")
Back to Journals » International Journal of Nanomedicine » Volume 20
Reactive Oxygen Species-Responsive Pyroptosis Nanoinitiators Promote Immune Cell Infiltration and Activate Anti-Tumor Immune Response
Authors Yan H , Liu Y, Wang M, Shu Z, Fang X , Li Z
Received 18 November 2024
Accepted for publication 23 March 2025
Published 2 April 2025 Volume 2025:20 Pages 4069—4084
DOI https://doi.org/10.2147/IJN.S503580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dongwoo Khang
Hongbo Yan, Yilun Liu, Maoshan Wang, Zhenbo Shu, Xuedong Fang, Zhongmin Li
Department of Gastrointestinal and Colorectal Surgery, China-Japan Union Hospital of Jilin University, Changchun, 130033, People’s Republic of China
Correspondence: Xuedong Fang, Email [email protected] Zhongmin Li, Email [email protected]
Background: Immunotherapy, particularly immune checkpoint inhibitors, has become the standard treatment strategy for diverse malignant tumors. However, the inadequate infiltration of immune cells in tumors coupled with the immunosuppressive tumor microenvironment severely hinders the efficacy of immunotherapy.
Methods: A poly(ethylene glycol)-block-poly(lysine) copolymer (mPEG-b-PLL) was prepared through ring-opening polymerization and deprotection, and thioketal (TK) was attached to the amino group of mPEG-b-PLL via the condensation reaction to obtain mPEG-b-PLL-TK. Doxorubicin (DOX) and decitabine (DAC) were encapsulated in mPEG-b-PLL-TK to prepare the pyroptosis nanoinitiator (NP/(DAC+DOX)). The drug release behavior, cellular uptake, pyroptosis-triggering performance, and cytotoxicity of NP/(DAC+DOX) were evaluated in vitro experiments. The in vivo pharmacokinetics and biodistribution of NP/(DAC+DOX) were assessed through fluorescence imaging and high-performance liquid chromatography analysis. CT26 and 4T1 tumor-bearing mouse models were established to evaluate the anti-tumor efficacy, pyroptosis-triggering performance, and immune activation effects of NP/(DAC+DOX).
Results: NP/(DAC+DOX) exhibited excellent reactive oxygen species (ROS)-responsive drug release behavior and could be effectively taken up by tumor cells. Experiments both in vitro and in vivo demonstrated that NP/(DAC+DOX) effectively triggered pyroptosis in tumor cells, which was attributed to the DOX-induced activation of caspase-3 and the upregulation of GSDME expression caused by DAC. Following intravenous administration, NP/(DAC+DOX) specifically aggregated in tumor tissues. NP/(DAC+DOX) significantly suppressed tumor growth and extended the survival time of tumor-bearing mice. Furthermore, NP/(DAC+DOX) promoted dendritic cell maturation, enhanced the infiltration of cytotoxic T lymphocytes within the tumor, and decreased the proportion of myeloid-derived suppressor cells.
Conclusion: This study developed a ROS-responsive pyroptosis nanoinitiator to precisely induce the pyroptosis of tumor cells, thereby enhancing intratumoral immune cell infiltration and activating anti-tumor immune responses.
Keywords: pyroptosis, reactive oxygen species-responsive nanomicelles, immune cell infiltration, immunotherapy
Introduction
Tumor immunotherapy is a revolutionary cancer treatment option that activates the patient’s immune system to recognize and attack cancer cells. In recent years, there has been significant progress in the development of different immunotherapy strategies, such as immune checkpoint inhibitors (ICIs), adoptive cell transfer therapy, and cytokine immunotherapy.1–3 Collectively, these techniques represent a new paradigm in cancer treatment. Among these, ICIs are the most widely used and have become the standard treatment for a variety of malignancies. A growing number of promising therapeutic benefits of ICIs have been reported. For example, a Phase II randomized trial of patients with early-stage or isolated lung parenchymal recurrent node-negative non-small cell lung cancer found that stereotactic ablative radiotherapy (SABR) applied in combination with nivolumab increased the 4-year event-free survival rate from 53% to 77% compared with SABR alone, which may enable some patients to follow a “surgery and chemotherapy free” treatment approach.4 Although ICIs exhibit powerful anti-tumor effects, a low response rate and serious side effects remain major limitations to their clinical application.1 In the case of metastatic colorectal cancer, for instance, immunotherapy is only effective in patients with high microsatellite instability or mismatch repair deficiency, who comprise only approximately 5% of the patient population.5 Therefore, enhancing the immunotherapy response rate is a clinical challenge that urgently needs to be addressed.
It has been found that immunotherapy-resistant tumors have no or very little T-cell infiltration. The tumor immune microenvironment can be classified into the following three different phenotypes based on the T-cell infiltration degree: immune-desert, immune-excluded, and immune-inflamed.6 The first two are also known as “cold tumors”, which are non-inflammatory tumors that are not sensitive to ICI therapy. The last type is alternatively called a “hot tumor”, which has abundant T-cell infiltration and effectively responds to immunotherapy. Intratumoral T-cell infiltration can serve as an important prognostic indicator of tumors.7 Therefore, promoting T-cell infiltration represents an important tool to improve immunotherapy efficacy. Furthermore, immunosuppressive cells and immunosuppressive factors in the tumor microenvironment (TME) make important contributions to tumor immune escape. For example, myeloid-derived suppressor cells (MDSCs) are important immunosuppressive cells in the TME. These cells have the ability to strongly inhibit the anti-tumor activities of T-cells and natural killer (NK) cells and stimulate regulatory T-cells, which leads to tumor progression. In addition, MDSCs enhance resistance to immune checkpoint inhibition.8
Pyroptosis, a recently reported mode of Gasdermin (GSDM)-dependent programmed cell death, manifests in the form of continuous cell swelling until the cell membrane ruptures, followed by the release of cellular contents, which then activates strong inflammatory and immune responses.9 When undergoing pyroptosis, tumor cells release tumor antigens as well as numerous pro-inflammatory factors, such as interleukin-1β (IL-1β), interleukin-18 (IL-18), adenine nucleoside triphosphate (ATP), and high-mobility-group protein B1 (HMGB1), which enhance antigen presentation in addition to immune cell infiltration and activation.10 In previous work, Shao et al designed a bioorthogonal chemical system to deliver GSDM protein to tumor cells in mice and found that less than 15% of tumor cells undergoing pyroptosis was sufficient to clear an entire 4T1 mammary tumor graft.11 Single-cell RNA sequencing (RNAseq) demonstrated that the proportions of CD4+ T cells, CD8+ T cells, NK cells, and M1-type macrophages in treated tumors rose significantly, while the proportions of monocytes, neutrophils, MDSCs, and M2-type macrophages declined. Lieberman et al reported that initiating tumor cell pyroptosis through enhancing the expression of Gasdermin E (GSDME) increased the number of tumor-infiltrating NK cells and CD8+ T cells and promoted their anti-tumor function. In addition, the elevated expression of GSDME promoted the phagocytosis of tumor cells by tumor-associated macrophages.12 Altogether, these findings suggest that pyroptosis holds great potential for application in anti-tumor immunotherapy. Research has demonstrated that certain chemotherapeutic drugs (eg, doxorubicin (DOX), cisplatin, and paclitaxel) can not only cause apoptosis but also induce pyroptosis through the caspase-3/GSDME pathway, and that converting chemotherapeutic drug-induced apoptosis into pyroptosis enhances the anti-tumor effects of these drugs.13 Unfortunately, GSDME generally has low expression in most tumors due to high methylation of the GSDME promoter, thus leading to the inability of chemotherapeutic agents to induce effective pyroptosis. As a result, administering chemotherapeutic agents (eg, DOX) and demethylating agents (eg, decitabine (DAC)) in combination acts synergistically to trigger strong tumor cell pyroptosis, thereby significantly enhancing T-cell infiltration in tumor tissues while simultaneously inhibiting the immunosuppressive TME. However, the optimal ratio of chemotherapeutic agents to demethylating agents is not clear, and the combination therapy suffers from a lack of targeting to tumor tissues and substantial side effects.
Nanotechnology represents a new approach for employing pyroptosis in anti-tumor immunotherapy.14 First, nanomaterials improve the biocompatibility and stability of pyroptosis-inducing drugs. Second, nanomaterials prolong drug circulation by limiting premature release and clearance by the immune system. Moreover, nanomaterials can selectively aggregate in tumor tissues due to the enhanced permeability retention (EPR) effect or active targeting and release pyroptosis-inducing drugs in response to precise stimuli from the TME, thereby avoiding damage to normal tissues or cells. Finally, multiple drugs can be loaded into nanomaterials at the same time, providing an effective vehicle for pyroptosis-based combination therapy. Importantly, nanomaterials can deliver multiple drugs to the tumor tissues in a fixed ratio, which improves the effectiveness of the combination therapy and reduces side effects.15
In this study, a reactive oxygen species (ROS)-responsive pyroptosis nanoinitiator was developed to induce tumor cell pyroptosis with precision and activate anti-tumor immune effects (Scheme 1). The findings revealed that the co-administration of DAC and DOX effectively induced pyroptosis in CT26 and 4T1 cells, and the most significant tumor cell pyroptosis occurred at DAC and DOX concentrations of 50 μg/mL and 10 µg/mL, respectively. A poly(ethylene glycol)-block-poly(lysine) copolymer (mPEG-b-PLL) was then prepared through ring-opening polymerization and deprotection, and thioketal (TK) was attached to the amino group of mPEG-b-PLL via the condensation reaction to produce mPEG-b-PLL-TK. DAC and DOX were loaded into mPEG-b-PLL-TK at a mass ratio of 5:1 in order to prepare drug-loaded nanomicelles (NP/(DAC+DOX)). NP/(DAC+DOX) displayed superior ROS-responsive drug release behavior and was efficiently taken up by the tumor cells. NP/(DAC+DOX) successfully induced pyroptosis in CT26 and 4T1 cells, likely due to the upregulated GSDME expression in tumor tissues caused by DAC, along with the activation of caspase-3 by DOX. The results of RNAseq verified the upregulated expression of genes related to pyroptosis and immune activation following NP/(DAC+DOX) administration. In vivo experiments demonstrated that NP/(DAC+DOX) could efficiently accumulate in tumor tissues and maintain an extensive circulation time. NP/(DAC+DOX) exhibited the optimum tumor suppression effect in CT26 and 4T1 tumor-bearing mice. Furthermore, NP/(DAC+DOX) promoted dendritic cell (DC) maturation, enhanced the intratumoral infiltration of T cells, and reduced the proportion of MDSCs, implying that NP/(DAC+DOX) activated the immune response and remodeled the tumor immune microenvironment. Therefore, triggering tumor cell pyroptosis represents an effective strategy to enhance anti-tumor immunotherapy, and the ROS-responsive pyroptosis nanoinitiator prepared in this study has broad clinical applications.
 |
Scheme 1 Schematic illustration of the preparation and immune activation mechanism of the ROS-responsive pyroptosis nanoinitiator (NP/(DAC+DOX)). |
Results and Discussion
Preparation and Characterization of ROS-Responsive Pyroptosis Nanoinitiators
Polyamino acid polymers are frequently employed in drug delivery due to their characteristics of good modifiability, biodegradability, biocompatibility, and abundant secondary structures. In this study, monomethoxy polyethylene glycol amine (mPEG-NH2) was utilized as an initiator, and mPEG-block-poly(N(ε)-benzyloxycarbonyl-L-lysine) (mPEG-b-P(Cbz-Lys)) was generated through the ring-opening polymerization of N(ε)-benzyloxycarbonyl-L-lysine N-carboxyanhydride (Cbz-Lys NCA), while mPEG-b-PLL was then produced via the deprotection of mPEG-b-P(Cbz-Lys) in trifluoroacetic acid and 33 wt% hydrogen bromide/acetic acid solution (Scheme S1A). The chemical structure of mPEG-b-PLL was obtained via proton nuclear magnetic resonance (1H NMR) spectrum (Figure S1A). TK was synthesized from anhydrous 3-mercaptopropionic acid and anhydrous acetone through a typical ketal reaction (Scheme S1B), and 1H NMR was conducted to verify its chemical structure (Figure S1B). mPEG-b-PLL-TK was then prepared through the condensation reaction between mPEG-b-PLL and TK catalyzed by diisopropylcarbodiimide/4-dimethylaminopyridine (DIC/DMAP) (Scheme S1C), and 1H NMR confirmed the successful synthesis of mPEG-b-PLL-TK (Figure S1C).
DAC and DOX were encapsulated in the mPEG-b-PLL-TK copolymer to prepare ROS-responsive nanoparticles (NP/(DAC+DOX)). The drug loading content (DLC) of NP/(DAC+DOX) was 12.21% for DAC and 2.34% for DOX. Meanwhile, the drug loading efficiency (DLE) for DAC and DOX were 53.7% and 51.5%, respectively. The hydrodynamic diameter (Dh) of NP/(DAC+DOX) detected by dynamic laser scattering was 169.07 ± 11.80 nm, which facilitated its accumulation in tumor tissue. The polydispersity index (PDI) and zeta potential of NP/(DAC+DOX) was 0.209 and −12.9 MV, respectively. Transmission electron microscopy (TEM) showed that NP/(DAC+DOX) had a uniform spherical morphology (Figure 1A).
 |
Figure 1 In vitro characterization of NP/(DAC+DOX). (A) TEM morphologies and Dhs of NP/(DAC+DOX). The release of (B) DAC and (C) DOX from NP/(DAC+DOX) in PBS and in PBS containing H2O2. The cell uptake of (D) DAC and (E) DOX after incubation with CT26 for 1 and 4 h. (F) In vitro cytotoxicity of DAC, DOX, DAC+DOX and NP/(DAC+DOX) on CT26 cells. The concentrations of DAC used were 7.8125, 15.625, 31.25, 62.5, 125.0 and 250.0 μg/mL. The concentrations of DOX used were 1.5625, 3.125, 6.25, 12.5, 25.0 and 50.0 μg/mL. All statistical data are presented as mean ± SD (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001). |
To evaluate the ROS-responsive drug release behavior, NP/(DAC+DOX) was incubated in PBS and in PBS containing 1.0 × 10–3 M hydrogen peroxide (H2O2). In PBS, DAC and DOX were slowly released from NP/(DAC+DOX), with total release rates of 48.41% and 53.69%, respectively, at 72 h. The total release rates of DAC and DOX within 72 h in PBS containing H2O2 were 62.70% and 64.99%, respectively (Figure 1B and C). These rates were significantly higher than those found in PBS, which demonstrated that H2O2 enhanced the drug release from nanoparticles. Because tumor cells contain significantly higher levels of ROS than normal cells, the superior ROS-responsiveness of NP/(DAC+DOX) ensures that it can release drugs selectively in tumor cells.16
In vitro Cellular Uptake and Cytotoxicity of NP/(DAC+DOX)
High-performance liquid chromatography (HPLC) was conducted to examine the cellular uptake of NP/(DAC+DOX). CT26 cells were alternately incubated with DAC+DOX and NP/(DAC+DOX). DAC+DOX and NP/(DAC+DOX) were both effectively internalized by tumor cells; however, the drug accumulation of NP/(DAC+DOX) in tumor cells was lower than that of DAC+DOX (Figure 1D and E), which could be due to the fact that free drugs are more easily diffused into tumor cells, while nanomedicine usually entered tumor cells via endocytosis.17 Notably, after 4 h of incubation, the uptake rate of NP/(DAC+DOX) by CT26 cells exceeded 65.00%, which supported its induction of pyroptosis and killing of tumor cells.
The cytotoxicity of polymer micelle, DAC, DOX, DAC+DOX, and NP/(DAC+DOX) toward CT26 or 4T1 tumor cells was assessed according to the Cell Counting Kit-8 (CCK-8) method. As expected, mPEG-b-PLL-TK exhibited no cytotoxicity towards CT26 cells, confirming its potential as a promising drug carrier (Figure S2). The results showed that the proliferation of CT26 cells was not significantly suppressed by DAC. In contrast, DOX, DAC+DOX, and NP/(DAC+DOX) exhibited stronger cytotoxicity with DOX half-maximal inhibitory concentrations (IC50s) of 5.66 ± 0.08 µg/mL, 2.67 ± 0.10 µg/mL, and 2.28 ± 0.05 µg/mL, respectively (Figure 1F). Similar results were observed in 4T1 cells. The DOX IC50s of DOX, DAC+DOX, and NP/(DAC+DOX) were 17.14 ± 1.39 µg/mL,13.64 ± 0.97 µg/mL, and 10.68 ± 0.57 µg/mL, respectively (Figure S3). Notably, NP/(DAC+DOX) showed the strongest cytotoxicity with the lowest IC50. Additionally, DAC+DOX showed greater cytotoxicity than free DAC and DOX, indicating that DAC and DOX have synergistic anti-tumor effects. DNA methylation is an epigenetic modification that regulates gene transcription and preserves the stability of the genome. As a DNA methyltransferase (DNMT) inhibitor, DAC regulates cellular genetics as well as metabolism at low doses (< 135 mg m–2 cycle–1) and induces cytotoxicity at high doses (> 500 mg m–2 cycle–1).18 We found that DAC exhibited only a slight inhibitory effect on the CT26 and 4T1 proliferation, even at the highest concentration (250 µg/mL), which was consistent with previous reports. The hepatotoxicity induced by DNMT inhibitors limits their utilization in the treatment of solid tumors.19 Therefore, our proposed approach of administering low-dose DAC in combination with the chemotherapeutic agent DOX to trigger tumor cell pyroptosis and promote immune cell infiltration may serve as a valuable reference to support the further development of effective therapeutic strategies.
Pyroptosis Evaluation in vitro
Cell swelling, the formation of large bubbles from the cytoplasmic membrane, and the rupture of the cell membrane are typical morphological characteristics of pyroptosis. In contrast, apoptosis is characterized by cell size reduction, nuclear condensation, chromatin fragmentation, cell membrane shrinkage, and the formation of apoptotic bodies, with the cell membrane remaining intact throughout the process.20 Based on previous studies, we established a therapeutic DOX concentration of 10 µg/mL.21 The CT26 cells underwent treatment with DAC and DOX in different mass ratios (1:1, 2.5:1, 5:1, and 10:1) for 24 h. Typical morphological features of pyroptosis were observed via light microscopy. The concentrations of lactate dehydrogenase (LDH) and adenosine triphosphate (ATP) in the supernatant were determined to examine the efficacy of pyroptosis induction. The results demonstrated that the 5:1 ratio achieved the most significant pyroptosis (Figure S4A–C). Although pyroptosis was also strongly induced at the ratio of 10:1, there was no significant difference compared with the mass ratio of 5:1. Considering the hepatic toxicity of DAC, the optimal ratio of DAC to DOX for inducing tumor cell pyroptosis was selected to be 5:1.
Following incubation with PBS, DAC, DOX, DAC+DOX, and NP/(DAC+DOX) for 24 h, an optical microscope was utilized to visualize tumor cells. Cells that exhibited pyroptosis were detected in the DOX, DAC+DOX, and NP/(DAC+DOX) treatment groups. Cells treated with NP/(DAC+DOX) showed the strongest degree of pyroptosis, while tumor cells treated with PBS exhibited no pyroptosis (Figures 2A and S5A). To more accurately assess pyroptosis in different treatment groups, we further calculated the pyroptosis index. For the DAC+DOX and NP/(DAC+DOX) treatment groups, the pyroptosis indices reached 5.84 ± 1.55% and 11.35 ± 2.92%, respectively. These values were notably higher than the pyroptosis index of the DOX group (2.22 ± 1.23%), demonstrating that combination therapy enhanced pyroptosis (Figure S6A). For 4T1 cells, the pyroptosis indices of the DOX, DAC+DOX, and NP/(DAC+DOX) groups were 1.23 ± 0.57%, 5.24 ± 1.28%, and 8.28 ± 0.76%, respectively (Figure S6B).
 |
Figure 2 Pyroptosis induced by NP/(DAC+DOX) in vitro. (A) Representative light microscopy and bio-TEM images of CT26 cells after different treatments. Red arrows indicate pyroptotic cells. Scale bars: 100 μm (light microscopy) and 2 μm (bio-TEM). The release of (B) LDH and (C) ATP from CT26 cells after different treatments. (D) Western blotting analysis of pyroptosis-related proteins (cleaved caspase-3, GSDME, and GSDME-N) in CT26 cells after different treatments. (E) The relative GSDME-N expression levels in CT26 cells after different treatments. All statistical data are presented as mean ± SD (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001). |
The morphology of CT26 and 4T1 cells receiving different treatments was also observed using biological transmission electron microscopy (bio-TEM). Tumor cell pyroptosis is characterized by cell swelling, loss of identifiable organelles, nuclear chromatin condensation, formation of bubble-like protrusions, and cell membrane rupture;22 these characteristics were observed in the DOX, DAC+DOX, and NP/(DAC+DOX) treatment groups, which was in accordance with the imaging results of optical microscopy. In contrast, the cell membranes and organelles of tumor cells treated with PBS and DAC were intact (Figures 2A and S5A).
Tumor cells were treated as above with different formulations for 24 h, after which the concentrations of inflammatory factors in the supernatant were determined using enzyme-linked immunosorbent assay (ELISA). ATP and LDH had significantly higher levels in the DAC+DOX and NP/(DAC+DOX) groups than in the control group, which verified that pyroptosis had occurred (Figures 2B, C, S5B and C). Compared with the control group of CT26 cells, the concentrations of LDH and ATP in the NP/(DAC+DOX) treatment group of CT26 cells were 1.32 and 1.51 times higher, respectively. DOX has been demonstrated to trigger pyroptosis through the caspase-3/GSDME signaling pathway. Caspase-3 is activated in response to DOX-induced cell death and subsequently cleaves GSDME into its N-terminal fragment (GSDME-N). The GSDME-N fragment then transfers to the cell membrane, forming pores that lead to pyroptosis. Western blot was utilized to determine the levels of cleaved caspase-3, GSDME, and GSDME-N (Figure 2D). Cleaved caspase-3 was expressed to a significantly higher degree in the DOX, DAC+DOX, and NP/(DAC+DOX) treatment groups compared with the control group because of the DOX-induced caspase-3 activation (Figure S7A). Compared with the control, the expression of GSDME was significantly enhanced in the DAC group, implying that demethylation is an effective approach to achieving the upregulation of GSDME (Figure S7B). Compared with PBS and DAC, the expression of GSDME-N in tumor cells was notably upregulated in the DOX, DAC+DOX, and NP/(DAC+DOX) treatment groups. The expression of GSDME-N was higher in the DAC+DOX and NP/(DAC+DOX) groups than in the DOX group, which confirmed that the combination treatment enhanced pyroptosis (Figure 2E).
Pharmacokinetics and Biodistribution of NP/(DAC+DOX) in vivo
To assess the in vivo pharmacokinetics of NP/(DAC+DOX), Sprague–Dawley (SD) rats were randomly divided into two groups. Either DAC+DOX or NP/(DAC+DOX) was injected via the tail vein at an equivalent DOX concentration of 3.0 mg (kg BW)−1 and a DAC concentration of 15.0 mg (kg BW)−1. Following the injection, blood samples were taken from the intraocular canthal vein at 15 min, 30 min, 1 h, 3 h, 12 h, 24 h, 48 h, and 72 h. The concentrations of DAC and DOX in plasma were detected through HPLC. The concentrations of DAC and DOX in the DAC+DOX group declined rapidly following injection and were almost undetectable after 48 h. In contrast, DAC and DOX concentrations in the NP/(DAC+DOX) group decreased slowly after 24 h and remained relatively high until 72 h (Figure 3A and B). The AUC0-∞ values of DAC and DOX rose from 132.17 and 711.37 µg·h mL−1, respectively, in the DAC+DOX group to 314.10 and 1899.55 µg·h mL−1, respectively, in the NP/(DAC+DOX) treatment group. The half-lives (t1/2z) of DAC and DOX were significantly extended from 19.79 and 17.49 h, respectively, in the DAC+DOX group to 67.12 and 41.00 h, respectively, in the NP/(DAC+DOX) treatment group. These findings imply that nanomicelles can effectively prevent the clearance of free drugs and significantly extend their blood circulation time.
 |
Figure 3 Pharmacokinetics and biodistribution of NP/(DAC+DOX) in vivo. The concentration of (A) DAC and (B) DOX in the serum of SD rats after injection of DAC+DOX or NP/(DAC+DOX). # represents the interruption of the HPLC signal. In vivo biodistribution of (C) DAC and (D) DOX 12 h after injection of DAC+DOX or NP/(DAC+DOX) in CT26 tumor-bearing mice. All statistical data are presented as mean ± SD (n = 3; *P < 0.05, **P < 0.01). |
The in vivo distribution of NP/(DAC+DOX) was evaluated in a subcutaneous CT26 tumor-bearing mouse model. This model was created through the subcutaneous injection of 1.0×106 CT26 cells into the right axilla of BALB/c mice. When the tumor volume reached approximately 80.0 mm3, either DAC+DOX or NP/(DAC+DOX) was injected into the mice via the tail vein. At 12 h post-injection, the tumors and major organs were collected and the concentrations of DAC and DOX were determined using HPLC (Figure 3C and D). The highest accumulation of DAC and DOX in both groups was found in the liver, which was unsurprising due to its known role in drug metabolism and elimination. Significantly higher drug concentrations were observed in the NP/(DAC+DOX) group compared with the free drug group, which was consistent with previous reports that nanoparticles tended to accumulate preferentially in the organs of the mononuclear phagocytosis system. Notably, the nanodrug delivery platform successfully increased the concentration of the drug within tumor tissues. Compared with the DAC+DOX treatment group, the intratumoral levels of DAC and DOX detected in the NP/(DAC+DOX) group were 1.33-fold and 2.78-fold greater, respectively.
To directly monitor the pharmacokinetics and biodistribution of nanomedicine, we fabricated NP/Cy5 by encapsulating the fluorescent dye cyanine 5 (Cy5) in mPEG-b-PLL-TK, then tracked its distribution in CT26 tumor-bearing mice using IVIS spectrum. NP/Cy5 exhibited rapid tumor accumulation at 1 h post-injection and sustained high retention levels in the tumor region at 24 h (Figure S8). In contrast, free Cy5 showed minimal accumulation in tumor tissue, with its fluorescence signal becoming nearly undetectable at 24 h. Tumor tissues and major organs were harvested and captured at 24 h post-injection. The results demonstrated significantly stronger fluorescence intensity of NP/Cy5 in tumor tissues compared to free Cy5 (Figure S9). The increased accumulation of nanomedicine in tumors is mainly due to the EPR effect, which provides a theoretical basis for its specific triggering of tumor cell pyroptosis and inhibition of tumor growth.
Anti-Tumor Effect and Pyroptosis Induction of NP/(DAC+DOX) in vivo
This work first determined the anti-tumor efficacy of NP/(DAC+DOX) using subcutaneous CT26 tumor-bearing mice. When the average tumor volume had increased to 80.0 mm3, mice underwent treatment with PBS, DAC, DOX, DAC+DOX, or NP/(DAC+DOX). Drugs were administered every 3 days via tail vein injection at a DOX dosage of 3.0 mg (kg BW)−1 and a DAC dosage of 15.0 mg (kg BW)−1. Compared with the control, the DOX, DAC+DOX, and NP/(DAC+DOX) treatments markedly inhibited tumor growth, and the tumor volume decreased from 1804.68 ± 188.48 mm3 in the control to 776.64 ± 199.91, 423.97 ± 99.76, and 225.66 ± 115.06 mm3 in the DOX, DAC+DOX, and NP/(DAC+DOX) treatments, respectively (Figure 4A and B). The administration of DAC alone did not suppress tumor growth. The lowest tumor weight was detected in the NP/(DAC+DOX) group (Figure 4C). The tumor inhibition rate of NP/(DAC+DOX) reached 87.33 ± 6.54%, which was significantly higher than the tumor inhibition rates of DOX (56.94 ± 9.40%) and DAC+DOX (76.60 ± 4.92%) (Figure 4D). Moreover, NP/(DAC+DOX) prolonged the average survival time of mice from 25.0 days in the control to 45.2 days (Figure 4E). The superior anti-tumor effect of NP/(DAC+DOX) was further confirmed using a subcutaneous 4T1 tumor-bearing model. Mice treated with NP/(DAC+DOX) exhibited the smallest tumor volume (Figure S10A and B) and the lowest tumor weight (Figure S10C). The NP/(DAC+DOX) group exhibited a significantly higher tumor inhibition rate (85.48 ± 3.43%) compared with the DAC+DOX group (69.03 ± 10.63%) (Figure S10D).
 |
Figure 4 In vivo anti-tumor effect of NP/(DAC+DOX) in CT26 tumor bearing mice model. (A) Tumor volume in CT26 tumor-bearing mice after treatment with PBS, DAC, DOX, DAC+DOX, or NP/(DAC+DOX). (B) Image of tumor and (C) tumor weight in different treatment groups on day 15, scale bar = 1.0 cm. (D) Tumor inhibition rate in different treatment groups. (E) Survival curves of mice in different treatment groups. (F) H&E, Ki-67, and TUNEL staining of tumor sections from mice receiving different treatments. Scale bar: 50 µm. All statistical data are presented as mean ± SD (n = 5; *P < 0.05, **P < 0.01, ***P < 0.001). |
Next, the tumors were dissected to conduct hematoxylin and eosin (H&E) staining and histological analysis. As shown in Figure 4F, the most severe disruption of tumor cell structure was observed in the NP/(DAC+DOX) group. The proportion of Ki-67-positive cells was significantly lower in the NP/(DAC+DOX) treatment group compared with the other groups, which demonstrated that NP/(DAC+DOX) suppressed the proliferation of tumor cells. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining indicated that NP/(DAC+DOX) treatment triggered the highest degree of DNA double-strand breakage. Similar results were shown in the histopathological analysis of 4T1 tumors, confirming the prominent inhibitory effect of NP/(DAC+DOX) on tumor growth (Figure S11).
To determine whether NP/(DAC+DOX) triggered tumor pyroptosis in vivo, serum from CT26 tumor-bearing mice was collected at the end of treatment to detect pyroptosis-related biomarkers. Compared with the control, LDH and ATP exhibited significantly increased concentrations in the NP/(DAC+DOX) group. The concentrations of LDH and ATP were also markedly elevated in the NP/(DAC+DOX) group compared with the DAC+DOX group (Figure S12). The expression of cleaved-caspase-3, GSDME, and GSDME-N was also detected in the tumor tissue. DAC upregulated GSDME expression and DOX activated caspase-3, which was in accordance with the results of the in vitro experiments (Figure S13A–C). GSDME-N was expressed at a much higher level in tumors subjected to NP/(DAC+DOX) treatment than in tumors treated with PBS, revealing that NP/(DAC+DOX) successfully induced tumor pyroptosis (Figure S13D).
Immune Activation of NP/(DAC+DOX) in vivo
Since the pyroptosis-induced release of pro-inflammatory factors contributes to immune cell recruitment and remodeling of the tumor immune microenvironment, the immune-activating effect of NP/(DAC+DOX) was further evaluated. Subcutaneous CT26 tumor-bearing mice underwent PBS, DAC, DOX, DAC+DOX, or NP/(DAC+DOX) treatment for 10 days, after which tumors and tumor-draining lymph nodes (TDLNs) were collected and subjected to flow cytometry.
T-cells play a crucial role in anti-tumor immunotherapy and function as essential components of the immune system that can identify and eliminate cancer cells. Compared with the control, the DAC+DOX and NP/(DAC+DOX) treatments markedly enhanced the proportions of CD3+, CD4+, and CD8+ T cells within the tumor (Figure S14). The percentages of CD3+ (Figure 5A), CD4+ (Figure 5B), and CD8+ (Figure 5C) T cells in the NP/(DAC+DOX) group were increased by 1.25-, 1.91-, and 1.93-fold compared with those of the control group, respectively, demonstrating that NP/(DAC+DOX) significantly promoted the intratumoral infiltration of T-cells. Similar findings were obtained for the TDLNs (Figures S15 and S16).
 |
Figure 5 NP/(DAC+DOX) activated the anti-tumor immune response in CT26 tumor-bearing mice. Frequencies of (A) CD3+, (B) CD4+, (C) CD8+ T cells, and (D) MDSCs in CD45+ cells in CT26 tumor tissues from mice receiving different treatments. (E) Frequencies of mature DCs in CD11c+ cells in TDLNs from mice receiving different treatments. Levels of (F) TNF-α, (G) IFN-γ, and (H) IL-10 in the serum of mice from different treatment groups. All statistical data are presented as mean ± SD (n = 5 for A- E; n = 3 for F-H; *P < 0.05, **P < 0.01, ***P < 0.001). |
MDSCs are a heterogeneous population of immune cells that can inhibit the activation of T-cells, thereby contributing to tumor immune evasion. Research has indicated that the inflammatory response and immune activation triggered by pyroptosis suppress MDSC recruitment and proliferation.23 NP/(DAC+DOX) reduced the proportion of intratumoral MDSCs from 10.13 ± 3.46% (control group) to 5.65 ± 1.45% (Figure 5D). In the TDLNs, the percentage of MDSCs in the NP/(DAC+DOX) group was the lowest among all groups (Figure S16D). DCs are essential in anti-tumor immunotherapy as they process and present tumor antigens to T-cells. IL-1β, IL-18, and HMGB1 released during tumor cell pyroptosis can promote DC maturation. In the TDLNs, the proportion of mature DCs in the NP/(DAC+DOX) group reached 23.28 ± 2.71%, which was significantly higher than the proportion in the control group (13.63 ± 4.59%) (Figure 5E).
To further verify the immune-activating function of NP/(DAC+DOX), tumor tissues from different groups were collected for immunofluorescence assay at the end of treatment. Consistent with the results of flow cytometry, CD3 and CD8 exhibited significantly higher immunofluorescence intensity in the NP/(DAC+DOX) group compared with the control group (Figures 6A and S17). Moreover, tumor tissues from the NP/(DAC+DOX) group expressed the lowest level of arginase-1 (Arg-1), a MDSC biomarker. Cytokines can modulate immune responses, thereby playing a pivotal role in anti-tumor immunotherapy. In addition to increasing the activation, proliferation, and differentiation of immune cells, such as T-cells and NK cells, cytokines also enhance the formation of a pro-inflammatory TME to overcome tumor-induced immune suppression. ELISA was performed to determine the serum cytokine levels of CT26 tumor-bearing mice in different treatment groups. In the NP/(DAC+DOX) group, the concentrations of TNF-α and IFN-γ were 3.46 and 4.02 times higher than those in the control group, respectively. By contrast, the level of IL-10 was significantly decreased in the NP/(DAC+DOX) group compared with the control group (Figure 5F–H). Similar results were detected in 4T1 tumor-bearing mice model (Figure 6B–D).
 |
Figure 6 Immunomodulatory effect of NP/(DAC+DOX) in 4T1 tumor-bearing mice. (A) CD3, CD8, and Arg-1 staining of tumor sections from 4T1 tumor-bearing mice receiving different treatments. Scale bar: 50 µm. Levels of (B) TNF-α, (C) IFN-γ, and (D) IL-10 in the serum of 4T1 tumor-bearing mice from different treatment groups. All statistical data are presented as mean ± SD (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001). |
These data suggest that NP/(DAC+DOX)-induced pyroptosis successfully engages the immune response and challenges the immunosuppressive TME.
Investigation of the Mechanisms Underlying NP/(DAC+DOX) Anti-Tumor Immunity
To further explore the mechanism by which NP/(DAC+DOX) activated anti-tumor immunity, CT26 cells treated with PBS and NP/(DAC+DOX) were collected for RNAseq. A total of 18,515 genes were analyzed, among which 4823 genes were upregulated and 5404 genes were downregulated in the NP/(DAC+DOX) group compared with the control group (Figure 7A and B). For further analysis, 602 differentially expressed genes that were significantly related to immune system were selected. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that the differentially expressed genes following NP/(DAC+DOX) treatment exhibited enrichment in the T cell receptor signaling pathway, the chemokine signaling pathway, and the NOD-like receptor signaling pathway (Figure 7C). Gene Ontology (GO) enrichment analysis indicated that genes related to programmed cell death and necroptotic process showed significant upregulation in the NP/(DAC+DOX) group (Figure 7D). The differentially expressed genes associated with pyroptosis were selected for heatmap analysis. This revealed that most genes involved in pyroptosis, such as GSDME, CHMP6, and HMGB1 were upregulated by NP/(DAC+DOX) (Figure 7E), suggesting that NP/(DAC+DOX) triggered pyroptosis. CHMP6 functions as a component of the endosomal sorting complexes required for transport, specifically the endosomal sorting complexes required for transport (ESCRT) III complex, which promotes endosome formation, enhances the degradation of damaged cell organelles, and repairs the vacuoles in the cellular membrane. CHMP6 regulates pyroptosis through its involvement in GSDM processing and the modulation of inflammatory responses.24 High mobility group box 1 (HMGB1) is a highly conserved nuclear protein that can be detected in all cell types. HMGB1 undergoes upregulation and extracellular release upon cellular injury or death, functioning as a pro-inflammatory signaling molecule. HMGB1 binds Toll-like receptor 4 to activate immune cells such as macrophages and DCs, thereby triggering inflammatory and immune responses.25
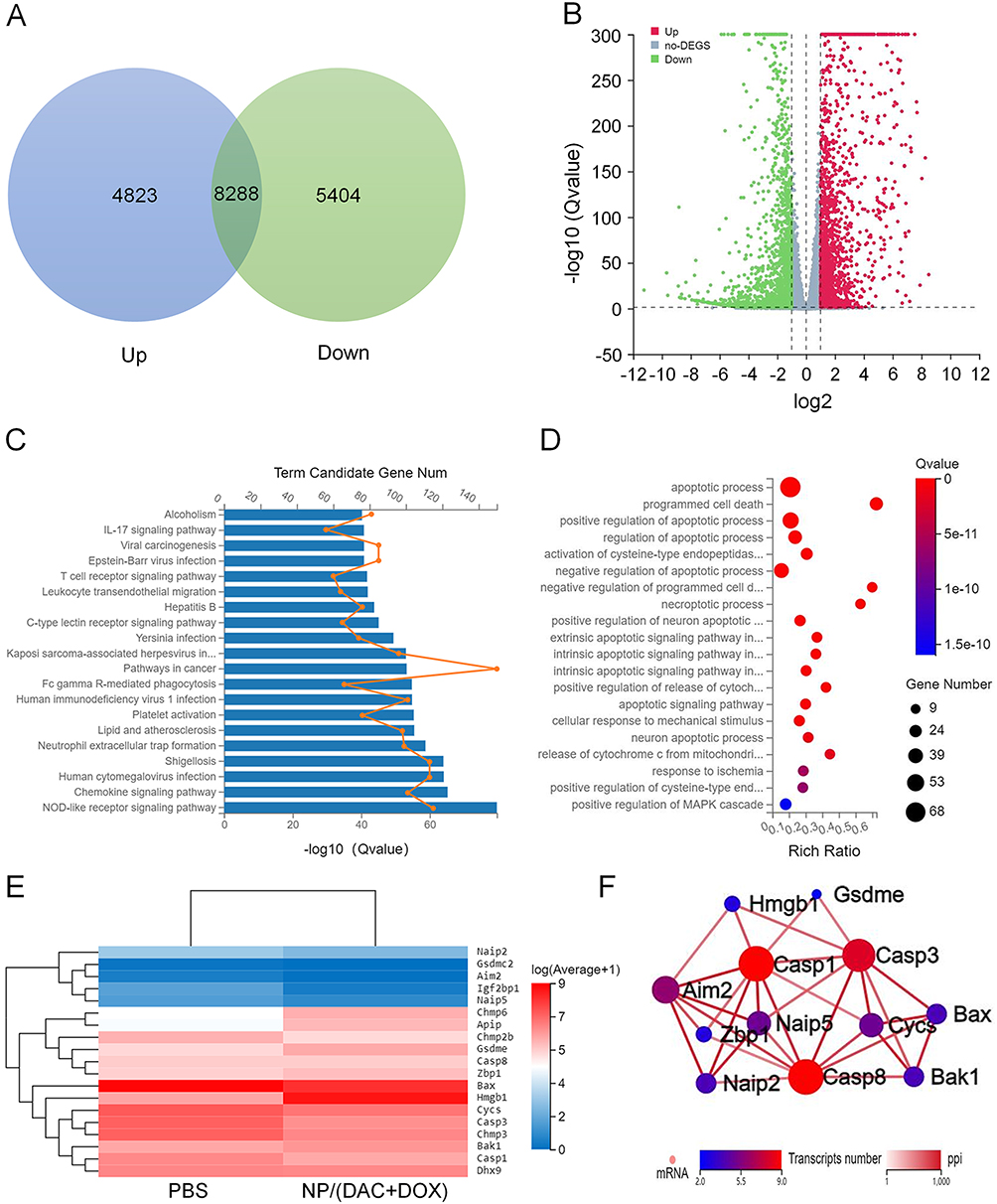 |
Figure 7 Anti-tumor mechanisms investigation via RNA sequencing. (A) Number and (B) volcano plot of the distributions of the downregulated or upregulated genes of NP/(DAC+DOX) treatment group, as compared to control group. (C) KEGG enrichment analysis of differentially expressed genes related to immune system after NP/(DAC+DOX) treatment. (D) GO enrichment analysis of differentially expressed genes related to programmed cell death after NP/(DAC+DOX) treatment. (E) Heatmap analysis of mRNA expression levels of genes involved in pyroptosis. (F) Interaction network of differential genes engaged in pyroptosis. |
Finally, we examined differential gene expression involved in pyroptosis to identify the protein-protein interaction (PPI) networks. These data confirm that NP/(DAC+DOX) triggers pyroptosis and activates inflammatory and immune responses (Figure 7F).
Biosafety of NP/(DAC+DOX)
Serious systemic side effects are an important factor limiting the application of combination immunotherapies. Weight changes and organ histology were assessed in mice receiving different treatments to evaluate the unintended side effects of treatment. Compared with the control, significant weight loss occurred in the DAC+DOX treatment group. However, mice subjected to NP/(DAC+DOX) treatment exhibited only minor changes in weight (Figure S18). In addition, the heart, liver, spleen, lung, and kidney were harvested from mice in different treatment groups for H&E staining. Myocardial cell degeneration, hepatocyte swelling and fatty degeneration, abnormal architecture in the white and red pulp of the spleen, alveolar edema and interstitial fibrosis, and tubular cell damage and glomerular abnormalities were observed in the DAC+DOX group. In contrast, the NP/(DAC+DOX) group exhibited negligible pathological changes in the examined organs. Therefore, NP/(DAC+DOX) exhibited relatively good biosafety in terms of peripheral organ effects, providing a foundation for future research into its safety and efficacy (Figures S19 and S20).
Conclusion
In this study, a ROS-responsive pyroptosis nanoinitiator (NP/(DAC+DOX)) was constructed to precisely induce tumor cell pyroptosis and activate the immune response. NP/(DAC+DOX) exhibited excellent ROS-responsive drug release behavior and could be effectively taken up by tumor cells. Experiments both in vitro and in vivo demonstrated that NP/(DAC+DOX) effectively triggered pyroptosis in tumor cells, which was attributed to the DOX-induced activation of caspase-3 and the upregulation of GSDME expression caused by DAC. Treatment with NP/(DAC+DOX) significantly decreased tumor growth and prolonged the survival time in both CT26 and 4T1 tumor-bearing mice. Flow cytometry and immunofluorescence analyses revealed that NP/(DAC+DOX) enhanced the maturation of DCs, promoted the tumor infiltration of T lymphocytes, and decreased the proportion of intratumoral MDSCs. RNAseq demonstrated that NP/(DAC+DOX) upregulated the expression of genes involved in pyroptosis, the inflammatory response, and immune activation. Altogether, this study provides a promising approach for cancer treatment and can serve as a theoretical and experimental reference for the application of pyroptosis-based immunotherapy strategies in clinical practice.
Funding
This work was financially supported by the National Natural Science Foundation of China (Grant No. 32471448), the Jilin Provincial Health Research Talent Special Project (Grant No. 2024SCZ79), the Science and Technology Development Program of Jilin Province (Grant Nos. YDZJ202301ZYTS103 and YDZJ202201ZYTS002), and the Norman Bethune Program of Jilin University (Grant No. 2022B15).
Disclosure
The authors declared that they have no conflicts of interest in this work.
References
1. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30(8):660–669. doi:10.1038/s41422-020-0343-4
2. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19(4):237–253. doi:10.1038/s41571-021-00588-9
3. Pan K, Farrukh H, Chittepu V, Xu HH, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022;41(1):119. doi:10.1186/s13046-022-02327-z
4. Chang JY, Lin SH, Dong W, et al. Stereotactic ablative radiotherapy with or without immunotherapy for early-stage or isolated lung parenchymal recurrent node-negative non-small-cell lung cancer: an open-label, randomised, Phase 2 trial. Lancet. 2023;402(10405):871–881. doi:10.1016/s0140-6736(23)01384-3
5. Ros J, Balconi F, Baraibar I, et al. Advances in immune checkpoint inhibitor combination strategies for microsatellite stable colorectal cancer. Front Oncol. 2023;13:1112276. doi:10.3389/fonc.2023.1112276
6. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349
7. Gerard CL, Delyon J, Wicky A, Homicsko K, Cuendet MA, Michielin O. Turning tumors from cold to inflamed to improve immunotherapy response. Cancer Treat Rev. 2021;101:102227. doi:10.1016/j.ctrv.2021.102227
8. Iglesias-Escudero M, Arias-González N, Martínez-Cáceres E. Regulatory cells and the effect of cancer immunotherapy. mol Cancer. 2023;22(1):26. doi:10.1186/s12943-023-01714-0
9. Li LS, Jiang MX, Qi L, et al. Pyroptosis, a new bridge to tumor immunity. Cancer Sci. 2021;112(10):3979–3994. doi:10.1111/cas.15059
10. Loveless R, Bloomquist R, Teng Y. Pyroptosis at the forefront of anticancer immunity. J Exp Clin Cancer Res. 2021;40(1):264. doi:10.1186/s13046-021-02065-8
11. Wang QY, Wang YP, Ding JJ, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579(7799):421–426. doi:10.1038/s41586-020-2079-1
12. Zhang ZB, Zhang Y, Xia SY, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579(7799):415–420. doi:10.1038/s41586-020-2071-9
13. Su LP, Chen YT, Huang C, et al. Targeting Src reactivates pyroptosis to reverse chemoresistance in lung and pancreatic cancer models. Sci Transl Med. 2023;15(678):eabl7895. doi:10.1126/scitranslmed.abl7895
14. Ji FJ, Shi CY, Shu ZB, Li ZM. Nanomaterials enhance pyroptosis-based tumor immunotherapy. Int J Nanomed. 2024;19:5545–5579. doi:10.2147/ijn.S457309
15. Dang Y, Guan J. Nanoparticle-based drug delivery systems for cancer therapy. Smart Mater Med. 2020;1:10–19. doi:10.1016/j.smaim.2020.04.001
16. Cao Z, Li D, Wang J, Yang X. Reactive oxygen species-sensitive polymeric nanocarriers for synergistic cancer therapy. Acta Biomater. 2021;130:17–31. doi:10.1016/j.actbio.2021.05.023
17. Rennick JJ, Johnston APR, Parton RG. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat Nanotechnol. 2021;16(3):266–276. doi:10.1038/s41565-021-00858-8
18. Fan J-X, Deng R-H, Wang H, et al. Epigenetics-based tumor cells pyroptosis for enhancing the immunological effect of chemotherapeutic nanocarriers. Nano Lett. 2019;19(11):8049–8058. doi:10.1021/acs.nanolett.9b03245
19. Hu CH, Liu XH, Zeng Y, Liu JQ, Wu F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin Clin Epigenet. 2021;13(1):166. doi:10.1186/s13148-021-01154-x
20. Tang R, Xu J, Zhang B, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. 2020;13(1):110. doi:10.1186/s13045-020-00946-7
21. Liang MY, Zhang MJ, Qiu W, et al. Stepwise size shrinkage cascade-activated supramolecular prodrug boosts antitumor immunity by eliciting pyroptosis. Adv Sci. 2022;9(26):2203353. doi:10.1002/advs.202203353
22. Zhou JY, Wang WJ, Zhang CY, et al. Ru(II)-modified TiO2 nanoparticles for hypoxia-adaptive photo-immunotherapy of oral squamous cell carcinoma. Biomaterials. 2022;289:121757. doi:10.1016/j.biomaterials.2022.121757
23. Wang H, Gao ZY, Jiao D, et al. A microenvironment dual-responsive nano-drug equipped with PD-L1 blocking peptide triggers immunogenic pyroptosis for prostate cancer self-synergistic immunotherapy. Adv Funct Mater. 2023;33(16):221449. doi:10.1002/adfm.202214499
24. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956–960. doi:10.1126/science.aar7607
25. Meng XL, Na RS, Peng X, et al. Musashi-2 potentiates colorectal cancer immune infiltration by regulating the post-translational modifications of HMGB1 to promote DCs maturation and migration. Cell Commun Signal. 2024;22(1):117. doi:10.1186/s12964-024-01495-z
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.