s disease. Neuropharmacology. 2018:
123. Zott B, Simon MM, Hong W, et al. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science. 2019;365:
124. Lleó A, Núñez-Llaves R, Alcolea D, et al. Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s disease cerebrospinal fluid. Mol Cell Proteomics. 2019;18(3):546–560. doi:10.1074/mcp.RA118.001290
125. Holler S, Köstinger G, Martin K, Schuhknecht G, Stratford KJ. Structure and function of a neocortical synapse. Nature. 2021;591(7848):111–116. doi:10.1038/s41586-020-03134-2
126. Sandvig I, Augestad IL, Håberg AK, Sandvig A. Neuroplasticity in stroke recovery. The role of microglia in engaging and modifying synapses and networks. Eur J Neurosci. 2018;47(12):1414–1428. doi:10.1111/ejn.13959
127. Wang F, Zhang C, Hou S, Geng X. Synergistic effects of mesenchymal stem cell transplantation and repetitive transcranial magnetic stimulation on promoting autophagy and synaptic plasticity in vascular dementia. J Gerontol a Biol Sci Med Sci. 2019;74(9):1341–1350. doi:10.1093/gerona/gly221
128. Koizumi S, Hirayama Y, Morizawa YM. New roles of reactive astrocytes in the brain; an organizer of cerebral ischemia. Neurochem Int. 2018;
129. Chidambaram SB, Rathipriya AG, Bolla SR, et al. Dendritic spines: revisiting the physiological role. Prog Neuropsychopharmacol Biol Psychiatry. 2019;92:161–193. doi:10.1016/j.pnpbp.2019.01.005
130. Bereczki E, Francis PT, Howlett D, et al. Synaptic proteins predict cognitive decline in alzheimer’s disease and lewy body dementia. Alzheimers Dement. 2016;12(11):1149–1158. doi:10.1016/j.jalz.2016.04.005
131. Millan MJ, Dekeyne A, Gobert A, et al. Dual-acting agents for improving cognition and real-world function in Alzheimer’s disease: focus on 5-ht6 and d3 receptors as hubs. Neuropharmacology. 2020;177(108099). doi:10.1016/j.neuropharm.2020.108099
132. Cuesta P, Ochoa-Urrea M, Funke M, et al. Gamma band functional connectivity reduction in patients with amnestic mild cognitive impairment and epileptiform activity. Brain Commun. 2022;4(2):fcac012. doi:10.1093/braincomms/fcac012
133. Kivisäkk P, Carlyle BC, Sweeney T, et al. Increased levels of the synaptic proteins psd-95, snap-25, and neurogranin in the cerebrospinal fluid of patients with alzheimer’s disease. Alzheimers Res Ther. 2022;14(1):58. doi:10.1186/s13195-022-01002-x
134. Staurenghi E, Cerrato V, Gamba P, et al. Oxysterols present in alzheimer’s disease brain induce synaptotoxicity by activating astrocytes: a major role for lipocalin-2. Redox Biol. 2021;39(101837). doi:10.1016/j.redox.2020.101837
135. González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz L. Involvement of astrocytes in alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci. 2017;10(427). doi:10.3389/fnmol.2017.00427
136. Ries M, Sastre M. Mechanisms of aβ clearance and degradation by glial cells. Front Aging Neurosci. 2016;8(160). doi:10.3389/fnagi.2016.00160
137. Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–263. doi:10.1038/nrn3898
138. Chun H, Lee CJ. Reactive astrocytes in alzheimer’s disease: a double-edged sword. Neurosci Res. 2018;
139. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:
140. Han RT, Kim RD, Molofsky AV, Liddelow SA. Astrocyte-immune cell interactions in physiology and pathology. Immunity. 2021;54(2):211–224. doi:10.1016/j.immuni.2021.01.013
141. Lee HG, Lee JH, Flausino LE, Quintana FJ. Neuroinflammation: an astrocyte perspective. Sci Transl Med. 2023;15(721):eadi7828. doi:10.1126/scitranslmed.adi7828
142. Oksanen M, Hyötyläinen I, Trontti K, et al. Nf-e2-related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human presenilin-1 mutated alzheimer’s disease astrocytes. Glia. 2020;68(3):589–599. doi:10.1002/glia.23741
143. Litvinchuk A, Wan YW, Swartzlander DB, et al. Complement c3ar inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and alzheimer’s disease. Neuron. 2018;100(6):1337–1353.e5. doi:10.1016/j.neuron.2018.10.031
144. Reemst K, Noctor SC, Lucassen PJ, Hol EM. The indispensable roles of microglia and astrocytes during brain development. Front Hum Neurosci. 2016;10(566). doi:10.3389/fnhum.2016.00566
145. Rahpeymai Y, Hietala MA, Wilhelmsson U, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. Embo J. 2006;25(6):1364–1374. doi:10.1038/sj.emboj.7601004
146. Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H. Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer’s disease. J Neurosci. 2016;36(2):577–589. doi:10.1523/JNEUROSCI.2117-15.2016
147. Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T. Overexpression of human s100b exacerbates cerebral amyloidosis and gliosis in the tg2576 mouse model of alzheimer’s disease. Glia. 2010;58(3):300–314. doi:10.1002/glia.20924
148. Shi ZM, Han YW, Han XH, et al. Upstream regulators and downstream effectors of nf-κb in alzheimer’s disease. J Neurol Sci. 2016;366:127–134. doi:10.1016/j.jns.2016.05.022
149. Lian H, Yang L, Cole A, et al. Nfκb-activated astroglial release of complement c3 compromises neuronal morphology and function associated with alzheimer’s disease. Neuron. 2015;85(1):101–115. doi:10.1016/j.neuron.2014.11.018
150. Wyss-Coray T, Yan F, Lin AH, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited alzheimer’s mice. Proc Natl Acad Sci U S A. 2002;99(16):10837–10842. doi:10.1073/pnas.162350199
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Dexmedetomidine Alleviates Neuropathic Pain via the TRPC6-p38 MAPK Pathway in the Dorsal Root Ganglia of Rats
Xu S, Yi Y, Wang Y, Wang P, Zhao Y, Feng W
Journal of Pain Research 2022, 15:2437-2448
Published Date: 19 August 2022
Protective Effects of Pioglitazone on Cognitive Impairment and the Underlying Mechanisms: A Review of Literature
Alhowail A, Alsikhan R, Alsaud M, Aldubayan M, Rabbani SI
Drug Design, Development and Therapy 2022, 16:2919-2931
Published Date: 31 August 2022
Neural Stem Cell-Derived Exosomes Improve Neurological Function in Rats with Cerebral Ischemia-Reperfusion Injury by Regulating Microglia-Mediated Inflammatory Response

Zhao X, Zhu J, Chen S, Liu R, Long T
Journal of Inflammation Research 2023, 16:3079-3092
Published Date: 24 July 2023
Non-Coding RNA in Microglia Activation and Neuroinflammation in Alzheimer’s Disease
He C, Li Z, Yang M, Yu W, Luo R, Zhou J, He J, Chen Q, Song Z, Cheng S
Journal of Inflammation Research 2023, 16:4165-4211
Published Date: 21 September 2023
Advances in the Understanding of the Correlation Between Neuroinflammation and Microglia in Alzheimer’s Disease
Yan H, Wang W, Cui T, Shao Y, Li M, Fang L, Feng L
ImmunoTargets and Therapy 2024, 13:287-304
Published Date: 12 June 2024