")
Back to Journals » International Journal of Nanomedicine » Volume 19
Puerarin-Loaded Liposomes Co-Modified by Ischemic Myocardium-Targeting Peptide and Triphenylphosphonium Cations Ameliorate Myocardial Ischemia-Reperfusion Injury
Authors Wang Y, Li F, Wei S, Li W, Wu J, Li S, Hu X, Tang T, Liu X
Received 7 May 2024
Accepted for publication 29 July 2024
Published 7 August 2024 Volume 2024:19 Pages 7997—8014
DOI https://doi.org/10.2147/IJN.S468394
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Krishna Nune
Yan Wang,1,2,* Fengmei Li,1,2,* Shanshan Wei,1,2,* Wenqun Li,1,2 Junyong Wu,1,2 Shengnan Li,1,2 Xiongbin Hu,1,2 Tiantian Tang,1,2 Xinyi Liu1,2
1Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, People’s Republic of China; 2Institution of Clinical Pharmacy, Central South University, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xinyi Liu, Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, People’s Republic of China, Tel +86-731-8529-2095, Email [email protected]
Purpose: Mitochondrial damage may lead to uncontrolled oxidative stress and massive apoptosis, and thus plays a pivotal role in the pathological processes of myocardial ischemia-reperfusion (I/R) injury. However, it is difficult for the drugs such as puerarin (PUE) to reach the mitochondrial lesion due to lack of targeting ability, which seriously affects the expected efficacy of drug therapy for myocardial I/R injury.
Methods: We prepared triphenylphosphonium (TPP) cations and ischemic myocardium-targeting peptide (IMTP) co-modified puerarin-loaded liposomes (PUE@T/I-L), which effectively deliver the drug to mitochondria and improve the effectiveness of PUE in reducing myocardial I/R injury.
Results: In vitro test results showed that PUE@T/I-L had sustained release and excellent hemocompatibility. Fluorescence test results showed that TPP cations and IMTP double-modified liposomes (T/I-L) enhanced the intracellular uptake, escaped lysosomal capture and promoted drug targeting into the mitochondria. Notably, PUE@T/I-L inhibited the opening of the mitochondrial permeability transition pore, reduced intracellular reactive oxygen species (ROS) levels and increased superoxide dismutase (SOD) levels, thereby decreasing the percentage of Hoechst-positive cells and improving the survival of hypoxia-reoxygenated (H/R)-injured H9c2 cells. In a mouse myocardial I/R injury model, PUE@T/I-L showed a significant myocardial protective effect against myocardial I/R injury by protecting mitochondrial integrity, reducing myocardial apoptosis and decreasing infarct size.
Conclusion: This drug delivery system exhibited excellent mitochondrial targeting and reduction of myocardial apoptosis, which endowed it with good potential extension value in the precise treatment of myocardial I/R injury.
Keywords: mitochondrial targeting, triphenylphosphonium cations, puerarin, ischemic myocardium-targeting peptide, liposomes
Introduction
Although the rapid advance in reperfusion therapy has provided a new era in the treatment of myocardial infarction (MI), MI remains the leading cause of death and disability worldwide.1 Timely reperfusion promotes rapid recovery of myocardial function, which is clinically important for the treatment of MI. However, reperfusion therapy may induce abnormal mitochondrial regulation and intense oxidative stress, further leading to massive myocardial apoptosis and enhanced infarct size, which is a common clinical myocardial ischemia/reperfusion (I/R) injury.2,3 The available literature indicates that the opening of the mitochondrial permeability transition pore (mPTP) plays a vital role in the pathological processes of myocardial I/R injury, and timely inhibition of mPTP opening could significantly reduce apoptosis in cardiomyocytes.4 However, the lack of mitochondrial targeting of most drugs severely compromises their expected efficacy at the lesion area. Therefore, it is urgent to construct a mitochondrial targeted drug delivery system for efficient delivery of drugs to myocardial mitochondria and inhibit the opening of mPTP to mitigate myocardial I/R injury.
Puerarin (PUE) is the main bioactive ingredient isolated from the roots of Radix Puerariae Lobatae and has been shown to alleviate myocardial I/R injury.5 However, there are some problems with PUE, such as water insolubility, lack of myocardial targeting and acute intravascular hemolytic adverse effects of PUE injection, which seriously affect the expected efficacy of the drug and its promotion in the clinic.6,7 Available literature indicated that PUE could inhibit the opening of mPTP in cardiomyocytes and reduce the burst release of mitochondrial ROS, thereby reducing myocardial I/R injury.8,9 It is assumed that the construction of a mitochondria-targeted drug delivery system for efficient delivery of PUE to the mitochondria will largely improve the effectiveness of the drug in alleviating myocardial I/R injury. Recently, Mitochondria-targeted nano-delivery systems have become a popular research topic as an efficient drug delivery and precision treatment modality. The advantages of these drug delivery systems were mainly in terms of enhanced water solubility, improved bioavailability and mitochondria-targeted delivery of drugs.10 However, it remains a great challenge to efficiently deliver the drugs to the site of mitochondrial lesions for full efficacy.
The literature shows that positively charged triphenylphosphonium (TPP) cations are readily attracted to negatively charged mitochondrial membranes, thereby facilitating the mitochondrial localization of TPP-modified drug delivery systems.11 Our previous results also showed that TPP-modified PUE-loaded micelles could target mitochondria and reduce isoproterenol-induced apoptosis in H9c2 cells.12 Based on the fact that the cationic TPP can cross the cell membrane and target into the mitochondria, researchers have developed a series of mitochondria-targeted drug delivery systems using TPP-modified nanocarriers. Recently, TPP based mitochondria-targeted nanocarriers, such as Tan-TPP, ANP/TPP-BN-LPNs, could accumulate in mitochondria via mitochondrial membrane potential and significantly alleviate myocardial I/R injury.13,14 However, efficient aggregation of drug delivery systems in the ischemic myocardium is critical for drug delivery to mitochondria, there are still some great challenges to solving this problem. The desired drug carriers first aggregate at the site of ischemic myocardium to form a drug reservoir for sustained accumulation and targeted drug delivery at the lesion site. In addition, the extremely fast blood flow during reperfusion makes it difficult for the heart to retain therapeutic drugs for long periods.15,16 Therefore, it is highly desirable to construct an efficient ischemic myocardial targeting delivery system to improve the ischemic myocardial targeting and retention effect of drugs.
Nowadays, the construction of targeted drug delivery systems for precise treatment based on the pathological characteristics of the lesion site has become a hot research topic. The blood vessels at the site of ischemic myocardium exhibit several pathological changes, such as poor vascular architecture, increased gap in the vessel wall, and enhanced permeability of the endothelial cell membrane. When liposomes reach the ischemic myocardial site, they could accumulate in the ischemic myocardium based on these physiological environmental characteristics of the ischemic myocardial site.17,18 On the contrary, the cells of normal blood vessel walls are tightly arranged and structurally intact, so there is little leakage of liposomes at normal vessel sites. Therefore, the liposome delivery system is able to target the drug to the ischemic myocardial site via the enhanced permeability and retention (EPR) effects, while the normal tissue lacks pathological changes, so the aggregation is relatively reduced. The current extensive literature has confirmed the excellent ischemic myocardial targeting of liposomes.19–21 Thus, the liposomes are expected to aggregate drugs at ischemic myocardial sites.
Recently, Kanki S et al reported that ischemic myocardium-targeting peptide (IMTP, amino acid sequence CSTSMLKAC) was highly specific for ischemic myocardium by binding to receptors located on the ischemic myocardial cell membrane.22 Based on the active targeting function of IMTP on ischemic myocardium, Vandergriff A and Wang X et al found that IMTP-modified exosomes could specifically target the ischemic myocardium and significantly improve the therapeutic effect of myocardial I/R injury.23 Thus, IMTP-modified nanocarriers are expected to promote tight binding of nanodrugs to ischemic cardiomyocytes, enhance targeting and retention at ischemic myocardial sites, and thus be able to withstand the impact of rapid blood flow during reperfusion.
Based on the ischemic myocardial targeting of liposomes, the enhanced ischemic cardiomyocyte targeting and retention of IMTP, and the mitochondrial targeting function of TPP, we attempt a multi-stage targeted drug delivery system of PUE-loaded liposomes co-modified by TPP and IMTP (PUE@T/I-L). It is expected that the PUE@T/I-L will first aggregate at the site of ischemic myocardium, then converge toward the myocardial cells, and finally target into the mitochondria of ischemic cardiomyocytes to promote full drug action at mitochondrial sites, thereby effectively reducing myocardial I/R injury (Scheme 1).
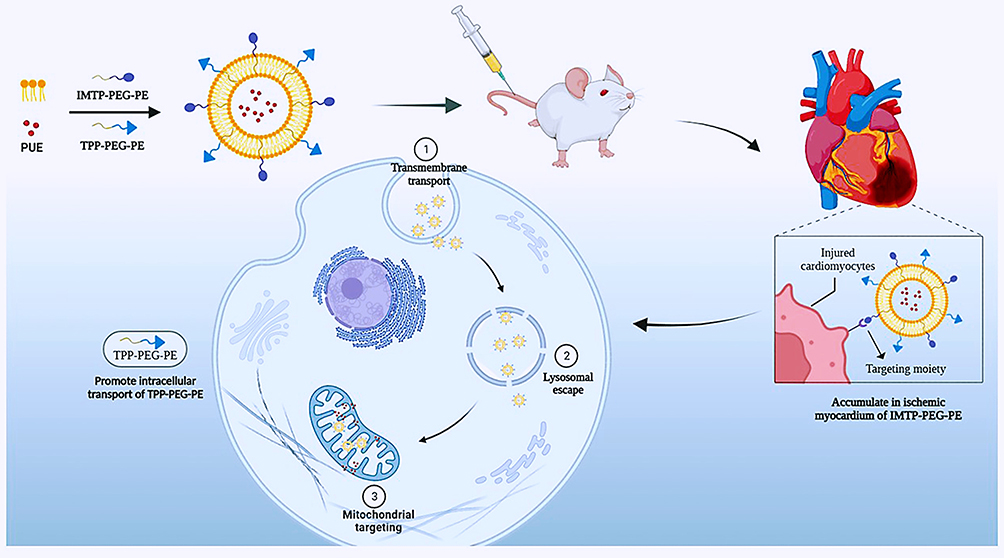 |
Scheme 1 Schematic illustrations for PUE@T/I-L stepwise targeting process to the mitochondria in ischemic cardiomyocytes. |
Materials and Methods
Materials
Cholesterol (Chol), soybean lecithin (SL) and (3-carboxypropyl) triphenylphosphonium bromide (CTPP) were purchased from Sigma-Aldrich. 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[succinimidyl Succinate ester(polyethylene glycol)2000] (DSPE-PEG-NHS) and 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)2000] (ammonium salt) (DSPE-PEG-NH2) were acquired from Xi’an ruixi Biological Technology Co., Ltd (Shanxi, China). PUE was purchased from Shandong Fangming Pharmaceutical Group Co., Ltd (Shandong, China). Ischemic myocardium-targeted peptide (IMTP, CSTSMLKAC) was synthesized by GL Biochem (Shanghai) Ltd (Shanghai, China). Coumarin-6 (C6) was provided by J&K Chemical Ltd (Shanghai, China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) and N-Hydroxysuccinimide (NHS) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China). 4’6-diamidino-2-phenylindole (DAPI), triethylamine (TEA), Mitotracker Red and Lysotracker Red were obtained from Invitrogen (Carlsbad, USA). Hoechst 33342, Mitochondrial Permeability Transition Pore (mPTP) Assay Kit and ROS Assay Kit were obtained from Beyotime Biotech (Jiangsu, China). Cell Counting Kit-8 (CCK-8) was purchased from MeilunBio (Dalian, China). H9c2 cells derived from rat myocardium were obtained from the American Type Culture Collection (Manassas, VA). C57BL/6J male mice and Sprague-Dawley (SD) female rats were provided by Hunan Slack Scene of Laboratory Animal Co., Ltd (Changsha, China).
Preparation of the Liposomes
In brief, PUE was entrapped into the mitochondria-targeted liposomes co-modified by TPP and IMTP (PUE@T/I-L). Soybean lecithin (SL), cholesterol (Chol), TPP-PEG-PE and IMTP-PEG-PE were mixed in a molar ratio of 52:26:14:8. The mixture and PUE were completely dissolved in chromatographic methanol and attached to the bottom of the vial after rotary evaporation, and a uniform film could be observed to form at the bottom of the vial. The film was hydrated by adding appropriate amount of distilled water or PBS at 37°C for 20 min and sonicated for 5 min. Finally, the lipid suspension was first filtered through a 0.22 μm microporous filter membrane and then extruded through a 100 nm pore size polycarbonate membrane to obtain a uniform particle size PUE@T/I-L. Similarly, PUE-loaded liposomes (PUE@L), PUE-loaded liposomes modified by TPP (PUE@T-L) and PUE-loaded liposomes modified by IMTP (PUE@I-L) were prepared by the same method described above and their formulations were shown in Table S1. Likewise, the coumarin-6 (fluorescent probes, C6) was used to replace PUE to prepare C6 labelled liposomes using similar methods as above.
H9c2 Cell Mode of Hypoxia/Reoxygenation
H9c2 cells were cultured with DMEM medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, and then incubated in a humidified cell culture incubator at 37°C and 5% CO2. When the growth density of H9c2 cells reached 80%, they were subjected to a hypoxia/reoxygenation (H/R) process that mimicked clinical myocardial I/R. The hypoxia/reoxygenation model of H9c2 cells was established as described previously.24,25 For hypoxia, H9c2 cells were cultured in DMEM medium without glucose and serum for 8 h with an incubation environment of 94% N2, 1% O2 and 5% CO2. At the end of hypoxia, H9c2 cells were exposed to 95% air and 5% CO2 for reoxygenation and incubated with the drugs at the initial stage of reoxygenation.
Cellular Uptake
H9c2 cells were initially grown as above and exposed to hypoxia for 8 h. They were then co-incubated with free C6 and four types of C6-labelled liposomes (C6@L, C6@T-L, C6@I-L and C6@T/I-L, 50 ng C6/mL) under the normal cell culture conditions. After incubation for 1 h and 4 h, H9c2 cells were washed twice with PBS, stained with DAPI for 10 min and fixed with 4% formaldehyde for 10 min at room temperature, and finally observed using fluorescence microscopy. Free C6 and four types of C6-labeled liposomes were co-incubated with normal H9c2 cells using the same experimental method, and their cellular uptake within normal cardiomyocytes was studied by fluorescence microscopy.
In flow cytometry experiments, H9c2 cells were exposed to H/R and treated with free C6, C6@L, C6@T-L, C6@I-L or C6@T/I-L under the same conditions described above. At 2 h of drug treatment, H9c2 cells were collected and washed. Intracellular C6 fluorescence intensity was measured by flow cytometry (BD LSR II, Biosciences) to quantify the cellular uptake of free C6 and C6-labelled liposomes by H9c2 cells. C6@T/I-L was co-incubated with normal H9c2 cells and H/R-injured H9c2 cells in the same way, and then the fluorescence intensity of C6@T/I-L uptake by H9c2 cells under these two conditions was quantified by flow cytometry.
Intracellular Distribution and Co-Location Analysis
H9c2 cells were cultured as described above and then exposed to hypoxic for 8 h. The hypoxic cells were immediately restored to reoxygenation and cultured with fresh DMEM medium containing free C6, C6@L, C6@T-L, C6@I-L, or C6@T/I-L for 1 h and 4 h. These cells were then fixed with 4% paraformaldehyde for 20 min, stained with Lysotracker Red (60 nM) or Mitotracker Red (200 nM), and finally incubated with DAPI solution (5 µg/mL) or Hoechst 33342 (5 µg/mL) for 10 min. The distribution of C6-labelled liposomes within the cells was photographed with an OLYMPUS IX73 inverted microscope (Olympus, JAPAN) and colocalization analysis was finished by the Fiji-Image J software.
Detection of Mitochondrial Permeability Transition Pore Opening
Following the method reported in the literature,26 Calcein AM and CoCl2 were used to detect the opening of mPTP according to the manufacturer’s instructions. Briefly, after 8 h of hypoxia, H9c2 cells were restored for reoxygenation and incubated with fresh DMEM containing free PUE, PUE@L, PUE@T-L, PUE@I-L or PUE@T/I-L (PUE equivalent dose: 20 μM) for 12 h. H9c2 Cells were then incubated with 1 μM calcein AM in a working solution at room temperature and then washed free of calcein and CoCl2. The cells were finally observed and photographed by fluorescence microscopy.
Measurement of ROS and Superoxide Dismutase (SOD)
ROS were detected by the DCFH-DA assay as described previously.25,27,28 After H9c2 cells were treated with hypoxia for 8 h, free PUE, PUE@L, PUE@T-L, PUE@I-L or PUE@T/I-L (PUE equivalent dose: 20 μM) were added at the beginning of reoxygenation and incubated for 12 h. 10 μM DCFH-DA was added and co-incubated with the cells for 30 min. The cells were then washed three times using 37°C DMEM to remove any remaining DCFH-DA. The fluorescence images of intracellular ROS were immediately acquired by the inverted fluorescence microscope. The DCFH-DA fluorescence was measured using the Image J software. The SOD activity was detected by Total Superoxide Dismutase Assay Kit with WST-8 (S0101, Beyotime Institute of Biotechnology, China). Briefly, H9c2 cells were seeded at a density of 2.5×105 cells/well in a 6-well plate at 37°C for 12 h, followed by hypoxia for 8 h, and then reoxygenated for 12 h with free PUE, PUE@L, PUE@T-L, PUE@I-L or PUE@T/I-L (PUE equivalent dose: 20 μM) cells were harvested with 0.25% trypsin and washed twice with PBS. SOD sample preparation solution was added at the rate of 100–200 µL per 1 million cells, the supernatant was taken as the sample to be tested and the absorbance was measured at 450 nm.
Determination of Cell Viability
Cell viability was measured using the CCK8 method. After hypoxia treatment of H9c2 cells in 96-well plates (1×104 cells/well) for 8 h, free PUE, PUE@L, PUE@T-L, PUE@I-L or PUE@T/I-L (PUE equivalent dose: 20 μM) were added at the beginning of reoxygenation. At the end of the 12 h incubation, the cells were washed three times with PBS. Then 10% CCK8 solution was added to the cells and the absorbance values of each group were measured at 450 nm after 1 h of incubation.
Hoechst Staining
H9c2 cells were cultured and treated with hypoxia for 8 h as above. At the beginning of reoxygenation of H9c2 cells, free PUE, PUE@L, PUE@T-L, PUE@I-L or PUE@T/I-L (PUE equivalent dose: 20 μM) were added and co-incubated for 12 h. At the end of drug treatment, H9c2 cells were fixed, washed twice with PBS and stained with Hoechst 33342 staining solution according to the manufacturer’s instructions (Beyotime, Haimen, Jiangsu, China). Image capture and changes in cell nuclei were performed using an OLYMPUS IX73 inverted microscope (Olympus, JAPAN). Apoptotic cells were defined by the condensation of nuclear chromatin or bright staining.29
Establishment of Myocardial I/R Injury Model and Treatment
Forty-eight male 10~12 week old C57BL/6J mice (22–27 g) (8 per group) were housed in 4 cages per cage in a 12-hour light-dark cycle at 25 ± 1°C and approved by the Laboratory Animal Ethics Committee of Xiangya Second Hospital of Central South University (IRB approval number 20220547). The mice mode of myocardial I/R injury was established according to the previous methods.30 Male C57 BL/6J mice were first deeply anesthetized with pentobarbital (50 mg/kg, i.p). The depth of anesthesia was tested by performing a withdrawal reflex test of at least 2 toes, with absence of reflex confirming sufficient anesthesia.31 The mice were then secured in a supine position with a thermostatic heating pad. The upper thoracic region was shaved and the tongue was retracted. A noninvasive tracheal cannula was then carefully inserted into the trachea of the mice, followed by connection of the catheter to a small animal ventilator. The tidal volume of mice was 1.8~2.2 mL and the respiratory rate was 110 breaths per minute. As reported in the classical method, temporary blockage of the left anterior descending artery causes myocardial ischemia and then the ligature is loosened to restore blood flow, which mimics myocardial ischemia/reperfusion injury in clinical patients.32,33 This method of ligating the left anterior descending artery is the most classic and closest to clinical myocardial ischemia available. A small oblique incision was made in the left chest wall skin of mice, and the chest wall muscles were carefully dissected and separated to expose the 4th intercostal space. The left anterior descending artery was ligated with 8–0 silk suture for 30 min. Ischemia was confirmed in all mouse by the appearance of discoloration of the heart surface.31 After 30 min, the ligature was released and reperfusion resumed for 24 h. The chest was closed under negative pressure. During the surgical operation, we tried to minimize heart exposure time and wound disinfection. Approximately 5 min prior to reperfusion, free PUE, PUE@L, PUE@I-L, and PUE@T/I-L were given to myocardial I/R-injured mice by tail vein injection at an administration dose of 15 mg/kg PUE. The sham operated animals were operated similarly but without ligation of the left anterior descending artery, and the myocardial I/R injury model group received saline. During the whole animal test, when the mice woke up and moved freely, the ventilator was then removed and the heating pad continued to be used to maintain the body temperature of the mice, which were then returned to the SPF laboratory animal environment with the isolation environment temperature controlled at about 25°C.
Cardiac Biomarker Enzymes
At the end of the experiment, the mice were anesthetized by 50 μg/g pentobarbital (i.p). and blood was sampled from retro-orbital venous sinus using the retro-orbital bleeding method.34 Briefly, the anesthetized mice were placed on their sides, insert a micro-hematocrit capillary tube into the corner of the eye socket underneath the eyeball, directing the tip at a 45-degree angle toward the middle of the eye socket. Apply gentle downward pressure and then release until the vein is broken and blood is visualized entering the pipette. Once the pipette is filled with blood, remove it from the orbital sinus. The collected blood samples were centrifuged at 3000 rpm for 10 min at room temperature, and the supernatant was aspirated using a pipette gun and transferred to a −80°C refrigerator for storage. The LDH and CK-MB activity of each sample was measured by automatic biochemical instruments (BS-240VET, Mindray Biochemical Co., Ltd., ShenZhen, China).
Determination of Myocardial Infarct Size
After 24 h of reperfusion, the hearts of C57BL/6J mice were rapidly excised under deep pentobarbital anesthesia,35 and the mice were executed by cervical dislocation. The obtained hearts were washed using 0.9% sodium chloride and frozen in a −80°C refrigerator for approximately 10 minutes. The hearts were then cut into 1 mm sized slices and stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC, Solarbio, Beijing, China) solution for 15 min under light-proof conditions, followed by fixation in 4% formaldehyde.13 The surviving myocardium was shown as red and the infarcted myocardium was shown as white, Infarcts were delineated from viable tissue (white versus red, respectively) and finally infarct size was calculated using Image J software as previously reported36,37. Infarct size (%) = (Infarct size/Viable size) ×100%.
Mitochondrial Morphology in Ischemic Myocardium
The preparation process of mitochondrial sections was performed as previously described.38 At 24 h after reperfusion, the anterior tissue of left ventricle was cut into 1mm3 small pieces and fixed in electron microscope fixative for 48 h, followed by incubation with 1% osmium acid for 2 h. After dehydration at room temperature, osmotic embedding, and polymerization at 60°C, the embedded samples were cut into 70 nm thick sections. The sections were stained with 2.5% uranyl acetate (8 min) and 2.6% lead citrate (8 min). Finally, the morphology of mitochondria in the stained sections was observed under TEM and images were taken.
Histological Analysis
The processing of histological (H&E and TUNEL staining) samples was performed according to the methods previously reported in the literature.39 Briefly, myocardial tissue was fixed in 4% paraformaldehyde, embedded in paraffin and cut into 5 μm thick sections. These sections were then stained with hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and histopathological changes were observed using light microscopy. Apoptosis of cardiomyocytes was expressed as the percentage of TUNEL-positive cells and calculated as TUNEL-positive cells/total cells × 100%.
Statistical Analysis
All data in this research were presented as mean ± standard deviation (SD) and analyzed using GraphPad Prism software (GraphPad software, Inc.; La Jolla, CA). One-way analysis of variance (ANOVA) with Tukey post hoc test was performed when comparing multiple groups. Statistical significance was considered at *p <0.05 and **p <0.01.
Result
Synthesis and Characterization of Functional Conjugates
The MALDI-TOF mass spectrometry and 1H NMR spectrum confirmed that IMTP-PEG-PE was synthesized. The IMTP was shown in black with a peak at 983 Da. The average mass peak of DSPE-PEG-NHS represented by the blue line was 2600 Da. After attaching the IMTP to DSPE-PEG-NHS, the average molecular weight of the final product IMTP-PEG-PE was exactly equal to the sum of the molecular weights of IMTP and DSPE-PEG-NHS, indicating that DSPE-PEG-NHS reacted quantitatively with IMTP in an equimolar ratio (Figure S1a). In addition, the chemical shift of IMTP (δ: 7.5–8.0 ppm) appeared in the 1H NMR spectrum of IMTP-PEG-PE, which also verified the success of IMTP conjugation with DSPE-PEG-NHS (Figure S1b).
The resulting TPP-functionalized polymer TPP-PEG-PE was shown in Figure S1c. TPP-PEG-PE was successfully synthesized as evidenced by the distinct peaks of the two peaks, namely the methylene group of the PEG (3.7 ppm) and the aromatic ring group of TPP (7.6–8.0 ppm).
Characterization of the Liposomes
Table S1 showed the analytical results of the compositions of lipids, particle size and zeta potential of the obtained PUE@T/I-L, PUE@T-L, PUE@I-L, and PUE@L. Overall, the dynamic laser scattering (DLS) measurement revealed that the particle size of all liposomes was in the range of 140–160 nm, with the smallest particle size for PUE@L and a slight increase in particle size for the modified liposomes. The particle size distribution of PUE@T/I-L was about 20 nm larger than that of PUE@L, which was due to the surface modification by TPP-PEG-PE and IMTP-PEG-PE, resulting in enlarged size of the particles (Figure 1b). The possible explanation for this is that the long-chain structure of TPP-PEG-PE and IMTP-PEG-PE stretches as much as possible in water and increases the particle size. As shown in the TEM images, it was clearly observed that the morphologies of the prepared PUE@T/I-L were generally spherical and uniformly dispersed (Figure 1a). The diameter of liposomes observed in TEM images was slightly smaller than that measured by DLS, which may be due to the difference in measurement methods, as DLS measured liposomes that were swollen and stretched as much as possible in water, while TEM measured dried liposomes. Overall, PUE@T/I-L particles of this size may exhibit lower rates of renal excretion and uptake by mononuclear phagocytes, which allows them to persist longer in vivo.40
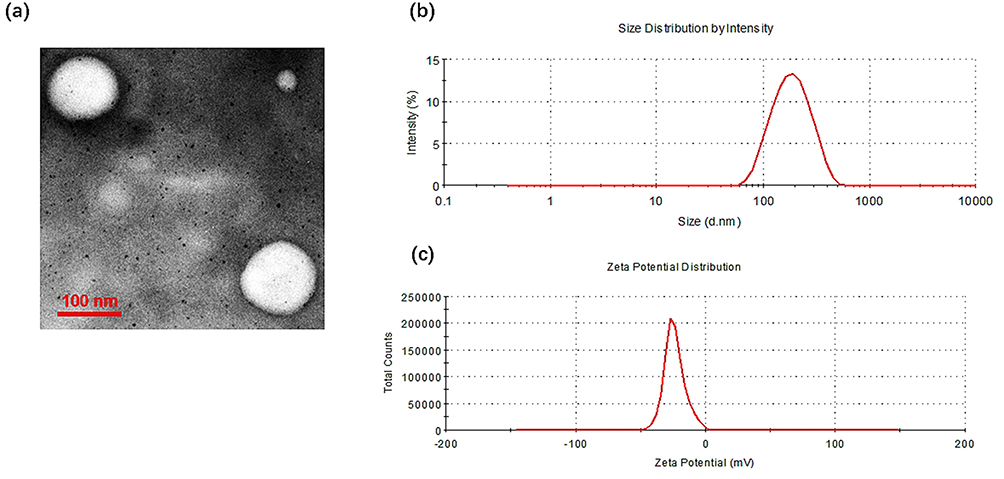 |
Figure 1 Physicochemical characterization of PUE@T/I-L. (a) Surface morphology by TEM; (b) Particle size distribution of PUE@T/I-L; (c) Zeta potential distribution of PUE@T/I-L. |
Despite the negligible difference in the average diameter of liposomes after TPP modification, their zeta potential dramatically altered. The mean zeta potential of PUE@L was - 42.7 ± 0.9 mV, while the PUE@T-L and PUE@T/I-L were −14.7 ± 0.3 mV and −27.8 ± 0.7 mV, respectively (Table S1). It was possible that the zeta potential was increased by the presence of the cationic TPP on the shell of PUE@T/I-L or PUE@T-L. Notably, the zeta potential of PUE@T/I-L was still significantly lower than that of PUE@T-L (Figure 1c), the possible reason was that the negatively charged IMTP cyclic peptide on the surface of the liposomes contributed to the relatively weak performance of the TPP cations. The average drug loading and encapsulation ratio of PUE@T/I-L were 6.19% and 85.2%, respectively.
The FT-IR result could confirm that the inner core of PUE@T/I-L was embedded with PUE. As shown in Figure S2, the IR spectral features of blank T/I-L and PUE@T/I-L behave relatively close to each other, while the characteristic peaks of PUE (shown by arrows) disappeared after it was loaded into the PUE@T/I-L, thus it could be presumed that PUE has been completely encapsulated in the liposome core.
In vitro Stability
The stability of liposomal formulations is a crucial issue in drug delivery, especially for functionally modified liposomes. As shown in Figure S3, During 60 days of storage time at 4°C, The appearance and size distribution of PUE@T/I-L did not change significantly during 60 days of storage at 4°C, and the zeta potential fluctuated only slightly, indicating that PUE@T/I-L had a fairly high thermodynamic stability.41 Moreover, PUE@T/I-L showed no appreciable variation in size and zeta potential after 24 h incubation with 10% FBS at 37°C, suggesting the excellent stability of PUE@T/I-L in serum. Taken together, PUE@T/I-L maintained essentially the same size and zeta potential over 24 h of incubation with 10% FBS, and it could be predicted that these liposomes are relatively stable during blood circulation and essentially maintain the intrinsic morphology of liposomes.42
In vitro Release
We investigated the in vitro release behavior of liposomes using a dialysis method. As shown in Figure S4a, more than 90% of PUE was released within 24 h. However, the liposomes loaded with PUE (PUE@L, PUE@I-L, PUE@T-L and PUE@T/I-L) were released relatively more during the first 16 h and then showed a slow release. These results suggested that drug depot effects could be achieved with PUE-loaded liposomes, resulting in sustained hydrophobic drug release. This sustained behavior may be explained by the slow degradation of polymeric materials, followed by the gradual dissociation and diffusion of PUE from the liposomal core. Notably, the modified liposomes (PUE@T-L, PUE@I-L and PUE@T/I-L) showed similar sustained behaviors, while PUE@L without modification exhibited relatively faster release profile, suggesting that drug release may have been hampered by TPP-PEG-PE or IMTP-PEG-PE coatings on liposome surfaces. Several similar reports in the literature have also shown that surface modification of nanocarriers could delay the release of drugs and result in a more consistent release pattern.43,44 It is speculated that the ligands applied on the surface of the modified nanoparticles may delay their release, resulting in a slower release of the modified nanoparticles than the unmodified nanoparticles.44 The relatively slow degradation of these modified polymeric materials may retard the release of PUE from the modified liposomes, which happens to be largely consistent with the previous studies.44
Hemolytic Activity
The hemolytic activity of free PUE and PUE-loaded liposomes (PUE@L, PUE@T-L, PUE@I-L and PUE@T/I-L) on rat erythrocytes was estimated by comparing the optical density of a solution containing the tested sample with the optical density of blood at 100% hemolysis. The experiments indicated that the hemolytic activity of all the tested samples at the concentration of PUE ranged from 18.75 to 300 μg/mL was relatively small (Figure S4b). Nevertheless, the hemolysis rate of free PUE showed a dose-dependent hemolytic activity. As reported in previous studies, high concentrations of PUE were associated with changes in membrane lipids, which may lead to obvious hemolysis.6,7 Therefore, there is a pressing need to reduce hemolysis by decreasing the direct contact of PUE with erythrocytes. Notably, the hemolysis of PUE-loaded liposomes showed a decreasing trend, which may be due to the reduced direct contact of PUE with erythrocytes after liposome encapsulation.
Cellular Uptake
The green fluorescent C6 was often used as a fluorescent probe for cellular uptake study.45 As shown in Figure 2, free C6-treated cells exhibited extremely weak green fluorescence. The possible reason is that C6 is extremely hydrophobic, and it is known that highly hydrophobic chemicals have difficulty entering cells in aqueous medium unless the cell culture medium contains high levels of DMSO, maintaining a high concentration of C6 around the cells.45 Compared with the C6@L group, the fluorescence intensity of liposome uptake by H9c2 cells in the C6@T-L and C6@I-L groups showed an increasing trend. A significantly stronger green fluorescence could be observed around the nucleus in the C6@T/I-L group, and it showed an enhancement trend with time, reaching the maximum intensity at 4 h, which was significantly stronger than the other groups. A possible explanation for this phenomenon is the attraction of TPP cations to negatively charged cell membranes and the role of IMTP in targeting binding receptors on H/R-injured H9c2 cells membranes,22 thus promoting cellular uptake. However, the fluorescence intensity of liposome uptake was significantly lower in normal H9c2 cells compared with H/R-injured cardiomyocytes, especially for C6@I-L (Figure S5), which is likely due to the reduced expression of some receptor proteins on the normal cardiomyocyte membrane, resulting in a reduced ability of IMTP to bind to normal H9c2 cells. In contrast, a small amount of fluorescence intensity was still visible in the C6@T/I-L and C@T-L groups, probably due to the attraction of TPP cations to negatively charged cell membranes, facilitating liposome uptake in normal H9c2 cells. It is speculated that PUE@T/I-L could preferentially enter the ischemic cardiomyocytes in vivo, while the drug uptake in the normal cardiomyocytes is relatively low.
 |
Figure 2 Cellular uptake behavior of free C6, C6@L, C6@I-L, C6@T-L and C6@T/I-L after co-culture with H/R-injured H9c2 cells for 1 h and 4 h. Scale bar: 100 μm. |
As depicted in Figure S6, compared with C6@L, the intracellular fluorescence intensity of the modified liposomes (C6@T-L, C6@I-L, C6@T/I-L) was significantly increased, especially for C6@T/I-L, indicating that lipophilic TPP cations and IMTP on the surface of C6@T/I-L could endow the liposomes with the ability to cross cell membrane barriers and promote cellular uptake. This stems from the synergistic effect of IMTP and TPP, where IMTP contributes to making the liposomes specific for the H/R-injured H9c2 cells membrane and TPP cations are readily attracted to negatively charged plasma membranes, thus facilitating transmembrane transport of PUE@T/I-L.23,46 Notably, the fluorescence intensity of C6@T/I-L was significantly stronger in H/R-injured H9c2 cells than in normal cardiomyocytes (Figure S7). The possible explanation is that the expression of some receptors on the membrane of H/R-injured H9c2 cells is elevated compared with normal conditions, so that IMTP binds more readily to H/R-injured cardiomyocytes, leading to a significant increase in C6@T/L uptake in these cells. It is also hypothesized that PUE@T/I-L could preferentially aggregate in ischemic cardiomyocytes.
Intracellular Distribution and Co-Location Analysis
After entering the cells, the drugs are easily captured and degraded by the lysosome as foreign substances, resulting in a decrease of drugs entering the mitochondria.47 Therefore, it is necessary to investigate the lysosomal escape property of C6@T/I-L. As depicted in Figure 3, the lysosomes were stained with Lyso-Tracker Red, which emitted red fluorescence. The merged images of green and red fluorescence appeared yellow, indicating that free C6 was trapped in the lysosomes at 4 h with the Pearson’s correlation coefficients (R) value of 0.51 ± 0.05. However, TPP-modified liposomes (C6@T-L and C6@T/I-L), even after 4 h of incubation, were not observed to significantly overlap with the lysosomes. The efficient lysosomal escape properties of C6@T-L and C6@T/I-L are attributed to the TPP cations on the liposome surface, which acts as a “proton sponge” upon entry into the lysosomes, increasing ATPase mediated proton and counter ion influx into the lysosome to maintain the organic physiological state and eventually contribute to lysosome expansion and rupture.46,47 Thus, TPP-modified liposomes successfully escaped lysosomal capture, making them accessible for efficient mitochondria-targeting delivery.
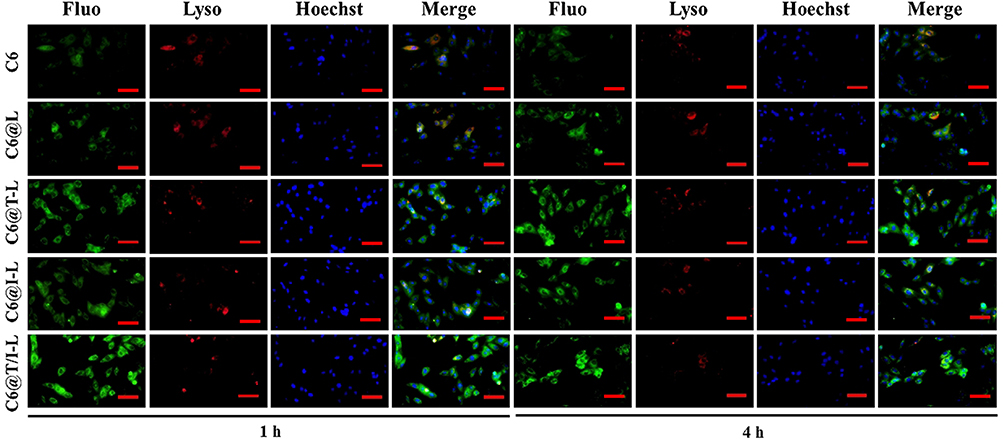 |
Figure 3 Lysosomal escape analysis. Fluorescence microscopy images of free C6, C6@L, C6@I-L, C6@T-L and C6@T/I-L incubated with H/R-injured H9c2 cells for 1 h and 4 h (Green marker for coumarin 6, blue marker for Hoechst-stained nuclei, and red marker for lysosomes). Scale bar: 100 μm. |
In order to investigate the mitochondria-targeting ability of C6@T/I-L, the green fluorescence of C6 was used to explore the subcellular distribution of C6@T/I-L. When the green fluorescent liposomes and the red fluorescent Mitotracker Red are co-localized, their overlap appears yellow color. The results shown in Figure 4 indicated that C6@L and C6@I-L treated groups appeared less bright yellow fluorescence, while bright yellow fluorescence was observed in H9c2 cells treated with C6@T/I-L and C6@T-L, thus inferring that there was a strong electrostatic attraction between the positively charged TPP and the negatively charged mitochondrial membrane, which promoted liposomes targeting to mitochondria. Especially at 4 h, C6@T/I-L and C6@T-L-treated cells showed a clear bright yellow fluorescence, suggesting that the modification of TPP cations on liposomes surface endowed them with mitochondria-targeting capacity. The co-localization efficiency of the drugs with mitochondria was further determined by quantitative analysis by Fiji-image J, the Pearson’s correlation coefficients (R) were often used to quantitatively assess the mitochondria-targeting property of the drug delivery system. Especially at 4 h, the R values of mitochondria with TPP-functional liposomes (C6@T/I-L and C6@T-L) were significantly increased compared to C6@L (R = 0.67 ± 0.02), with 0.87 ± 0.02 and 0.83 ± 0.02, respectively. The result indicated that TPP cations play a very important role in the delivery of TPP-modified liposomes to mitochondria. The reason may be that TPP has lipophilic three phenyl ligands coupled with a positive charge on phosphorus, which enhances the tight binding of the drug carrier to the negatively charged mitochondrial membrane and thus facilitates mitochondrial localization.48 It is worth noting that the R value of C6@T-L was slightly lower than those of C6@T/I-L, the reason was potentially IMTP promoted the fusion of liposomes with the cardiomyocyte membrane,23 resulting in relatively easier access to mitochondria.
 |
Figure 4 Mitochondria-targeting analysis. Fluorescence microscopy images of free C6, C6@L, C6@I-L, C6@T-L and C6@T/I-L incubated with H/R-injured H9c2 cells for 1 h and 4 h (Green marker for coumarin 6, blue marker for DAPI-stained nuclei, and red marker for mitochondria). Scale bar: 100 μm. |
PUE@T/I-L Reduces Mitochondria-Dependent Apoptosis in H9c2 Cells
The effect of PUE@T/I-L on mPTP opening in H/R-injured H9c2 cells was assessed using the mitochondrial Calcein-AM assay and the results were shown in Figure S8a. Compared with the control group, the fluorescence intensity was significantly lower in the H/R group, thus indicating that H/R induced the opening of mPTP in H9c2 cells. Drug treatment could inhibit the opening of mPTP and increase the fluorescence intensity of H/R-injured H9c2 cells. Among them, the PUE@T/I-L showed the most significant inhibition of mPTP opening. The possible reason was that IMTP promoted cellular uptake and mitochondrial targeting of TPP cations, facilitating the accumulation of the drug at the mitochondrial sites and fully exploiting the effect of PUE in inhibiting mTPP opening, thus enhancing the efficacy to a great extent.
As drug inhibition of mPTP opening increases, there is a corresponding decrease in intracellular ROS release. Figure S8b indicated the effect of PUE@T/I-L treatment on ROS level in H/R-injured H9c2 cells. The fluorescence intensity was significantly increased in the H/R group compared with the control group, and it was hypothesized that H/R stimulation induced the opening of mPTP in H9c2 cells, resulting in large intracellular ROS.49 The drug administration groups could inhibit ROS level in the H/R-injured H9c2 cells, and PUE@T/I-L showed the most significant reduction in mitochondrial ROS levels. Superoxide dismutase (SOD) is an enzyme that scavenges oxygen radicals and could assist in the scavenging of excess ROS,50 so the assay of SOD level could reflect the antioxidant activity. Figure S8c showed the effect of PUE@T/I-L treatment on SOD levels in H/R-injured H9c2 cells. The results indicated that PUE@T/I-L performed best in increasing SOD activity.
The CCK-8 assay suggested that drug treatment increased the viability of H9c2 cells with H/R injury. In particular, PUE@T/I-L showed the best performance in increasing cell viability (Figure S8d). These results suggested that PUE@T/I-L treatment significantly reduced apoptosis in H/R-injured H9c2 cells. Hoechst staining results could also further verify the advantage of PUE@T/I-L treatment in reducing the apoptosis rate of H/R-injured H9c2 cells. As shown in Figure S9a and S9b, H/R stimulation increased the number of Hoechst positive cells with condensed and fragmented fluorescent nuclei, which was inhibited by treatment with PUE or PUE loaded liposomes (PUE@L, PUE@T-L, PUE@I-L and PUE@T/I-L). Among them, PUE@T/I-L showed the lowest ratio of Hoechst positive cells. The possible reason for this experimental result was the synergistic effect of IMTP and TPP cations, which facilitated the drug transport to the cellular mitochondria. With a large amount of PUE inhibiting mTPP opening, reducing the release of ROS levels and increasing SOD activity, PUE@T/I-L largely reduced the apoptosis of H9c2 cells and improved the survival of cardiomyocytes.
PUE@T/I-L Attenuate Myocardial I/R Injury
CK-MB and LDH activities are often used clinically as important diagnostic indicators of myocardial enzymes to assess the extent of myocardial I/R injury.51 As shown in Figure 5a, CK-MB and LDH activities were significantly increased in the I/R group compared with the sham group. Free PUE, PUE@L, PUE@I-L and PUE@T/I-L reduced the release of CK-MB and LDH in the myocardial tissue of I/R-injured mice and showed different degrees of inhibition of CK-MB and LDH activities. Among them, PUE@T/I-L showed the strongest inhibitory effect on the elevated CK-MB and LDH activities in mice with myocardial I/R injury, indicating that the degree of myocardial injury was significantly attenuated after PUE@T/I-L treatment. Infarct size is a typical index for visual assessment of the extent of myocardial injury. TTC staining also confirmed the advantage of PUE@T/I-L in reducing myocardial infarct size (Figure 5b). Compared with PUE@L and PUE@I-L treated groups, PUE@T/I-L treatment showed better effect in reducing myocardial infarct size (Figure 5c). This may be explained in terms of the composition of the carrier system. Firstly, liposomes had a better EPR effect at the ischemic myocardial site (Figure S10a) and could accumulate drugs in the ischemic myocardium. The literature reported that liposome extravasation was most likely limited by their large size, resulting in liposomes being more persistent in ischemic myocardium and often located within or near the vasculature.52,53 Secondly, IMTP promoted tight binding of PUE@T/I-L to ischemic cardiomyocytes and induced cellular uptake of the drugs. Finally, the presence of positive and negative charge attraction between TPP cations and negatively charged mitochondrial membranes could induce a 100–500 fold increase in the ability of drug carriers to target mitochondria (Figure S10b).54 For these reasons, PUE@T/I-L could maximize the aggregation of PUE at the mitochondrial effector site of ischemic cardiomyocytes, inhibit the opening of mPTP and largely improve the efficacy of the drugs.
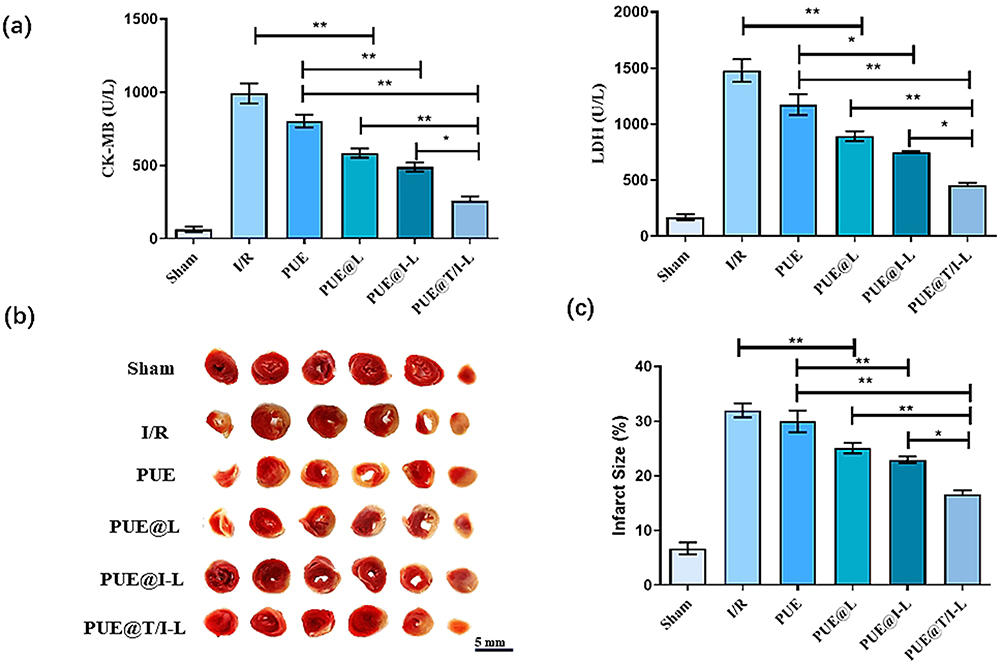 |
Figure 5 The protective effects of PUE@T/I-L in a mouse myocardial I/R injury mode. (a) The activities of CK-MB and LDH in different groups of mice after 24 h of reperfusion. (b) Representative infarcted myocardium in each group base on TTC staining. Those that appear red represent normal myocardium, while those that appear white represent infarcted myocardium. (c) Quantitative assessment of infarct size in each group. (**p < 0.01, *p < 0.05). |
In addition, microscopic morphological changes of myocardial mitochondria in each group could be observed from TEM (Figure 6). The myocardial fibers in the sham group showed neatly arranged tightly, and these mitochondrial membranes were also relatively intact, with clear cristae and normal morphology. However, in the I/R group, cardiac myocytes showed signs of myofilament disorder, marked damage to mitochondrial membranes, reduction or disappearance of mitochondrial cristae, and swelling and rounding of some mitochondrial morphologies.55 After drug treatment, the degree of myocardial mitochondrial damage appeared to be reduced. In particular, PUE@T/I-L may significantly inhibit the abnormal opening of mPTP, reduce the degree of mitochondrial damage, prompt mitochondrial morphology to be basically close to normal. It is speculated that the combined effect of IMTP and TPP cations can promote drug targeting to ischemic myocardial mitochondria, thus better facilitating drug action at the mitochondrial effector sites and maintaining normal mitochondrial morphology.
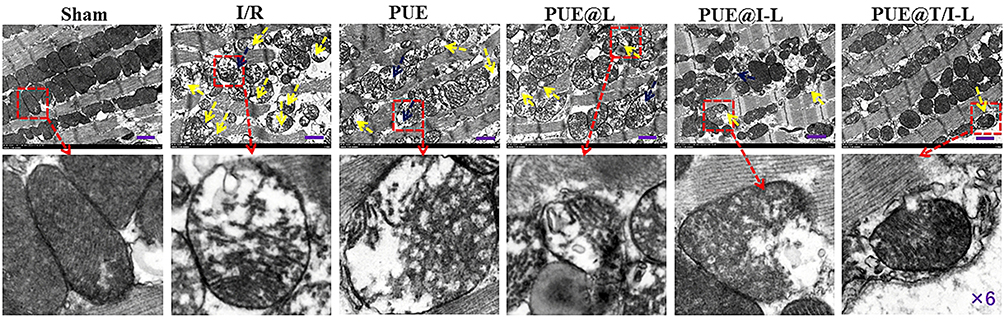 |
Figure 6 TEM images of mitochondrial features in each group of myocardial sections (Mitochondrial swelling appearing as severe deformation is indicated by blue arrows, and mitochondrial membrane rupture appearing as severe damage is indicated by yellow arrows). Scale bar: 1 μm. |
The H&E staining in Figure 7a showed the changes of myocardial micromorphology in each group. Myocardial tissue in the sham group was neatly and regularly arranged, with intact myocardial fibers and no breaks. However, the myocardial tissues of mice in the I/R group exhibited severe cellular disarrangement with swollen and broken myocardial fibers. Some pathological changes were observed in the myocardium of the free PUE-treated group of mice, which may be related to the poor myocardial targeting and bioavailability of PUE, resulting in low drug concentration in the ischemic myocardium. In contrast, the degree of myocardial histopathological damage was significantly reduced in the PUE@T/I-L and PUE@I-L treated groups. Especially in the PUE@T/I-L group, the myocardial tissue was neatly organized and shifted toward normalization. TUNEL staining was performed to quantify the apoptosis of cardiac myocytes in paraffin-embedded sections. As depicted in Figure 7b, the number of TUNEL-positive cells was significantly increased in the I/R group. Compared with the other administered groups, the quantitative results indicated that both PUE@I-L and PUE@T/I-L groups showed fewer apoptotic cells, and PUE@T/I-L were more effective than PUE@I-L in reducing the rate of I/R-induced apoptosis in cardiomyocytes (Figure 7c). Mitochondria damage was an important pathological mechanism in the process of apoptosis in cardiac myocytes. Based on the following aspects, PUE@T/I-L could effectively deliver the drug to the mitochondria, thus enhancing the apoptosis-reducing effect of the drug on cardiomyocytes. With larger particle size, liposomes have a better EPR effect at the ischemic myocardial site, which can aggregate the drug first at the ischemic myocardial site. IMTP then promotes the cellular uptake of PUE@T/I-L by ischemic cardiomyocytes, and finally the mitochondrial targeting of TPP cations facilitates the drug transport to the mitochondrial sites.3,56 In this way, PUE@T/I-L targeted large amounts of PUE into mitochondria to inhibit the opening of mPTP and suppress the cascade effect of caspases, thereby reducing apoptosis in cardiomyocytes.
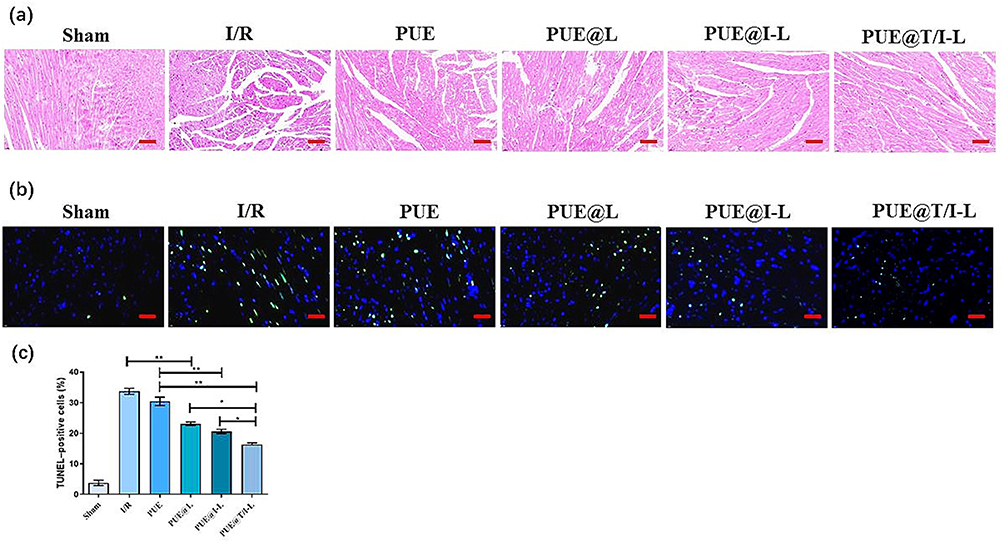 |
Figure 7 The protective effects of PUE@T/I-L in a mouse model of myocardial I/R injury. (a) Representative H&E-stained micrographs; (b) Representative TUNEL-stained micrographs. Green represents TUNEL-stained apoptotic nuclei, blue represents DAPI-stained nuclei; (c) Statistical analysis of TUNEL-positive cells in different groups. Scale bar: 100 μm. (**p < 0.01, *p < 0.05). |
Discussion
Mitochondrial dysfunction has been identified as a significant pathogenesis of myocardial I/R injury and could induce massive apoptosis in cardiomyocytes.57 In the early stage of myocardial I/R, a large amount of ROS is generated in the mitochondria, prompting the opening of mPTP on the mitochondrial membrane, which causes cytochrome C to be released into the cytoplasm from the mitochondria, triggering Caspase-dependent apoptosis in cardiomyocytes.58 In this regard, effective inhibition of mPTP opening and reduction of cardiomyocyte apoptosis at the early stage of reperfusion could be a promising therapeutic strategy for the treatment of myocardial I/R injury.
Current studies have found that PUE could inhibit the opening of mPTP and regulate the physiological function of mitochondria,59 but the lack of myocardial mitochondrial targeting severely limits the clinical efficacy of the drug. In recent years, TPP cations have received increasing attention in drug delivery systems as a candidate molecule that can drive nanomedicine targeting to mitochondria via electrostatic interactions.60,61 However, mitochondrial targeting of TPP alone is insufficient, as the continuous washout of pulsatile blood flow during reperfusion severely impairs drug retention at the ischaemic myocardial site. We prepared TPP cations and IMTP dual-modified liposomes to deliver PUE efficiently into the mitochondria of ischemic cardiomyocytes, giving full play to the drug’s role in inhibiting mPTP opening. The prepared PUE@T/I-L showed a spherical structure with small particle size, which facilitated drug carrier uptake by cells and intracellular transport. Quantitative uptake by flow cytometry showed that the uptake of C6@T/I-L in H/R-injured H9c2 cells was more than that in normal cells, which was due to the targeting of IMTP to H/R-injured cardiomyocytes and relatively weak binding to normal cells.23 This result indicates that PUE@T/I-L could deliver drugs to lesions in ischemic myocardium with relatively little in normal tissue, which not only improves the selectivity of the drug but also reduces the adverse effects. Based on the binding of IMTP to receptors on the membrane of H/R-treated cardiomyocytes and the subsequent mitochondrial targeting of TPP cations, C6@T/I-L actively targeted into the mitochondria of H/R-injured H9c2 cells, and these results were significantly better than those of the other groups. In addition, C6@T/I-L successfully escaped lysosomal capture via the “proton sponge” effect of TPP cations, facilitating more drug targeting into the mitochondria.62 Similarly, our cellular pharmacodynamic results also confirmed the advantages of PUE@T/I-L, which could significantly inhibit the opening of mPTP in H/R-injured cardiomyocytes, reduce the level of ROS, and increase the level of SOD, thus largely enhancing the drug’s inhibition of cardiomyocyte apoptosis. The possible explanation is that the combined targeting effect of IMTP and TPP cations prompted a large amount of PUE to accumulate in the mitochondria of H/R-injured cardiomyocytes, giving full play to the drug’s advantage of accurately regulating the function of damaged mitochondria at the subcellular level. This result indicated that PUE@T/I-L could reduce mitochondria-dependent apoptosis in H/R-injured H9c2 cells. In addition, the results of animal efficacy studies in myocardial I/R injury mouse model also showed that PUE@T/I-L significantly inhibited cardiomyocyte apoptosis and reduced the degree of mitochondrial damage in ischemic cardiomyocytes, thereby reducing myocardial infarct size. Statistical results showed that the effect of PUE@T/I-L in reducing the degree of myocardial I/R injury was also significantly better than that of other groups. In this regard, IMTP binds to receptors on the membrane of ischemic cardiomyocytes and promotes liposomes first accumulate at the site of ischemic myocardium.22 TPP cations are considered to be a potent mitochondrial targeting moiety due to their high lipophilicity and stable cationic charge,63 Based on the combined action of the dual-modified ligands, the efficacy of PUE@T/I-L can therefore be enhanced to a large extent. Taken together, the results of both cellular and animal tests indicated that PUE@T/I-L delivered more PUE to the mitochondria, thereby enhancing the drug inhibition of apoptosis in ischemic cardiomyocytes.
In addition, although the positively charged TPP has been reported to have a risk of hemolysis,64 the PUE@T/I-L also exhibited minor hemolysis, suggesting that they were highly biocompatible for intravenous injection. It was speculated that the steric hindrance of IMTP on the surface of liposomes may reduce the direct binding of erythrocytes to TPP. In addition, negatively charged liposomes may attenuate the positive potential effect of TPP, resulting in PUE@T/I-L remaining negatively charged. In a physiological environment, both erythrocytes and plasma proteins are negatively charged, which may reduce the interaction with negatively charged PUE@T/I-L based on charge repulsion. For these reasons, PUE@T/I-L may reduce PUE-induced hemolysis and are likely to reduce the clinical adverse effects of acute intravascular hemolysis with PUE injection.65,66
The pathological mechanisms of myocardial I/R injury are very complex, in addition to mitochondria-associated apoptosis of cardiomyocytes, there are also a variety of pathological mechanisms, such as ferroptosis and pyroptosis, which are worthy of our subsequent development of some novel drug-carrying systems for in-depth study.67
Conclusion
In summary, we successfully developed PUE@T/I-L co-modified with IMTP and TPP cations and achieved mitochondria-targeted delivery of PUE. In vitro cellular pharmacodynamic results showed that PUE@T/I-L significantly inhibited the opening of mPTP, reduced ROS levels and increased SOD activity, thereby increasing the survival of H/R-damaged H9c2 cells and reducing apoptosis. The results of animal pharmacodynamic experiments indicated that PUE@T/I-L significantly reduced myocardial infarct size, restored normal mitochondrial morphology, and decreased cardiomyocyte apoptosis in a mouse model of myocardial I/R. Altogether, these results suggested that PUE@T/I-L could target ischemic cardiomyocyte mitochondria, inhibited mitochondria-dependent apoptosis and had great potential in enhancing drug reduction of myocardial I/R injury.
Abbreviations
I/R, ischemia-reperfusion; PUE, puerarin; IMTP, ischemic myocardium-targeting peptide; TPP, triphenylphosphonium; ROS, reactive oxygen species; SOD, superoxide dismutase; H/R, hypoxia-reoxygenated; MI, myocardial infarction; mPTP, mitochondrial permeability transition pore; EPR, enhanced permeability and retention; Chol, Cholesterol; SL, soybean lecithin; CTPP, (3-carboxypropyl) triphenylphosphonium bromide; C6, Coumarin-6; FBS, fetal bovine serum; TTC, 2,3,5-triphenyltetrazolium chloride; H&E, hematoxylin and eosin; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; DLS, dynamic laser scattering; TEM, Transmission electron microscope; CCK-8, Cell Counting Kit-8; SD, Sprague-Dawley; PUE@L, PUE-loaded liposomes; PUE@T-L, PUE-loaded liposomes modified by TPP; PUE@I-L, PUE-loaded liposomes modified by IMTP; PUE@T/I-L, PUE-loaded liposomes modified by TPP and IMTP; C6@L, C6-loaded liposomes; C6@T-L, C6-loaded liposomes modified by TPP; C6@I-L, C6-loaded liposomes modified by IMTP; C6@T/I-L, C6-loaded liposomes modified by TPP and IMTP.
Data Sharing Statement
The datasets used or analyzed during the current study available from the corresponding author on reasonable request.
Ethical Approval Statement
All animals were housed in a specific pathogen free environment (ventilated room, 25 ± 1°C, 55-65% humidity, 12 h light/dark cycle) and had free access to standard water and food (SYXK 2022-0012). Ligation of left anterior descending artery is now a commonly used model for constructing myocardial ischemia-reperfusion injury to simulate myocardial ischemia-reperfusion in clinical patients. No other alternative models are available. The mice were anesthetized by 50 μg/g pentobarbital (i.p.). The procedures for care and use of animals were in accordance with the “Regulations of Hunan province on the administration of laboratory animals”, “the ARRIVE guidelines” and “Guiding principles in the Care and Use of Animals” (China). The study protocol was approved by the Laboratory Animal Ethics Committee of Xiangya Second Hospital of Central South University. (IRB approval number: 20220547).
Acknowledgments
This research work is supported by the National Natural Science Foundation of China (81673614), Hunan Provincial Natural Scientific Foundation (No. 2024JJ8126, 2020JJ4128), Health Research Project of Hunan Provincial Health Commission (No. W20243065), Fundamental Research Funds for the Central Universities of Central South University (No. 2024ZZTS0995).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declared no conflicts of interest in this work.
References
1. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92–100. doi:10.1172/JCI62874
2. Hausenloy DJ, Yellon DM. Targeting myocardial reperfusion injury--the search continues. N Engl J Med. 2015;373(11):1073–1075. doi:10.1056/NEJMe1509718
3. Biswas S, Dodwadkar NS, Deshpande PP, Torchilin VP. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J Control Release. 2012;159(3):393–402. doi:10.1016/j.jconrel.2012.01.009
4. Zhou M, Yu Y, Luo X, et al. Myocardial ischemia-reperfusion injury: therapeutics from a mitochondria-centric perspective. Cardiology. 2021;146(6):781–792. doi:10.1159/000518879
5. Han Y, Wang H, Wang Y, Dong P, Jia J, Yang S. Puerarin protects cardiomyocytes from ischemia-reperfusion injury by upregulating LncRNA ANRIL and inhibiting autophagy. Cell Tissue Res. 2021;385(3):739–751. doi:10.1007/s00441-021-03463-2
6. Yue PF, Hai-Long Yuan HL, Zhu WF, et al. The study to reduce the hemolysis side effect of puerarin by a submicron emulsion delivery system. Biol Pharm Bull. 2008;31(1):45–50. doi:10.1248/bpb.31.45
7. Hou SZ, Su ZR, Chen SX, et al. Role of the interaction between puerarin and the erythrocyte membrane in puerarin-induced hemolysis. Chem Biol Interact. 2011;192(3):184–192. doi:10.1016/j.cbi.2011.03.007
8. Gao Q, Pan HY, Bruce IC, Xia Q. Improvement of ventricular mechanical properties by puerarin involves mitochondrial permeability transition in isolated rat heart during ischemia and reperfusion. Conf Proc IEEE Eng Med Biol Soc. 2005;2005:5591–5594. doi:10.1109/IEMBS.2005.1615753
9. Gao Q, Pan HY, Qiu S, et al. Atractyloside and 5-hydroxydecanoate block the protective effect of puerarin in isolated rat heart. Life Sci. 2006;79(3):217–224. doi:10.1016/j.lfs.2005.12.040
10. Liu B, Yan W, Luo L, et al. Macrophage membrane camouflaged reactive oxygen species responsive nanomedicine for efficiently inhibiting the vascular intimal hyperplasia. J Nanobiotechnology. 2021;19(1):374. doi:10.1186/s12951-021-01119-5
11. Han M, Vakili MR, Soleymani Abyaneh H, Molavi O, Lai R, Lavasanifar A. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol Pharm. 2014;11(8):2640–2649. doi:10.1021/mp500038g
12. Li WQ, Wu JY, Xiang DX, et al. Micelles loaded with puerarin and modified with triphenylphosphonium cation possess mitochondrial targeting and demonstrate enhanced protective effect against isoprenaline-induced H9c2 cells apoptosis. Int J Nanomed. 2019;14:8345–8360. doi:10.2147/IJN.S219670
13. Wang J, Zhang S, Di L. Acute myocardial infarction therapy: in vitro and in vivo evaluation of atrial natriuretic peptide and triphenylphosphonium dual ligands modified, baicalin-loaded nanoparticulate system. Drug Deliv. 2021;28(1):2198–2204. doi:10.1080/10717544.2021.1989086
14. Zhao YP, Wang F, Jiang W, et al. A mitochondrion-targeting tanshinone IIA derivative attenuates myocardial hypoxia reoxygenation injury through a SDH-dependent antioxidant mechanism. J Drug Target. 2019;27(8):896–902. doi:10.1080/1061186X.2019.1566338
15. Horwitz LD, Kaufman D, Keller MW, Kong Y. Time course of coronary endothelial healing after injury due to ischemia and reperfusion. Circulation. 1994;90(5):2439–2447. doi:10.1161/01.cir.90.5.2439
16. Wexler L, Bergel DH, Gabe IT, Makin GS, Mills CJ. Velocity of blood flow in normal human venae cavae. Circ Res. 1968;23(3):349–359. doi:10.1161/01.res.23.3.349
17. Lipinski MJ, Albelda MT, Frias JC, et al. Multimodality imaging demonstrates trafficking of liposomes preferentially to ischemic myocardium. Cardiovasc Revasc Med. 2016;17(2):106–112. doi:10.1016/j.carrev.2016.01.003
18. Matoba T, Koga JI, Nakano K, Egashira K, Tsutsui H. Nanoparticle-mediated drug delivery system for atherosclerotic cardiovascular disease. J Cardiol. 2017;70(3):206–211. doi:10.1016/j.jjcc.2017.03.005
19. Levchenko TS, Hartner WC, Torchilin VP. Liposomes for cardiovascular targeting. Ther Deliv Apr. 2012;3(4):501–514. doi:10.4155/tde.12.18
20. Dasa SSK, Suzuki R, Gutknecht M, et al. Development of target-specific liposomes for delivering small molecule drugs after reperfused myocardial infarction. J Control Release. 2015;220(Pt A):556–567. doi:10.1016/j.jconrel.2015.06.017
21. Torchilin VP. Immunoliposomes and PEGylated immunoliposomes: possible use for targeted delivery of imaging agents. Immunomethods. 1994;4(3):244–258. doi:10.1006/immu.1994.1027
22. Kanki S, Jaalouk DE, Lee S, Yu AY, Gannon J, Lee RT. Identification of targeting peptides for ischemic myocardium by in vivo phage display. J Mol Cell Cardiol. 2011;50(5):841–848. doi:10.1016/j.yjmcc.2011.02.003
23. Wang X, Chen Y, Zhao Z, et al. Engineered exosomes with ischemic myocardium-targeting peptide for targeted therapy in myocardial infarction. J Am Heart Assoc. 2018;7(15):e008737. doi:10.1161/jaha.118.008737
24. Lei D, Li B, Isa Z, Ma X, Zhang B. Hypoxia-elicited cardiac microvascular endothelial cell-derived exosomal miR-210-3p alleviate hypoxia/reoxygenation-induced myocardial cell injury through inhibiting transferrin receptor 1-mediated ferroptosis. Tissue Cell. 2022;79:101956. doi:10.1016/j.tice.2022.101956
25. Qiu Z, He Y, Ming H, Lei S, Leng Y, Xia ZY. Lipopolysaccharide (LPS) aggravates high glucose- and hypoxia/reoxygenation-induced injury through activating ros-dependent NLRP3 inflammasome-mediated pyroptosis in H9C2 cardiomyocytes. J Diabetes Res. 2019;2019:8151836. doi:10.1155/2019/8151836
26. Kong HL, Li ZQ, Zhao YJ, et al. Ginsenoside Rb1 protects cardiomyocytes against CoCl2-induced apoptosis in neonatal rats by inhibiting mitochondria permeability transition pore opening. Acta Pharmacol Sin. 2010;31(6):687–695. doi:10.1038/aps.2010.52
27. Yu W, Sun S, Xu H, Li C, Ren J, Zhang Y. TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics. 2020;10(24):11244–11263. doi:10.7150/thno.46883
28. Zhai M, Li B, Duan W, et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. 2017;63(2):e12419. doi:10.1111/jpi.12419
29. Li W, Wu J, Zhang J, et al. Puerarin-loaded PEG-PE micelles with enhanced anti-apoptotic effect and better pharmacokinetic profile. Drug Deliv. 2018;25(1):827–837. doi:10.1080/10717544.2018.1455763
30. Xu H, Cheng J, Wang X, et al. Resveratrol pretreatment alleviates myocardial ischemia/reperfusion injury by inhibiting STIM1-mediated intracellular calcium accumulation. J Physiol Biochem. 2019;75(4):607–618. doi:10.1007/s13105-019-00704-5
31. Vandergriff A, Huang K, Shen D, et al. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics. 2018;8(7):1869–1878. doi:10.7150/thno.20524
32. Gao E, Lei YH, Shang X, et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107(12):1445–1453. doi:10.1161/CIRCRESAHA.110.223925
33. Zhang G, Wang X, Li C, et al. Integrated stress response couples mitochondrial protein translation with oxidative stress control. Circulation. 2021;144(18):1500–1515. doi:10.1161/CIRCULATIONAHA.120.053125
34. Charles L, Agius T, Filz von Reiterdank I, et al. Modified tail vein and penile vein puncture for blood sampling in the rat model. J Vis Exp. 2023;196:e65513. doi:10.3791/65513
35. Stumpner J, Redel A, Kellermann A, et al. Differential role of Pim-1 kinase in anesthetic-induced and ischemic preconditioning against myocardial infarction. Anesthesiology. 2009;111(6):1257–1264. doi:10.1097/ALN.0b013e3181bdf9f4
36. Li L, Wang Y, Guo R, et al. Ginsenoside Rg3-loaded, reactive oxygen species-responsive polymeric nanoparticles for alleviating myocardial ischemia-reperfusion injury. J Control Release. 2020;317:259–272. doi:10.1016/j.jconrel.2019.11.032
37. de Couto G, Gallet R, Cambier L, et al. Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation. 2017;136(2):200–214. doi:10.1161/CIRCULATIONAHA.116.024590
38. Li Y, Wu J, Yang M, et al. Physiological evidence of mitochondrial permeability transition pore opening caused by lipid deposition leading to hepatic steatosis in db/db mice. Free Radic Biol Med. 2021;162:523–532. doi:10.1016/j.freeradbiomed.2020.11.009
39. Okumura M, Ichihara H, Matsumoto Y. Hybrid liposomes showing enhanced accumulation in tumors as theranostic agents in the orthotopic graft model mouse of colorectal cancer. Drug Deliv. 2018;25(1):1192–1199. doi:10.1080/10717544.2018.1475517
40. Lee CK, Atibalentja DF, Yao LE, Park J, Kuruvilla S, Felsher DW. Anti-PD-L1 F(ab) conjugated PEG-PLGA nanoparticle enhances immune checkpoint therapy. Nanotheranostics. 2022;6(3):243–255. doi:10.7150/ntno.65544
41. Wu H, Zhu L, Torchilin VP. pH-sensitive poly(histidine)-PEG/DSPE-PEG co-polymer micelles for cytosolic drug delivery. Biomaterials. 2013;34(4):1213–1222. doi:10.1016/j.biomaterials.2012.08.072
42. Dong Z, Guo J, Xing X, Zhang X, Du Y, Lu Q. RGD modified and PEGylated lipid nanoparticles loaded with puerarin: formulation, characterization and protective effects on acute myocardial ischemia model. Biomed Pharmacother. 2017;89:297–304. doi:10.1016/j.biopha.2017.02.029
43. Zhang S, Li J, Hu S, Wu F, Zhang X. Triphenylphosphonium and D-alpha-tocopheryl polyethylene glycol 1000 succinate-modified, tanshinone IIA-loaded lipid-polymeric nanocarriers for the targeted therapy of myocardial infarction. Int J Nanomed. 2018;13:4045–4057. doi:10.2147/IJN.S165590
44. Shao Y, Luo W, Guo Q, Li X, Zhang Q, Li J. In vitro and in vivo effect of hyaluronic acid modified, doxorubicin and gallic acid co-delivered lipid-polymeric hybrid nano-system for leukemia therapy. Drug Des Devel Ther. 2019;13:2043–2055. doi:10.2147/DDDT.S202818
45. Rivolta I, Panariti A, Lettiero B, et al. Cellular uptake of coumarin-6 as a model drug loaded in solid lipid nanoparticles. J Physiol Pharmacol. 2011;62(1):45–53.
46. Choi YS, Kwon K, Yoon K, Huh KM, Kang HC. Photosensitizer-mediated mitochondria-targeting nanosized drug carriers: subcellular targeting, therapeutic, and imaging potentials. Int J Pharm. 2017;520(1–2):195–206. doi:10.1016/j.ijpharm.2017.02.013
47. Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci U S A. 2012;109(40):16288–16293. doi:10.1073/pnas.1210096109
48. Lu J, Li R, Mu B, et al. Multiple targeted doxorubicin-lonidamine liposomes modified with p-hydroxybenzoic acid and triphenylphosphonium to synergistically treat glioma. Eur J Med Chem. 2022;230:114093. doi:10.1016/j.ejmech.2021.114093
49. Yao H, Xie Q, He Q, et al. Pretreatment with panaxatriol saponin attenuates mitochondrial apoptosis and oxidative stress to facilitate treatment of myocardial ischemia-reperfusion injury via the regulation of Keap1/Nrf2 activity. Oxid Med Cell Longev. 2022;2022:9626703. doi:10.1155/2022/9626703
50. Zhang YJ, Lu SJ, Wang HY, Qi QL. Effects of electroacupuncture with dexmedetomidine on myocardial ischemia/reperfusion injury in rats. Ann Palliat Med. 2022;11(9):2916–2922. doi:10.21037/apm-22-969
51. Sun B, Liu S, Hao R, Dong X, Fu L, Han B. RGD-PEG-PLA delivers MiR-133 to infarct lesions of acute myocardial infarction model rats for cardiac protection. Pharmaceutics. 2020;12(6):575. doi:10.3390/pharmaceutics12060575
52. Paulis LE, Geelen T, Kuhlmann MT, et al. Distribution of lipid-based nanoparticles to infarcted myocardium with potential application for MRI-monitored drug delivery. J Control Release. 2012;162(2):276–285. doi:10.1016/j.jconrel.2012.06.035
53. Geelen T, Paulis LE, Coolen BF, Nicolay K, Strijkers GJ. Passive targeting of lipid-based nanoparticles to mouse cardiac ischemia-reperfusion injury. Contrast Media Mol Imaging. 2013;8(2):117–126. doi:10.1002/cmmi.1501
54. Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 2008;1777(7–8):1028–1031. doi:10.1016/j.bbabio.2008.03.029
55. Zhang CX, Cheng Y, Liu DZ, et al. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J Nanobiotechnology. 2019;17(1):18. doi:10.1186/s12951-019-0451-9
56. Kang JH, Ko YT. Enhanced subcellular trafficking of resveratrol using mitochondriotropic liposomes in cancer cells. Pharmaceutics. 2019;11(8):423. doi:10.3390/pharmaceutics11080423
57. Wang J, Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. 2020;10(10):1866–1879. doi:10.1016/j.apsb.2020.03.004
58. Morciano G, Bonora M, Campo G, et al. Mechanistic role of mPTP in ischemia-reperfusion injury. Adv Exp Med Biol. 2017;982:169–189. doi:10.1007/978-3-319-55330-6_9
59. Li F, Wang Y, Li W, et al. Enhanced protection against hypoxia/reoxygenation-induced apoptosis in H9c2 cells by puerarin-loaded liposomes modified with matrix metalloproteinases-targeting peptide and triphenylphosphonium. J Liposome Res. 2023;33(4):378–391. doi:10.1080/08982104.2023.2193845
60. Shueng PW, Yu LY, Hou HH, Chiu HC, Lo CL. Charge conversion polymer-liposome complexes to overcome the limitations of cationic liposomes in mitochondrial-targeting drug delivery. Int J Mol Sci. 2022;23(6):3080. doi:10.3390/ijms23063080
61. Zhang J, Gu L, Jiang Y, et al. Artesunate-nanoliposome-TPP, a novel drug delivery system that targets the mitochondria, attenuates cisplatin-induced acute kidney injury by suppressing oxidative stress and inflammatory effects. Int J Nanomed. 2024;19:1385–1408. doi:10.2147/ijn.S444076
62. Zhang L, Cao X, Chen J, et al. Co-delivery of siBcl-2 and PTX with mitochondria-targeted functions to overcoming multidrug resistance. Int J Pharm. 2024;654:123970. doi:10.1016/j.ijpharm.2024.123970
63. Shi X, Lv G, Sun X, Cao D, Wang G, Chang Y. Amphiphilic copolymer and TPGS mixed magnetic hybrid micelles for stepwise targeted co-delivery of DOX/TPP–DOX and image-guided chemotherapy with enhanced antitumor activity in liver cancer. RSC Adv. 2017;7(41):25694–25701. doi:10.1039/C7RA00597K
64. Kuznetsova DA, Gaynanova GA, Vasileva LA, et al. Mitochondria-targeted cationic liposomes modified with alkyltriphenylphosphonium bromides loaded with hydrophilic drugs: preparation, cytotoxicity and colocalization assay. J Mater Chem B. 2019;7(46):7351–7362. doi:10.1039/c9tb01853k
65. Zhao GJ, Hou N, Cai SA, et al. Contributions of Nrf2 to puerarin prevention of cardiac hypertrophy and its metabolic enzymes expression in rats. J Pharmacol Exp Ther. 2018;366(3):458–469. doi:10.1124/jpet.118.248369
66. Jiang FQ, Yuan Y, Xiang ZJ, et al. Effects of chloride channels on hemolysis induced by puerarin injection. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2018;34(5):441–444449. doi:10.12047/j.cjap.5681.2018.100
67. Xiang Q, Yi X, Zhu XH, Wei X, Jiang DS. Regulated cell death in myocardial ischemia-reperfusion injury. Trends Endocrinol Metab. 2024;35(3):219–234. doi:10.1016/j.tem.2023.10.010
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.