")
Back to Journals » International Journal of Nanomedicine » Volume 20
Red Blood Cell Membrane Vesicles for siRNA Delivery: A Biocompatible Carrier With Passive Tumor Targeting and Prolonged Plasma Residency
Authors Della Pelle G , Markelc B , Bozic T, Šribar J, Krizaj I, Zagar Soderznik K, Hudoklin S, Kreft ME , Urbančič I, Kisovec M , Podobnik M, Kostevšek N
Received 4 November 2024
Accepted for publication 4 February 2025
Published 15 March 2025 Volume 2025:20 Pages 3269—3301
DOI https://doi.org/10.2147/IJN.S504644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sachin Mali
Giulia Della Pelle,1,2 Bostjan Markelc,3 Tim Bozic,3 Jernej Šribar,4 Igor Krizaj,4 Kristina Zagar Soderznik,1 Samo Hudoklin,5,* Mateja Erdani Kreft,5,* Iztok Urbančič,6 Matic Kisovec,7 Marjetka Podobnik,7 Nina Kostevšek1
1Department for Nanostructured Materials, Jožef Stefan Institute, Ljubljana, 1000, Slovenia; 2Jožef Stefan International Postgraduate School, Ljubljana, 1000, Slovenia; 3Department of Experimental Oncology, Institute of Oncology Ljubljana, Ljubljana, 1000, Slovenia; 4Department of Molecular and Biomedical Sciences, Jožef Stefan Institute, Ljubljana, 1000, Slovenia; 5Institute of Cell Biology, Faculty of Medicine, University of Ljubljana, Ljubljana, 1000, Slovenia; 6Laboratory of Biophysics, Condensed Matter Physics Department, Jožef Stefan Institute, Ljubljana, 1000, Slovenia; 7Department of Molecular Biology and Nanobiotechnology, National Institute of Chemistry, Ljubljana, 1000, Slovenia
*These authors contributed equally to this work
Correspondence: Giulia Della Pelle, Jozef Stefan Institute, Jamova Cesta 39, Ljubljana, 1000, Slovenija, Tel +393356577520, Email [email protected]
Background: Despite many advances in gene therapy, the delivery of small interfering RNAs is still challenging. Erythrocytes are the most abundant cells in the human body, and their membrane possesses unique features. From them, erythrocytes membrane vesicles can be generated, employable as nano drug delivery system with prolonged blood residence and high biocompatibility.
Methods: Human erythrocyte ghosts were extruded in the presence of siRNA, and the objects were termed EMVs (erythrocyte membrane vesicles). An ultracentrifugation-based method was applied to select only the densest EMVs, ie, those containing siRNA. We evaluated their activity in vitro in B16F10 cells expressing fluorescent tdTomato and in vivo in B16F10 tumor-bearing mice after a single injection.
Results: The EMVs had a negative zeta potential, a particle size of 170 nm and excellent colloidal stability after one month of storage. With 0.3 nM siRNA, more than 75% gene knockdown was achieved in vitro, and 80% was achieved in vivo, at 2 days PI at 2.5 mg/kg. EMVs mostly accumulate around blood vessels in the lungs, brain and tumor. tdTomato fluorescence steadily decreased in tumor areas with higher EMVs concentration, which indicates efficient gene knockdown. Approximately 2% of the initial dose of EMVs was still present in the plasma after 2 days.
Conclusion: The entire production process of the purified siRNA-EMVs took approximately 4 hours. The erythrocyte marker CD47 offered protection against macrophage recognition in the spleen and in the blood. The excellent biocompatibility and pharmacokinetic properties of these materials make them promising platforms for future improvements, ie, active targeting and codelivery with conventional chemotherapeutics.
Keywords: biomimetic, RNA interference, EPR effect, nanomedicine, melanoma
Graphical Abstract:

Introduction
Red blood cells (RBCs) are the most abundant cells in the human body.1 From a pharmaceutical point of view, they have been overlooked for a long time, being considered merely hemoglobin carriers, until “nanorevolution” alighted new interest in biomimetic nanocarriers. They can be harvested from the patient’s body, suitably treated,2 and reinjected, thereby obtaining an allergy-free drug carrier that can persist in the blood flow for several weeks. They, and their derivatives, fall within the ever-growing category of biomimetic carriers.3,4
As of 2023, the full proteome of red blood cells5 and their membrane-derived vesicles6 and full transcriptome7 have been intensely characterized. Moreover, the RBC membrane, and its underlying cytoskeleton, is a unique scaffold with interesting rheological and biological properties: extreme deformability8 combined with good mechanical resistance to shear force;9 the RBC membrane also possesses the CD47 marker, colloquially known as the “don’t eat me” marker for macrophage recognition,10 which, in aging RBCs, is instead triggered by phosphatidylserine exposure on the outer leaflet.11 CD47, which is also present on naturally released extracellular vesicles [10,11], enables RBC-derived carriers to achieve long blood circulation times and systemic activity. RBCs have also rarely been used as therapeutic nucleic acid carriers.1 Since the discovery of RNA interference,12 the research and development of therapeutic RNAs have become a new challenge in personalized molecular medicine, despite only a handful of drugs being approved by the European Medicine Agency and the Federal Drug Agency.13 Indeed, as of 2022, five siRNA drugs have been approved by the US FDA, and, later, by EMA: the already mentioned Patisiran (Onpattro), Givosiran (Givlaari), Lumasiran (Oxlumo), Inclisiran (Leqvio), and Vutrisiran (Amvutta), all directed to the liver. Nanomedicine runs in strict correlation with personalized medicine, with the number of papers concerning small interfering RNA delivery skyrocketing in the last fifteen years and with more than 20,000 papers in 2022 about siRNA delivery (data from14). Furthermore, the discovery of extracellular vesicles (EVs)15 as naturally occurring microinterfering RNA carriers fueled research on lipidic, biomimetic, neutral delivery systems for therapeutic nucleic acids (TNAs) as safe replacements for cationic lipofection. Unfortunately, the route for the clinical translation of EVs as small interfering RNA carriers is challenging and steep because of their difficult recovery, purification, low encapsulation efficiency and presence of off-target miRNAs.16 On the other hand, red blood cells are a large source of EVs.17 Therefore, the advantages of using RBC as starting material for a nanocarriers, compared with synthetic alternatives, are numerous: lack of recognition from phagocytic cells due to the presence of CD47 co-receptor; presence, in the membrane proteome, of the protein machinery for membrane fusion/cell uptake typical of EVs, but without the collection and purification hurdles;6,18 the possibility of using patient’s own blood, as it happened for a some approved formulations that uses red blood cells as carriers;1,19 the presence of an RNAse inhibitor, that allows for long-term storage of ribonucleic TNA;20,21 the negative surface charge, due to red blood cell sialic acid glycocalyx, reduces the interaction with plasma protein, prolonging circulation time, tuning the composition and abundance of protein corona components.22,23 For an enhanced targeting and a specific tissue-tropism, red blood cell surface modification has been extensively characterized and successfully achieved, both via “hitchhiking”, due to electrostatic interactions in the glycocalyx, and via covalent coupling: the reader can then refer to the following review.24 Furthermore, they share the advantages of other “neutral” TNA carriers, such as liposomes and micelles: the lack of cationic charges, typical of polymers and dendrimers such as PEI25 and PAMAM,26,27 notoriously eliciting apoptotic cascades.28–30
In the present work, we used red blood cells as the starting material for a neutral therapeutic nucleic acid (TNA) delivery system, which we named erythrocyte membrane vesicles (EMVs). Our TNA of choice is a siRNA because of its compactness and well-established clinical applications.27 Nevertheless, siRNA are double-stranded RNA molecules, behaving as PAMPS within macrophages (pathogen-associated molecular pattern), being recognized by toll-like receptor 3.31 SiRNAs are capable of activating multiple immune system pathways, including interferon 1 cascade, composed of roughly 2000 genes; finally, a mitochondrial apoptosis is initiated via dsRNA dependent caspase-binding domains in important RNA helicases and transducers such as MDA5 and RIG-I.32 Lastly, siRNA has a very short residency time in plasma, being rapidly cleared away by kidney filtration.33 For its innate cytotoxicity and brief permanence in blood, encapsulation of siRNAs within nanodrugs is advisable and a thriving field of study.34
As a proof-of-concept, we loaded an anti-tdTomato siRNA into EMVs obtained via sonication and extrusion of red blood cell membranes. tdTomato (tandem-dimer Tomato) is an orange-emitting fluorescent protein, derived from coral Discosoma sp.35 Despite being used as a coating material,36–38 this is the first report of an erythrocyte membrane being used as a liposomal-like formulation, reduced to object of 100–200 nm, for a TNA delivery, that does not involve any other nanoparticle actor ie a core shell structure, where the TNA is embedded within the core and the red blood cell membrane acts as a camouflaging material. We characterized the interactions of the payload and erythrocyte membranes via superresolution fluorescence microscopy and cryo- and freeze-fracture TEM. We greatly optimized the purification strategy, reaching 67% encapsulation efficiency, and were able to select and use only the carriers containing the TNA for transfection, removing the unloaded and unprotected molecules, together with cell debris not shaped into EMVs. Moreover, we optimized a long-term freeze‒drying storage strategy. EMVs have been proven to be a successful, inexpensive, and safe delivery system for siRNAs both in vitro and in vivo as demonstrated in a tdTomato-expressing murine melanoma cell line in vitro and in a subcutaneous murine tumor model in vivo, with excellent pharmacokinetic properties. At low doses, they elicited significant knockdown of target genes as early as 24 hours postinjection, whereas EMVs were still present in the blood circulation two days postinjection. Despite relying solely on passive targeting, more than 15% of the injected dose was present in the tumor tissue at 48 hours postinjection, effectively silencing the target gene in the proximity of major and minor blood vessels. Moreover, the mean circulation time of EMVs exceeds 4–5 times that of a naked, unmodified siRNA,39,40 and their main clearance route is via renal filtration with low liver accumulation.
Materials and Methods
Materials and Chemicals
Red blood cells were obtained from the Blood Transfusion Centre of Slovenia (Ethical approval number 0120–592/2020/7), which belong to two young anonymous males, A RhD+ blood type, supplemented with CPD-SAGM preservative, 70% v/v hematocrit, accounting for 4•109 RBCs/mL, and stored in 1 mL aliquots at −80°C without additional cryopreservatives. For EMVs preparation, we used phosphate-buffered saline tablets obtained from Sigma Aldrich (P4417) in diethyl-pyrocarbonate-treated Milli-Q water. Trisodium citrate dihydrate, RNase A (catalog number 10109142001) and Amicon centrifugal devices (30, 10 and 4 kDa NMWL, EMD Millipore), as well as fluorescent dyes (CM-DiI, C7000, and Vybrant DiO, V22886), were purchased from Sigma Aldrich. For the Stewart assay, ferric chloric hexahydrate (F2877) and ammonium thiocyanate (A7149) were purchased from Sigma Aldrich, whereas the heart lipid extract (171201), which was used as a standard, was obtained from Avanti Polar lipids. The Bradford reagent was prepared according to the traditional recipe41 (Coomassie Brilliant Blue R-250, B7920, ethanol and phosphoric acid were obtained from Sigma Aldrich), and bovine serum albumin (A9418, Sigma Aldrich) at a stock concentration of 1 mg/mL was used as a standard. The polycarbonate extrusion membranes were 19 mm wide, 800 nm wide (610009), and 200 nm wide (610006), and the support filters (610014) were all purchased from Avanti Polar and installed on an Avanti Polar mini extruder. Anti-TdTomato-siRNA was purchased from Thermo Fisher as a duplex oligo with the following sequence: 5’rGrCrC rArCrC rArCrC rUrGrU rUrCrC rUrGrU rATT- 3′; 3′rUrArC rArGrG rArArC rArGrG rUrGrG rUrGrG rCGG-5′; “r” stands for ribonucleotide to distinguish it from DNA overhangs), whereas Cy-5 siRNA was later obtained from Integrated DNA Technologies (USA). All the labware, including tips and Falcon and Eppendorf tubes, was RNAse/pyrogenase free, and the buffers were DEPC-treated. Advanced Dulbecco’s modified MEM (ADMEM), Advanced Roswell Park Memorial Institute 1640 medium (RPMI-1640), and MCDB 131 media were purchased from Gibco (Thermo Fisher Scientific, VA, US), and FGM™-2 Fibroblast Growth Medium-2 BulletKit™ was purchased from Lonza (Basel, CH). All media except FGM™-2 were supplemented with GlutaMAX (100x, Gibco) and penicillin‒streptomycin (100x, Sigma‒Aldrich, Merck, Darmstadt, Germany). ADMEM and ARPMI were supplemented with 5% fetal bovine serum (FBS; Gibco). MCDB 131 was supplemented with 10% FBS (Gibco), 10 ng/mL epidermal growth factor (EGF; Gibco), 1 µg/mL hydrocortisone (Sigma‒Aldrich) and 10 mm glutamine (GlutaMAX; Gibco). The MycoAlertTM PLUS Mycoplasma Detection Kit was purchased from Lonza (Basel, CH). The B16F10 (CRL-6475), CT26 (CRL-2638), 4T1 (CRL-2539), and Hulec-5a (CRL-3244) cell lines were purchased from ATCC (VA, US), the NHLF cell line was purchased from Lonza (Basel, CH), and the B16F10-tdTomato line was a kind gift from Dr. Muriel Golzio (The Institute of Pharmacology and Structural Biology, IPBS-Toulouse). The cells were grown in a humidified 5% CO2 atmosphere at 37°C in a cell culture incubator. The Presto Blue reagent (A13261) and Hoechst 33342 solution (62249) were purchased from Thermo Fisher Scientific, and the MemBrite® Fix 488/515 Cell Surface Staining Kit from Biotium (#30093-T, Fremont, CA, USA) were used.
Preparation of Erythrocyte Membrane Vesicles
Erythrocytes were depleted of hemoglobin via the hypotonic lysis method.42 The cells were incubated overnight with 40 mL/each mL of erythrocytes at 4°C, as first established by,43 and later centrifuged at 11,000 rpm with a benchtop Minispin Eppendorf centrifuge. The pellet was washed four times to allow hemoglobin release. One milliliter of original red blood cell suspension was finally diluted four times in buffer containing siRNA at a suitable concentration, incubated at 37°C under gentle shaking, sonicated with an ultrasonic processor for 10 seconds (Sonics Vibra cell VC-505; pulses: 1 s on, 1 s off; 20% amplitude) for 10s, and subsequently extruded at 37°C with an Avanti Polar mini extruder, which was previously thoroughly rinsed with DEPC water via an 800 nm membrane 5 times and a 200 nm membrane 20 times. The final suspension was stored at 4°C.
Protein and Lipid Quantification
A Bradford assay was used to evaluate the protein concentration, with bovine serum albumin used as a standard. The Bradford reagent was prepared according to previous methods,41 and the assay-to-sample ratio was 30:1 v/v. All measurements were performed in triplicate. The absorbances at 450 and 595 nm were recorded, and a calibration curve was built from their ratio. A Stewart assay was used to quantify the phospholipid content,44 with heart lipid extract (Avanti Polar) used as a standard reference.
Selection of the Best Purification Method
First, for size exclusion chromatography, siRNA-EMVs were loaded into a PD10 G25 Sephadex column (GE Healthcare) as reported elsewhere for similar purposes;45 the sample was recovered after the addition of 5–6 mL of 1× PBS (Figure 1). Second, Amicon 30 kDa centrifugal units were used: 500 µL of the sample was loaded into a 0.5 mL tube and centrifuged at 13,000 rpm for 5 minutes. Subsequently, the retentate and eluate were redispersed in the original volume and used for RNA quantification. Third, we tested common ultracentrifugation-based purification techniques for siRNA recovery and EMVs integrity (details in the), ie, a sucrose gradient, regular ultracentrifugation, and density-changing centrifugation.46
 |
Figure 1 (a) Overview of the protocol used for the preparation of DCC-siRNA-EMVs. (b) Size (nm) and RNA concentration (µg/mL) after ultracentrifugation. The data refer to n=3 experiments. The initial concentration of siRNA, before DCC, was 10 µg/mL, resulting in a 67% encapsulation efficiency. (c) Concentrations of protein, lipids, and siRNA among the 5 top fractions. p≤0.01 **. |
Density Changing Ultracentrifugation
Density changing centrifugation (DCC) has proven to be the best and easiest method for obtaining high concentrations of siRNA-loaded EMVs, and it is described in detail here. To select only loaded EMVs and to separate correctly shaped ones from protein aggregates and unloaded siRNA, we adapted an easy purification protocol by.46 Ultracentrifuge tubes (3 mL, Beckman Coulter Life Sciences, catalog number 355635) were filled with 2 mL of 10% (m/v) trisodium citrate (TC) dissolved in PBS, and the sample to be separated was gently pipetted on top. This allows selection on the basis of the mixed effects of changed solubility and density rather than time-consuming sucrose gradient ultracentrifugation. Heavier and denser EMVs will be found in the upper layer of the tube with decreasing siRNA concentration while approaching the bottom, where protein and other cellular debris, together with unloaded siRNA, will be pelleted. The tubes were centrifuged in a Beckman Coulter Optima XPN-90 ultracentrifuge equipped with an SW 60 Ti swinging-bucket rotor for 40 minutes at 4°C and 100.000 × g (27.200 rpm). Eight 0.25 mL fractions were then collected and subjected to analysis. The six top fractions were determined to contain siRNA via a Quantifluor RNA assay (see next section). The TC was removed with an Amicon centrifugal device, 10 kDa NMWL (EMD Millipore, UFC8010). Finally, the DCC-siRNA-EMVs were resuspended in PBS and stored at 4°C until further use.
Encapsulation Efficiency Determination
On the basis of at least three replicates, the siRNA concentration was determined for every collected fraction via the following formula:

A Quantifluor RNA system (Promega) and a Quantus spectrophotometer were used to evaluate the RNA concentration according to the manufacturer’s specifications.
Particle Size and Zeta Potential Determination
The hydrodynamic diameter, polydispersity index (PDI), and zeta potential of the empty and siRNA-containing EMVs were determined via a zeta potential and particle size analyzer Litesizer (Anton Paar, Austria) equipped with a built-in volume-weighted algorithm in proprietary software (Kalliope). Omega cuvettes were used for both analyses. The backscattering angle (175°) was used for EMVs particle size determination.
Encapsulation Stability
Amicon 0.5 mL 30 NMWL devices were used to quantify spontaneous RNA leakage from EMVs after siRNA encapsulation. Two hundred microliters of each sample was loaded in the centrifugal device and centrifuged at 13,500 rpm on a Minispin Eppendorf tabletop centrifuge for 10 minutes until it was reduced to a final volume of 60 µL. The eluate was resuspended to the initial volume with PBS and used for RNA quantification. Between measurements, the samples were kept at 4°C.
RNase A Stability Assay
To evaluate the nuclease resistance of EMVs, three different concentrations of RNAse A (50, 5, and 0.5 µg/mL) were tested on naked RNA, followed by centrifugation of EMVs and ultracentrifugation of EMVs for 24 and 2 hours, with two independent preparations. After incubation at 37°C for the desired amount of time, the samples were run on a 2% agarose gel, and the same amount of siRNA in each well was carefully pipetted to avoid dilution bias. In accordance with the manufacturer’s procedures, the gels were subsequently stained with SYBR Gold stain and visualized in a transilluminator.
Ultrastructure Characterization
CryoTEM
Four kinds of samples were analyzed:
- Empty EMVs not subjected to density-changing centrifugation (DCC)
- Empty EMVs, DCC
- Loaded EMVs with 10 µg/mL siRNA, not subjected to DCC
- LoadedEMVs with 10 µg/mL siRNA, not subjected to DCC
Two hundred microliters of each typology were used as the starting material, and a 3x concentrated sample of each centrifuged sample was obtained. The samples for cryoTEM were prepared via Vitrobot Mark IV (Thermo Fischer Scientific, Waltham, MA, USA). Quantifoil® R 2/2, 200 mesh holey carbon grids (Quantifoil Micro Tools GmbH, Großlöbichau, Germany) were glow-discharged for 60s at 20 mA and positive polarity in an air atmosphere (GloQube® Plus, Quorum, Laughton, UK). Vitrobot conditions were set to 4°C, 95% relative humidity, a Blot time of 6 s and a Blot force of 2. Three microliters of the suspension were applied to the grid, blotted, and vitrified in liquid ethane. The samples were visualized with a 200 kV Glacios microscope (Thermo Fischer Scientific, Waltham, MA, USA). Micrographs were analyzed for size distribution via ImageJ software and GraphPad 9.0 for statistical analysis. Size analysis was performed on n>6 20.000x micrographs.
Freeze-Fracturing EM
The same set of samples analyzed with cryoTEM was also used for freeze-fracturing EM. Suspensions of EMVs were mixed 1:1 with 60% glycerol and incubated for 30 minutes at 4°C. Using a pipette, 1.5 μL samples were transferred to gold-stub well-type carriers and frozen in a LN2-cooled freezer. The frozen samples were immediately transferred to a freeze-fracture device (BALZERS, BAF200), fractured (knife temperature of −150°C), shadowed with carbon-platinum (nominal angle of 45°) and further strengthened with carbon (nominal angle of 90°). The obtained replicas were cleaned overnight in a sodium hypochlorite solution, subsequently collected on copper microscopic grids and examined with a CM100 transmission electron microscope (Philips, Netherlands) running at 80 kV.
Negative Staining Conventional Transmission Electron Microscopy
EMV suspensions were fixed for 30 minutes in 1:1 2.5% glutaraldehyde cacodylate buffer (pH 7.3, 0.008 M) to match the osmolarity of the PBS solution. Briefly, 5 µL of suspension was placed on Formvar-coated copper grids (150 mesh, TEMFCF150CU50, Sigma Aldrich) for 10 minutes, and excess liquid was gently removed with Whatman filter paper. The grids were briefly washed with Milli-Q water by placing them on drops, and the excess liquid was removed. Finally, the grids were placed over a drop of UA-zero negative stain (Alpha Aesar) for 45 seconds and subsequently air-dried. Sample grids were examined with a JEOL JEM-2100 instrument operating at an accelerating voltage of 200 kV. The samples were imaged at 20,000x to 40,000x magnification.
Superresolution Fluorescence Microscopy
DCC-siRNA-EMVs and siRNA-EMVs were imaged on a superresolution confocal microscope47 equipped with a STED laser. Glass slides and coverslips were first coated with 0.1% polylysine (P8920, Sigma Aldrich) by gently dipping them in the solution, followed by a brief wash with 1× PBS. Finally, 20–50 µL of CM-DiI-labeled Cy5-siRNA-EMVs were spread on the slide surface with a coverslip and subsequently sealed with nail polish. The slides were imaged a maximum of two hours after mounting. Imaging of the samples was performed on an Olympus IX83 microscope with a 60 × water-immersion objective (NA 1.2), custom-upgraded by Abberior Instruments with 561 nm and 640 nm excitation lasers (120 ps pulse length, 50 μW mean power), a 775 nm STED laser (1.2 ns pulse length, 170 mW mean power), and two avalanche photodiodes for fluorescence detection in the ranges of 580–625 nm and 655–720 nm, as defined by prefilters and a dichroic. All imaging was performed at an 80 MHz frequency with a 1.1 A.U. pinhole. To compare the resolution between confocal and STED microscopy, a field of view of 5 μm × 5 μm was imaged with a 33 nm pixel size. The dwell time was 10 μs for confocal microscopy and 30 μs for STED microscopy, and the pinhole size was 0.6 AU. Other parameters (gating and laser powers) were tuned for each probe separately to achieve the best possible resolution. Superimposition of the Cy-5 and DiI signals was performed via the Colocalization Finder ImageJ plugin.
In Vitro Assays
Cytotoxicity Assay
For the in vitro cytotoxicity assay, 1000 cells/well in 100 µL of appropriate cell culture medium for each cell line were seeded in a 96-well plate (VWR, PA, US) and allowed to grow overnight. The next day, the medium was exchanged with medium containing 4.5 µg/mL EMVs protein and incubated for 72 h. Then, 10 µL/well Presto Blue reagent (A13261, Thermo Fisher Scientific) was added to the cells, which were incubated at 37°C for an hour when the emitted fluorescence was read with a Cytation 1 system (BioTek Instruments, VT, USA). The measured fluorescence intensity was then normalized to that of the control group without the EMVs.
In Vitro Internalization Assay
The cells were seeded in 12-well chamber slides (#81201, iBidi, Germany) in 100 µL of appropriate cell culture medium for each cell line. The next day, the media was changed to 100 µL of appropriate media for each cell line containing 0.7 nmol of Cy-5 siRNA loaded in DCC-EMVs. At 24, 48 and 72 h after incubation, the cells were washed with 1x HBSS and stained with the MemBrite® Fix 488/515 Cell Surface Staining Kit (Biotium) following the manufacturer’s instructions. Briefly, the cells were incubated for 5 min at 37°C in 100 µL/well of presoliding solution, followed by a 10 min incubation at 37°C with 100 µL/well of MemBrite® Fix 488/515 stain. The cells were then washed twice with 1x HBSS and fixed with 4% PFA (#12606, Cell Signaling Technology, Massachusetts, USA) for 15 min at 37°C. Afterwards, the cells were washed twice with PBS for 5 min and stained with 3 µg/mL Hoechst solution (Hoechst 33342, Thermo Fisher Scientific) in PBS for 15 min at RT to label the nuclei. The cells were subsequently washed twice with PBS at RT for 5 min, the silicon wells were removed, and the slides were mounted in Prolong Glass Antifade Mount (Thermo Fisher Scientific) for 24 h at RT and then sealed with nail polish. Imaging was performed with an LSM 800 confocal microscope (Carl Zeiss, Oberkochen, Germany) with a 20× objective (NA 0,8). Hoechst 33342, Alexa Fluor 488, CM-DiI and Cy-5 dyes were excited with lasers with excitation wavelengths of 405, 488, 561 and 640 nm, respectively. The emitted light was collected sequentially with a Gallium Arsenide Phosphide (GaAsP) detector via a variable dichroic and filtered at the following wavelengths: 410–545 nm (Hoechst 33342), 488–545 nm (Alexa Fluor 488), 565–620 nm (CM-DiI), and 645–700 nm (Cy-5).
Protease Activity Determination
A Pierce colorimetric protease assay (Thermo Scientific, 23263) was used according to the manufacturer’s instructions to evaluate the hypothetical protease activity of the RBCs. While the trypsin positive control assay was carried out at pH 8, RBC ghosts and DCC-EMVs were assayed at pH 3 at a known concentration of proteins. The final absorbance was read at 450 nm and normalized to the intrinsic absorbance of the sample. The data were further processed in GraphPad 9.0.
Evaluation of RNA Interference Ability in Vitro
DCC-siRNA-EMVs were mixed 1:1 with Optimem medium (#31985070, Thermo Fisher Scientific), and the volume corresponding to 30 picomoles of siRNA/well was added to B16F10-TdTomato cells. The empty control volumes used were subjected to density-changing centrifugation as well and calibrated according to protein content to add the same amount of EMVs to the cell culture. The experiment was carried out on a 24-well culture plate, in which 50,000 cells/well were seeded the day before transfection. After 48 hours, total RNA extraction was carried out with a VWR RNA Gold kit, and the relative amount of RNA was quantified with an Agilent Cytation 5 with a Take3 plate for nucleic acid quantification. Retrotranscription (Nippon Genetics, Scriptase II, LS563) was carried out according to the manufacturer’s specifications. Tdtomato mRNA silencing was evaluated via RT‒PCR (Table 1 for primer sequence), with n = 6 replicates each of a final volume of 20 µL, on Eppendorf Twin Tec 96-well plates. Beta-actin was used as a calibrator. The delta-delta CT method was used to calculate the relative expression of the Tdtomato gene. The GraphPad 9 built-in ROUT algorithm for outlier recognition was used to clean the Ct data.
 |
Table 1 RT‒qPCR Primer Sequences |
Freeze-Drying Optimization
To optimize long-term storage stability, EMVs were freeze dried in the presence of different cryoprotectants, their RNA retention, particle size, and Z-potential were measured, and their ultrastructures were evaluated via conventional transmission electron microscopy (TEM). To maintain the osmolarity between the inner section of the EMVs and the outer buffer, 0.83X diluted PBS was used when 5% disaccharides (ie, sucrose and trehalose) were present. PVP-40 (polyvinyl-pyrrolidone average molecular weight of 40.000 Da) was also tested as a cryoprotectant at a 2.5% w/v concentration and was not considered to influence the osmolarity balance. For each formulation, at least three replicates were prepared. Lyophilization was carried out in a Martin Christ freeze drier for 24 hours, and the chamber temperature was approximately −20°C. Every replicate sample was weighed, and the samples were stored at room temperature for one month. For comparison, the samples were stored at 4°C without any additives.
CM-DiI/Vybrant DiO Labeling of EMVs
CM-DiI (550/564 ex/em), a highly lipophilic dye, inserts spontaneously in double layers. It was found to optimally stain erythrocyte ghost membranes at a 1 µM concentration. The ghosts were incubated with the dye according to the manufacturer’s recommendations. Briefly, the suspension was first incubated at 37°C under vigorous shaking for 15 minutes and then for another 15 minutes at 4°C. Afterwards, the suspension was pelletized, and the supernatant, containing the excess dye, was removed. The same procedure was applied for Vybrant DiO (483/501 ex/em) labeling.
In Vivo Experiments
Injection and Biodistribution of Fluorescently Labeled DCC-siRNA-EMVs
All experiments were approved by the Ministry of Agriculture, Forestry and Food of the Republic of Slovenia (permission no. U34401-35/2020/8). Up to six mice were housed per cage in specific pathogen-free conditions in an IVC rack system (Animal Care Systems Inc., Revere Parkway, USA) with a 12 h light‒dark cycle, food and water ad libitum, and cage enrichment (VWR). Female 6–8-week-old C57BL/6NCrl mice (Charles River, Lecco, Italy) were used in the experiments. Tumors were induced by subcutaneously injecting 3×105 B16F10-tdTomato cells in 100 µL of 0.9% NaCl into the right flank of each mouse. The tumor volume was determined by measuring the tumor volume with a digital caliper and calculating the tumor volume via the formula for ellipsoid (V = a × b × c × π/6, where a, b, and c are perpendicular tumor diameters). Animal weight was monitored three times a week as a sign of well-being. When the tumors reached a volume of 50–60 mm3, Vybrant-DiO-labeled DCC-siRNA-EMVs containing Cy5-labeled anti-tdTomato siRNA were injected via the caudal vein into each mouse. A total of 30 µg of Cy5-siRNA (1.5 mg/kg weight) was administered to each mouse in 100 µL of 1x PBS. The mice were randomly divided into four groups: the control group (n=4), 2 hours after injection (n=5), 24 hours after injection (n=5) and 48 hours after injection (n=5). At 10 min after EMV injection, 100–200 µL of blood was collected from the retro-orbital plexus of the mouse eye, and whole blood was obtained immediately before sacrifice. The plasma was immediately separated from the whole blood via BD Microtainer SST Blood Collection Tubes (Beckton Dickinson). For quantification, the mice were assumed to have 1.65 mL of blood in total on the basis of their weight. The samples were stored at −80°C until further use. At the time of mouse sacrifice, the spleen, lungs, brain, kidneys, tumor, heart and liver were collected and weighed, and one half of each organ was flash frozen and processed for RNA extraction by homogenizing the organ in liquid nitrogen with a mortar and pestle. The resulting powder was collected and stored at −80°C until further use. The other half of each organ/tumor was fixed in 4% paraformaldehyde (PFA, Thermo Fisher Scientific) overnight at 4°C and dehydrated in 30% sucrose overnight at 4°C before being embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, VWR) and stored at −80°C for further use.
Biodistribution and Pharmacokinetics via stem‒loop PCR
RNA extraction was performed with a mirVANA MagMax kit (Thermo Fisher, A27828) from 10–15 mg of fine tissue powder and a total of 100 µL of plasma from each mouse, and a total of 5 ng was used for reverse transcription with a TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher, 4366596) with custom primers against anti-TdTomato siRNA (TaqMan Small Assay, Applied Biosystems, 4440418). A total of 1.33 µL of cDNA was used in each stem‒loop qPCR. Thermal cycling was performed according to the manufacturer’s specifications. The amount of anti-TdTomato siRNA guide strand in plasma and tissues was subsequently quantified via TaqMan real-time PCR with the help of a siRNA calibration curve (the procedure is described below). All samples were run in triplicate, and a nontemplate well was added to every reaction.
For standard curve generation, the following procedure was carried out. Five nanograms of both Cy5-siRNA and unlabeled siRNA were used as templates for reverse transcription from a stock of 1000 ng/mL, and then, serial dilutions of its cDNA were used in qPCR to generate a standard curve. For tissue quantification, values in nanograms obtained from the standard curve were divided for the qPCR, reverse transcription, and RNA extraction dilution factors and multiplied by the original amount of total RNA obtained from RNA extraction.

Noncompartmental analysis of pharmacokinetic parameters relative to those of plasma was performed via PKSolver Excel add-in,48 with linear uplog down settings. All pharmacokinetic parameters refer to the terminal half-life of the siRNA.
Tissue Sample Preparation for Fluorescence Microscopy
From OCT-embedded tissues, 14 µm thick sections were cut on a Leica CM1850 cryostat and collected on SuperFrost Plus slides (Epredia, Netherlands, J7800AMNZ), with a final n=3 sections on each slide. The slides were stored at −20°C until further use. Later, the nuclei were stained with 3 µg/mL Hoechst 33342 nuclear stain, the sections were mounted with ProLong Glass Antifade Mountant (Thermo Fisher, P36982), and the cover slip (#1.5) was sealed with transparent nail polish. The slides were subsequently imaged on an Axio Observer 7 equipped with a Colibri 7 LED light source (UV (385 nm), Violet (430 nm), Blue (475 nm), Green (555 nm), Yellow (590 nm), Red (630 nm), Far Red (735 nm) and a Hamamatsu Orca Flash 4.0 V3 camera. Emitted light was imaged through the following filters: Filter set 38 hE eGFP shift free (Zeiss), Filter set 43 hE Cy 3 shift free (Zeiss), Filter set 50 Cy 5 shift free (Zeiss) and Filter Set 112 hE LED (Zeiss).
Evaluation of RNA Interference Ability in Vivo
For the in vivo silencing experiment, B16F10 tumor-bearing mice were divided into three experimental groups and injected with 1) 100 µL of PBS; 2) 100 µL of naked anti-TdTomato siRNA, for a total of 2.5 mg/kg body weight; or 3) 100 µL of DCC-siRNA-EMVs, for a total of 2.5 mg/kg body weight, as indicated by the siRNA content. After 48 hours, the mice were sacrificed, and the tumors were excised, flash-frozen in liquid nitrogen and ground to a fine powder with a mortar and pestle. RNA extraction was carried out with a mirVANA MagMax kit (Thermo Fisher) from 10–15 mg of fine tissue powder. TdTomato gene knockdown was quantified similarly to that in vitro, and the presence of the guide strand of anti-TdTomato siRNA was assayed as described in the previous sections. Images of tdTomato-fluorescent B16F10 tumors were taken at the beginning of the experiment (t=0), at 24 hours and at 48 hours with a SteREO Lumar. A V12 fluorescence microscope (ZEISS, Jena, Germany) was used. The mean fluorescence intensity of the tdTomato protein in tumors was quantified with ImageJ, the appropriate region of interest (ROI) area was selected, and the Yen threshold algorithm49 was used to minimize disturbance by mouse hair. For tdTomato MFI quantification in frozen sections, with n = 5 for each mouse, images of the tdTomato channel were taken with a 20x objective. Images were subjected to the above-described process via the Huang thresholding algorithm.
Ex Vivo Red Blood Cell and DCC-siRNA-EMV Incubation With Inverted Confocal Microscopy
DCC-EMVs were prepared as previously described, with DiO-labeled ghosts, and their final protein concentration was 0.54 mg/mL. The mice were anesthetized via isoflurane inhalation, and blood was collected via cardiac puncture in 3.2% sodium citrate, after which the mice were sacrificed. The collected blood was then centrifuged at 100 × g for 10 min at RT and washed twice in 1x PBS. The washed red blood cells (RBCs) were counted via a CytoSMART cell counter (AXION Biosystems) and resuspended in RPMI medium containing GlutaMAX (Gibco, Thermo Fisher Scientific, VA, US) and penicillin/streptomycin at a concentration of 1×106 RBCs/mL. The cell culture medium did not contain fetal bovine serum. The erythrocytes were labeled with Vybrant-DiD according to the manufacturer’s instructions with a final dye concentration of 5 µM (Invitrogen, Thermo Fisher Scientific, VA, US). Subsequently, 100 µL of the prepared RBCs were transferred to 96-well plates (96-well plates µCLEAR®, BLACK, Greiner), mixed with 10 µL of DCC-EMVs and incubated at 37°C in a humidified incubator at 5% CO2 for 24 or 48 h. The interaction between mouse RBCs and EMVs was observed via inverted confocal laser scanning microscopy (CLSM, Axio Observer Z1 LSM 710, ZEISS, Germany) with a Plan-Apochromat 63x/1.40 Oil DIC M27 objective. The two dyes CM-DiO and Vybrant-DiD were excited simultaneously with argon (488 nm) and helium–neon (633 nm) lasers, and the emitted light was collected through emission bandpass filters set.
Statistical Analysis
All the statistical analyses were performed via GraphPad 9, with p≤0.05 *p≤0.01 **p ≤ 0.001 ***p ≤ 0.0001 ****. For cryo-TEM EMV size, postfreeze drying siRNA retention and EV size, qRT‒PCR fold changes (in vitro and in vitro), and in vivo tumor tdTomato MFI variation, a normality test was carried out. A parametric (two- or one-way ANOVA) or nonparametric (Kruskal‒Wallis) test was chosen to test the statistical significance of the results. In instances where multiple comparisons testing was needed, the Tukey’s test for multiple comparisons was utilized. Where present), the data were log10 transformed to highlight the differences.
Results
Comparison of Different Techniques for Removing Unencapsulated siRNA
EMVs were prepared via the hypotonic lysis method42 and then loaded with siRNA via incubation, sonication and extrusion steps. Before any purification, the vesicles had a median particle size of 192 nm (Table 2). To establish actual encapsulation efficiency and remove unloaded siRNA, five different methods were tested (Supplementary Material for protocols), ie, sucrose gradient, regular ultracentrifugation, and density-changing centrifugation.46 The final apparent encapsulation efficiency, an estimate of the required time and most significant drawbacks are shown in Table 3.
 |
Table 2 Particle Size Distributions of Different Preparations of EMVs After Freeze Drying and Cryoprotectant Removal. In Light Gray, Empty EMVs; in White and Dark Gray, Noncentrifuged siRNA-EMVs and DCC-siRNA-EMVs, Respectively |
 |
Table 3 Efficiency of the Encapsulation of siRNAs Into EMVs via Different Techniques |
The use of PD10 desalting columns led to interesting results: the encapsulation efficiency, that is, multiple recoveries of EMVs and siRNA, reached 90%, suggesting an interaction between the two (Figure S1). The siRNA-EMVs were recovered after the addition of 4/5 mL, and no other amount of siRNA was detected after complete elution of the column (~20 mL). While unbound siRNA is supposed to be eluted later than EMVs, this was not the case since, after the addition of 6 mL of recovery, the total initial dose of siRNA was almost complete, with only 0.3–0.5% of the initial siRNA amount being eluted after EMV recovery. EMVs recovery was confirmed by DLS analysis and RNA quantification with a Quantifluor kit.
Both size exclusion purification and the use of Amicon centrifugal units pose the problem of maintaining sterility. Both can allow the recovery of weakly externally complexed siRNA and imperfectly shaped EMVs. Moreover, while the preparation of sucrose gradient columns is laborious and time consuming, regular ultracentrifugation does not discriminate among EMVs of different densities. We then decided on density changing centrifugation (DCC), with 10% w/v trisodium citrate dissolved in PBS, since it was proven to be the most repeatable and reliable technique and capable of ensuring the isolation of siRNA internally loaded with EMVs.46
DCC-siRNA-EMV Characterization
EMVs were loaded with different amounts of siRNA (2.5–12 µg/mL) and subjected to purification via density-changing ultracentrifugation, as schematically shown in Figure 1a. Eight fractions of equal volume from the centrifugation tubes were collected, the TC was replaced with 1× PBS, and the RNA concentration was evaluated with a Quantifluor RNA kit. The average encapsulation efficiency was 67.21 ± 3.36% in six fractions (as a result of 6 independent experiments), with a particle size ranging from 170–190 nm and a PDI ranging from 0.01–0.02. The size was found to increase from the top to the bottom, and vice versa for RNA quantity (Figure 1b), indicating that the method is effective in selecting EMVs on the basis of their density alone. The last fraction arose from the resuspension of the pellet, consisting of membrane remnants not shaped into vesicles and unloaded siRNA. Notably, RBC ghosts, prior to coextrusion with siRNA and subsequent postprocessing, were free from endogenous miRNAs and other RBC-related RNAs.50,51
The overall phospholipid concentration in the original sample not subjected to ultracentrifugation was found to be 0.240 ± 0.09 mg/mL via the Stewart assay, whereas the protein concentration was 0.46 mg/mL ± 0.097 (determined via the Bradford assay), which is in line with the literature for erythrocyte membranes, where 40% of the dry weight is composed of lipids, including 40% cholesterol, and approximately 60% is composed of proteins and sugars. Starting from one mL of a solution of 70% v/v erythrocyte ghosts diluted in 4 mL of PBS, the dry mass (protein, lipid and salt) obtained was 10.63 mg ±0.9. To further prove the solidity of this purification method for EMVs, we evaluated the phospholipid and protein concentrations in the five top fractions (See Protein and lipid quantification) in a separate experiment, ie, those with the highest concentration of RNA (Figure 1b and c), with 1--5 fractions.
The mixed effect analysis revealed (p < 0.01) that the siRNA content was the only source of variation among the different fractions (Figure 1c), decreasing from top to bottom, whereas the lipid and protein concentrations remained constant. The lipid/protein ratio slightly increased after DCC, from approximately 0.5, as in extruded ghosts, to 0.8, indicating a loss of protein net content. The zeta potential was also measured for every fraction and was found to be constant throughout the sample, accounting for −11.2 ± 0.2 mV in the presence of 10% TC at pH = 7.4. In line with the literature,46 the zeta potential was re-established to precentrifugation values after 10% TC removal, which was −17.7 ± 3.4 mV, confirming that the DCC-siRNA-EMVs were right-side out.42 We then decided to save the top six fractions for further experiments, combining them, eventually concentrating the EMV suspension, which was needed for further analysis. The particle size of all the siRNA-containing fractions combined was slightly lower than 200, with a value of 185.4 ± 6.7 and a PDI of 0.192 ± 0.015. DCC-siRNA-EMVs also shared a similar protein profile with parental erythrocyte ghosts and with the pellet resulting from DCC, as established with SDS‒PAGE (s, Figure S2).
Microscopic Characterization of siRNA-EMVs: CryoTEM, Freeze-Fracture TEM and STED Microscopy
DCC-siRNA-EMVs resulting from ultracentrifugation were imaged via cryo-electron microscopy to determine whether ultracentrifugation was effective at reducing the amount of unloaded EMVs and membrane debris, which could harm the stability of the colloidal suspension. Both empty and loaded (siRNA concentration before DCC was 12 µg/mL) EMVs were assayed before and after ultracentrifugation. Compared with extruded EMVs, cryoTEM imaging (Figure 2a) has indeed proven our premises to be true, showing centrifuged samples as clearer from membrane debris, which are not correctly rearranged into EMVs, when compared to only extruded EMVs. Many rod-shaped objects were also present, which is consistent with literature.52,53 Such objects are most likely EMVs sitting on their side, being EMVs biconcave as confirmed by conventional TEM (Figures 3 and 5, yellow arrows). Therefore, EMVs can be assumed to be relatively flat (1–10 nm thick), discoidal nanoparticles, as already reported for similar objects.53 Image analysis via ImageJ (Figure 2b) revealed that loaded and centrifuged EMVs were significantly larger than empty EMVs were (one-way ANOVA, p < 0.001), confirming that their internal content was the only selection factor in DCC. All four size distributions followed a lognormal distribution (p < 0.001, Figure 2b) according to the Kolmogorov‒Smirnov and Anderson‒Darling tests. Multilamellar EMVs are extremely rare and hard to spot (Figure 2a, inset).
 |
Figure 2 (a) CryoTEM micrographs of EMVs. The scale bar is 200 nm, and it refers to the insert too. Yellow arrows are pointing at EMVs sitting on the side, appearing as rod-like objects. (b) Relative frequencies of the diameters in the analyzed cryoTEM micrographs. Measurements were carried out on 10 micrographies for each category, for a total of n > 200 measurements per typology. One-way ANOVA was performed to assess the statistical significance of the distributions. p≤0.01 **, p ≤ 0.001 *** (c) Freeze fracture TEM micrograph of empty and DCC-siRNA EMVs. For higher magnification images (left), the scale bar is 50 nm. In the right image, a wider field of DCC-siRNA-EMVs is shown (scale bar = 500 nm). The internal content is recognizable as aggregate material (green arrows). |
 |
Figure 3 TEM micrographs of freeze-dried, noncentrifuged samples negatively stained with UA Zero. EMVs appear as discoidal, flat objects. Yellow arrows are pointing at EMVs sitting on the side. |
 |
Figure 4 Fluorescence superresolution microscope images of siRNA-EMVs. (a) Wide-field image of DCC-siRNA-EMVs; scale bar = 1 µm. (b) Close-up image of an EMV, Cy-5 and CM-DiI signals colocalized, scale bar = 250 nm; (c) non-DCC-siRNA-EMVs, scale bar = 1 µm. |
To investigate the internal side of EMVs and DCC-siRNA-EMVs and better appreciate their surface structure, we also conducted freeze-fracturing electron microscopy on the same set of samples (Figure 2c). The surface structure was consistent with that of EMVs obtained via a similar technique54 and with red blood cell extracellular vesicles,55 being rough and wrinkled but overall circular and regular. When DCC-siRNA-EMVs were imaged, a clot of solid material was found in most of the fractured vesicles (Figure 2c, inner). While aggregation has already been reported56 in the case of sonicated siRNA loaded inside extracellular vesicles, this is the first time, to our knowledge, that such clots can be visualized via electron microscopy techniques.
To verify the identity of such clots, we conducted several tests on a confocal microscope equipped with a STED laser,47 which labels EMV membranes with CM-DiI dye and loads them with Cy-5-conjugated siRNA. The average size, as measured by CM-DiI fluorescence signals, was consistent with previously reported data (<200 nm, Figure S3). Furthermore, Cy-5 is prone to create nonfluorescent H-aggregates57 and self-quench its fluorescence when it is found at high concentrations, as occurs in the lumen of 100–150 nm DCC-siRNA-EMVs. Indeed, the DiI and Cy-5 signals were hardly perfectly superimposed (Pearson Rr parameter = −0.00075, over the average of 10,200x200 pixels/20x20 µm images). EMVs, in which the two signals overlap (Figure 4b), are extremely difficult to spot. CLSM also confirmed the efficient removal of RBC membrane debris in the DCC-siRNA-EMV sample (Figure 4a), considering the lack of CM-DiI-labeled aggregates, which are present in noncentrifuged samples (Figure 4c). This finding further supports the observation via cryoTEM (Figure 2a) of a cleaner, monodisperse colloidal suspension. Finally, the Cy-5 siRNA signal seemed to be concentrated only within EMVs in samples subjected to DCC (Figure 4a and c), confirming the total removal of unencapsulated siRNA molecules.
 |
Figure 5 (a) Percentage of siRNA payload loss over the course of four weeks for DCC and siRNA-EMVs. Data are reported over n=3 replicates. (b) Ultrastructural evolution of negatively stained siRNA-EMVs (left) and DCC-siRNA-EMVs (right). REd arrows indicate pancake-shaped EMVs, which appear as rods via cryo-EM. (c) Size evolution of siRNA-EMVs stored at 4 °C. Above the bars, the percentage of recorded events belonging to a specific population is shown. (d) Percentage of phospholipid weight loss in siRNA-EMVs, stored at 4 °C. (e) Agarose gel (2 % w/v) for free siRNA, DCC-siRNA-EMVs, and non-ultracentrifuged siRNA-EMVs exposed to different concentrations of RNAse A. (f) siRNA retention in siRNA-EMVs after freeze drying. The marked results are significant compared with those of the PBS control for the siRNA concentration. p ≤0.05 * p ≤ 0.01 ** p ≤ 0.001 *** p ≤ 0.0001 ****. |
Encapsulation Stability Over Time and Effects on the Storage of EMVs
To assess the eventual spontaneous release of cargo by EMVs upon long-term storage at 4°C, two identical sets of samples were evaluated (initial siRNA concentration of 10 µg/mL). One was subjected to DCC, while the other was not. To remove unencapsulated siRNA, the noncentrifuged set of samples was ultrafiltered immediately after extrusion at 30 kDa via Amicon devices. At fixed intervals, 100 µL (n=3 for each time point) of each replicate of both sets of samples were subsequently loaded into an Amicon 30 kDa centrifugal filter device. The eluate and retentate were resuspended to the original volume, and the siRNA concentration was evaluated with a Quantus RNA fluorometric assay over one month of storage at 4°C. The payload loss was much greater for nonultracentrifuged EMVs (57% after 4 weeks, Figure 5a), suggesting continuous leakage of the payload from EMVs or slow decomplexation of siRNA from RBC debris. However, in the case of the DCC-siRNA-EMVs, leakage started to be noticeable after only one month (Figure 3a), accounting for 10% of the initial amount by the end of the experiment. Noncentrifuged EMVs were also assayed for phospholipid loss (Figure 5d): a consistent amount of phospholipid was found in the eluate of Amicon 15 NMWL centrifugal devices, which increased with increasing incubation time. EMVs and DCC-siRNA-EMVs were also evaluated via TEM (Figures 5b and S5 for uranyl acetate comparison), whereas for DCC-siRNA-EMVs, no morphological or ultrastructural changes were noticeable; for non-DCC-EMVs, the increase in size was evident in the TEM micrographs, with the appearance of “pancake-like” objects (red arrows, in Figure 5b) and a general loss of a stable structure and the appearance of smaller EMVs, probably due to further vesiculation. The size evolution of the siRNA-EMVs was also assayed via DLS (Figure 5c). Even if quite stable, the volume-weighted diameter of the EMVs increased over time, causing a slight increase both in size and in the population abundance of objects larger than 400 nm. Nonetheless, after one month, the regular-sized EMV population (less than 120 nm) still represented the majority of the objects, accounting for 71% of the measured events. We therefore speculated that an already reported protease activity present in the red blood cell membrane is responsible for self-digestion, loss of structure and, finally, spontaneous budding of the membrane in the absence of ATP.58,59 The digestion of integral membrane proteins such as Band 3 proteins is indeed involved in aging, which includes aspecific vesiculation due to disassembly of the cytoskeleton.60,61 The presence of such proteases, which are active both in self-digestion and toward casein, was confirmed via a Pierce protease assay on EMVs but was not detectable or extremely faint on DCC-siRNA-EMVs (s, Figure S4), likely being less represented in the population of objects selected via density-changing centrifugation.
Density-changing centrifugation again effectively removed the cellular debris and stabilized the overall colloidal suspension: no significant changes in terms of the hydrodynamic radius or fusion or fragmentation of EMVs were observed, confirming what was previously observed for siRNA retention.
RNase A Protection Assay
To test the RNA accessibility of siRNA-loaded EMVs, we carried out an RNase A resistance assay. Briefly, we incubated a known amount of siRNA-EMVs with 0.5, 5, or 50 µg/mL RNAse A for 2 hours or 24 hours at 37°C and then run the appropriate volume of each sample on a 2% agarose electrophoresis gel. The volumes to be loaded were carefully calculated to run the same initial amount of siRNA-EMVs. The starting concentrations of siRNA were 10, 8.5 and 10 µg/mL for naked siRNA, DCC-siRNA EMVs, and siRNA-EMVs, respectively. For DCC-siRNA-EMVs, ie, siRNA-EMVs subjected to density-changing centrifugation, bands were present even after 24 hours (Figure 5e), in the presence of 5 µg/mL bovine pancreatic RNase A and after two hours at 50 µg/mL. Surprisingly, the noncentrifuged siRNA-EMVs also presented a band at 5 µg/mL after two hours of incubation, while the corresponding free siRNA band was absent.
Freeze-Drying and Storage Optimization
To establish a long-term optimal storage procedure, DCC-siRNA-EMVs, siRNA-EMVs and empty EMVs were freeze dried in the presence of different common cryo-/lyoprotectants: sucrose, trehalose and PVP40. In the case of the siRNA-EMVs, the DCC and noncentrifuged samples were equally freeze-dried and characterized. Disaccharides were added to a diluted PBS solution at a 50 mm concentration, while PVP40 was used at 2.5 w/v %.62 The same volume of PBS was used as a negative control. After one month of storage at room temperature, the freeze-dried samples (n=6 for each type) were rehydrated to the original volume, and the cryoprotectant was removed via Amicon device ultrafiltration (4 kDa). Particle size, zeta potential (Table 2, Table 4), and siRNA retention (Figure 5f) were assessed, and their ultrastructures were evaluated via conventional TEM preparations with a UA-Zero negative stain (Figure 3). Upon rehydration, all the samples were immediately dissolved in DEPC water except for the negative control, which contained numerous insoluble clots and a foggy appearance. Indeed, PBS is not the most suitable buffer for freeze drying both exosomes and extracellular vesicles since it is subjected to changes in pH during drying and after rehydration;63 after rehydration, the pH also shifted from 7.4 to 6.5. Nonetheless, when freeze-dried with only disaccharides, all the samples retained their original size, zeta potential, and siRNA cargo (Table 2, Figure 5f). The presence of cryo-lyoprotectant increased siRNA retention in DCC-siRNA-EMVs dried, only, in the presence of the same volume of PBS 1x, with almost 100% of recovery in presence of disaccharides, ie trehalose and sucrose. Indeed, payload retention reached 99%, for DCC samples in the presence of disaccharides, indicating excellent colloidal stability. PVP40 is another well-known and widely used not-penetrative cryo/lyoprotectant,62,64 whose mechanism relies on the modification of the collapse temperature of the structure65 by increasing the glass transition temperature of the colloidal suspension and water sequestration properties.66 We therefore chose to test this nontoxic and well-known polymer to extend the shelf-life of freeze-dried EMVs. Counterintuitively, before cryoprotectant removal, samples freeze-dried in the presence of PVP40 presented a much lower zeta potential (−23.5 ± 3.1 mV and −23.3 ± 3.8 mV for trehalose + PVP40 and sucrose + PVP40, respectively) and lower siRNA payload retention after rehydration than samples freeze-dried in the presence of disaccharides (even if overall higher than that of the negative control, ie, PBS). After centrifugal dialysis and PVP40 removal, the zeta potential of both sets of samples returned to their original values (Table 4), despite the particle size still being significantly larger (p < 0.001 in all cases). This finding is consistent with what has already been reported for extracellular vesicles and exosomes,62,63 ie, a reduction in the number of nanoparticles, possibly originating from their fusion with each other, and the substantial impossibility of complete polymer removal; therefore, the unsuitability of PVP40 as a cryoprotective for this type of object is assessed. On the other hand, trehalose or sucrose alone did not significantly affect the size of any type of EMVs sample (Table 2).
 |
Table 4 Z-Potentials of Freeze-Dried DCC-siRNA-EMVs After Cryo-Lyoprotectant Removal |
Given that siRNA was retained after freeze drying, the best-performing cryo-lyoprotectant was sucrose at a 50 mm concentration. Moreover, we used TEM to assess the suitability of the above-said cryo-lyoprotectant with TEM, observing the ultrastructures of EMVs. The samples (DCC and not DCC) were negatively stained with UA-zero (Figure 3 and S6 for freeze-drying only in the presence of PBS). Exceptionally interesting were also the ultrastructure differences highlighted by the staining: UA-zero complexation with membranes was lower in the presence of PVP40, and where trehalose was present, both for DCC and not samples, an organic and soft shield around the vesicles can be observed. This feature was not highlighted in previous reports on cell-derived vesicles freeze-dried in the presence of PVP polymers62 negatively stained with uranyl acetate, although a similar electron-dense area was observed around organic objects. Owing to their safety and cost-effectiveness, disaccharides have been proven to be the best cryo-/lyoprotectant for RBC membrane-derived vesicles.
 |
Figure 6 Internalization of DCC-siRNA-EMVs by B16F10, CT26 and NHLF cells over three days. The cell nuclei were labeled with Hoechst 33342 (blue), and the membranes were labeled with MemBrite® Fix 488/515 stain (green). The membranes of EMVs were stained with CM-DiI (Orange), while they were loaded with Cy-5-labeled siRNA (red). The scale bar is 50 µm. |
In Vitro Activity of DCC-siRNA EMVs
Cytotoxicity and Internalization of siRNA-EMVs by Different Cell Lines
We investigated whether DCC-siRNA-EMVs at a final concentration of 4.5 µg/mL EMV protein (1:66 dilution), corresponding to 30 nM siRNA, had any toxic effects on multiple cell lines: NHLF human lung fibroblasts, Hulec 5a human lung microvascular cells, CT26 murine colorectal carcinoma, B16F10 murine melanoma, and 4T1 murine mammary carcinoma. These conditions were chosen to mimic an in vitro knockdown experiment. No signs of cytotoxicity were detected in any of the tested cell lines (Figure 7a). To determine whether the DCC-siRNA-EMVs internalized B16F10, CT26 and NHLF cells were incubated with them (Figure 6). The vesicles were loaded with Cy-5-labeled siRNA, and a total of 0.7 nM/well of Cy-5 siRNA/well was incubated with the cells. Additionally, the EMV membranes were marked with CM-DiI. After 48 hours of coincubation, the CM-DiI signal was present in both the plasma and inner membranes of the cells, suggesting membrane fusion between the cells and EMVs, together with the presence of Cy-5 in the cytoplasm. Overall, these findings indicate that EMVs are actively internalized by normal and tumor cells rather than just adhering to the cell surface. As a proof of concept for our carriers, we chose B16F10 cells, a well-characterized murine melanoma cell line that showed robust membrane fusion with CM-DiI-labeled EMVs, as a model for further experiments.
 |
Figure 7 (a) Viability of various cell lines treated with 4.5 µg/mL DCC-siRNA-EMVs; (b) log10 of in vitro tdTomato (fold change) at a final concentration of 0.3 nanomolar. The data are expressed as the means ± SDs. The percentage of knockdowns ± SDs is reported below the graph. p ≤ 0.001 ***, p ≤ 0.0001 ****. |
In Vitro Knockdown Effect of siRNA-EMVs
The tdTomato-expressing B16F10 murine melanoma cell line was selected to evaluate the ability of the siRNAs to silence the tdTomato fluorescent protein expression. To prove the robustness of our storage protocol, we also tested sucrose-freeze-dried DCC-siRNA-EMVs. The silencing efficiency was compared with that of RNAiMAX, the most efficient nonviral transfection agent on the market, and the same final amount of anti-tdTomato siRNA was added to the cells, corresponding to 30 picomoles/well (for a final siRNA concentration of 30 nM in each well). Total RNA extraction was performed after 48 hours. Real-time PCR revealed circa 80% knockdown efficacy in the case of the DCC-siRNA-EMVs (Figure 7b), whereas the FD samples retained approximately 50% of their activity. Transfection with EMVs did not impact β-actin expression, since none of the samples shown were significantly different from the control Ct (Figure S7). The reduced activity of sucrose freeze-dried DCC-siRNA-EMVs can be attributed to the eventual loss of the right-sidedness of EMVs, protein unfolding, lipid loss upon rehydration, and early cargo release. In contrast, RNAiMAX caused almost complete knockdown of the target gene at the same dose.
In Vivo Experiments: Biodistribution, Pharmacokinetic and Knockdown Effects
Biodistribution and Pharmacokinetics of DCC-siRNA-EMVs
To determine the biodistribution of DCC-siRNA-EMVs in mice, we coupled stem‒loop PCR of the siRNA guide strand with fluorescence microscopy of thin tissue sections. For this purpose, we subcutaneously induced tdTomato-B16F10 tumors in C57BL/6 mice. When the tumors reached 50–60 mm3, we intravenously injected 100 µL of DCC-siRNA EMVs, whose membranes were labeled with Vybrant-DiO and loaded with Cy-5-labeled passenger strand siRNA (Figure S8 for the TaqMan calibration curve). We first determined the pharmacokinetic parameters relative to those of the DCC-siRNA-EMVs (Table 5). Upon caudal vein injection, siRNA-EMVs were rapidly distributed among body compartments, with 40% of the total injected dose (ID) per mL of blood remaining in circulation after 10 minutes (Figure 8a). After two days, approximately 1.5% ID/mL was still present. The mean residence time67 was 18 hours, where a commonly reported value for a free siRNA is 2–4 hours;40,68 distribution in the tissue phase was extremely fast, with an apparent volume of distribution (Vz) of 20 mL (or 1000 L/kg of mouse weight). This behavior can be attributed to the low affinity of EMVs for plasma proteins, leading to faster tissue distribution, as observed in other nanoparticle-assembled siRNA treatments.69,70 Overall, the distribution half-life was comparable to that of other stealth, neutral siRNA delivery systems,36,40 confirming the potential of our delivery system.
 |
Table 5 Basic Pharmacokinetic Parameters Relative to siRNA-EMVs Administered at 1.5 mg/kg, Calculated With the PK Solver 2.0 Excel Add-in |
 |
Figure 8 (a) Plasma concentration of the anti-tdTomato siRNA lead strand. For calculation of the injected dose (ID), the mice were assumed to have 1.6 mL of blood. (b) Nanograms of anti-tdTomato siRNA lead strand for each gram of organ, as obtained via interpolation of stem‒loop qPCR data from tissues with a calibration curve (Figure S8). |
We then proceeded to determine the presence of the siRNA lead strand in various organs. At an injected dose of 1.5 mg/kg of weight of anti-tdTomato siRNA (30 μg/mouse), the earliest clearance organ for DCC-siRNA-EMVs appeared to be the spleen (Figure 8b). Similar to Liang et al,71 the lungs were shown to be one of the main accumulation sites for EMVs, although after administration (ie, 24 hours), the presence of the lead strand of anti-tdTomato siRNA in the kidneys was already substantially lower than that of free siRNA.33 While the concentration of the lead strand of anti-tdTomato siRNA remained constant in the liver, it increased over two days in tumors, reaching 4000 ng/g ± 335 of siRNA/g tumor tissue. Therefore, the DCC-siRNA-EMVs appeared to have eluded accumulation in the main reticuloendothelial site organ, ie, the liver: at 48 hours, the ratio of the tumor siRNA/liver siRNA was 1.83. Such behavior can be ascribed to the presence of CD47 on EMVs, masking effect on phagocytic SIRPα + Kupffer cells, as observed elsewhere72,73. While the occurrence of high accumulation in heavily perfused organs such as the heart and lungs was predictable, we were surprised by the abundance of siRNAs in the brain.
To investigate this phenomenon, we analyzed the distribution in organs in detail by tracking Cy-5 (conjugated at the 5′ end of the anti-tdTomato passenger strand) and DiO fluorescence, which was used to label EMV membranes. Taken together, these findings confirmed the molecular data and strongly suggested that EMVs rapidly entered the blood circulation and promptly accumulated in tissues. As early as 2 h, the accumulation of the Cy-5 and DiO signals was greater in proximity to the blood vessels and more abundant in highly perfused organs, such as the lungs (Figure 9), brain (Figure 14)) and heart (Figure S10). In the lungs, after 24 hours, the Cy-5 signal had a homogenous distribution, suggesting deeper tissue penetration from blood vessels over time (Figure 9), reflecting the molecular data. In the kidneys (Figure 10), the main filtration organs for the nanoparticles, Cy-5 and DiO fluorescence, appeared to be homogenously distributed, without any greater accumulation in either the glomeruli or tubules. No preference for any lobe can be noted in the liver, despite the Cy-5 signal being mostly concentrated in the proximity of sinusoids (Figure S9) and around larger blood vessels. Within 24 hours, the Cy-5 signal is present around small arteriolae in the brain but not in tissues, as EMVs are likely unable to cross the tight endothelium of the blood–brain barrier; the high density of brain capillaries74 (Figure 8 and 14), therefore, explains the strong accumulation determined via qRT‒PCR. The absence of the Cy-5 signal can be noted in the spleen white pulp (Figure 11) and in the marginal zone, while strong accumulation can be observed in the red pulp, which is devoted to recycling old erythrocytes;75 EMVs, therefore, eluded capture by the lymphoid tissue of white pulp, which is composed of T and B lymphocytes and macrophages.75 On the other hand, the B16F10 tdTomato-treated tumors presented a disorganized and chaotic structure, with moderate autofluorescence in the green channel around blood vessels and in connective tissue and heterogeneously distributed blood vessels. In the controls, a vast majority of the cells presented fluorescence in the tdTomato channel (Figure 12 and 13).
 |
Figure 9 Distribution of Cy-5/DiO DCC-siRNA-EMVs at different time points in frozen sections of mouse lungs via siRNA-Cy-5 (red) and DiO (green) signals in frozen sections of lungs. Nuclei were labeled with Hoechst 33342 (blue). The scale bar is 100 µm. |
 |
Figure 10 Distribution of Cy-5/DiO DCC-siRNA-EMVs at different time points in frozen sections of mouse kidneys via siRNA-Cy-5 (red) and DiO (green) signals. Nuclei were labeled with Hoechst 33342 (blue). The scale bar is 100 µm. |
 |
Figure 11 Distribution of Cy-5/DiO DCC-siRNA-EMVs at different time points in frozen sections of the mouse spleen via siRNA-Cy-5 (red) and DiO (green) signals. Nuclei were labeled with Hoechst 33342 (blue). The scale bar is 100 µm. |
In tumors, despite being not the main accumulation site, the Cy-5 signal is greater in the tumor rim and within a layer of 9–10 cells from the closest capillary, which is consistent with the enhanced permeation and retention theory,76 and it slowly diffuses farther from blood vessels over 48 hours. Indeed, while EMV tissue penetration is limited to 3–4 cells from the closest blood vessel at the 2-hour time point, the Cy-5 signal distribution appears to be much more homogenous at 48 hours (Figure 12). Overall, as indicated by the qRT‒PCR data, the Cy-5 and DiO signals continued to increase over time (Figures 13 and 12). Interestingly, extravasated erythrocytes, which occur in solid melanomas due to defective blood vessels,77 are widespread in tumor tissue and are fluorescent in the Cy-5 channel (Figure 13) as early as 2 h post-administration but are positive but fainter in the FITC channel.
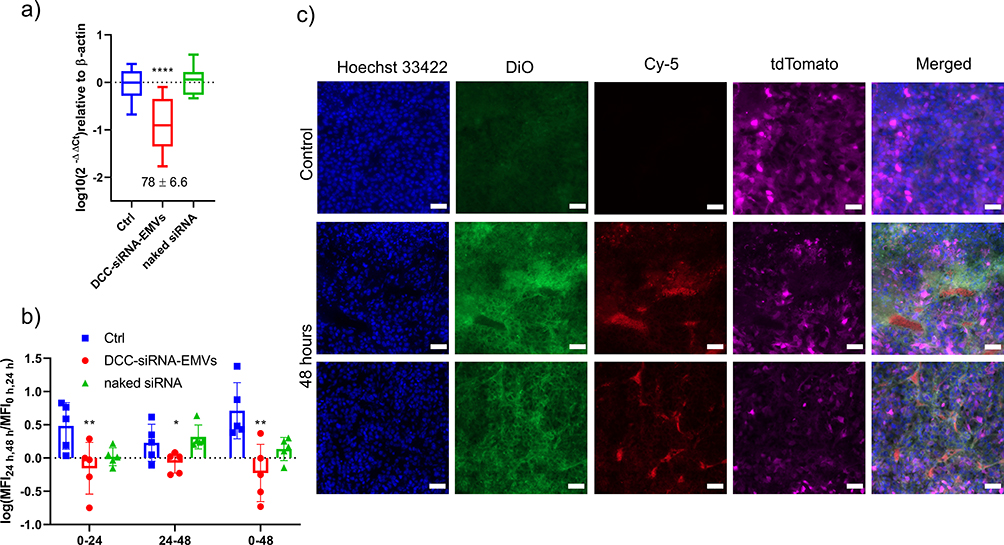 |
Figure 12 Summary of the knockdown activity of systemically delivered DCC-siRNA-EMVs. (a) Log10 of the tdTomato fold change in vivo at the 2.5 mg/kg dose compared with the basal expression of the gene in control tumors, as analyzed with the Kruskal‒Wallis nonparametric t-test. p ≤ 0.0001 **** (b) Log10 of the tdTomato median fluorescence intensity variation at 0‒48 hours and 24‒48 hours in control, DCC-siRNA-EMV-, and naked siRNA-treated mice. p≤0.05 *, p≤0.01 ** (c) Tumor tissue sections with blood vessel details at 0 and 48 hours. Red blood cells were unanimously positive in the Cy-5 channel at 48 hours and fainter in the DiO channel. The scale bar is 50 μm. |
 |
Figure 13 Distribution of Cy-5/DiO DCC-siRNA-EMVs within B16F10-tdTomato tumors via frozen sections of B16F10-tdTomato tumors at different time points after the injection of DCC-siRNA-EMVs with nuclei labeled with Hoechst 33342 (blue), Vybrant DiO (green), Cy-5 siRNA (red) and tdTomato expression (magenta). The scale bar is 100 µm. |
From 24 hours onward, the circulating erythrocytes in the blood vessels in the kidneys (Figure 10), heart, brain, lungs and liver showed the same behavior. The Cy-5/DiO erythrocytes were absent in every organ in the control group. In tumors, areas of extravasated erythrocytes 20–40 μm in diameter were completely labeled with Cy-5 48 hours after administration and were weaker in the DiO channel. Although red fluorescence of erythrocytes has been previously reported 78 and hemoglobin fluorescence has been evaluated by fluorescence microscopy with two-photon excitation, 79 the phenomenon we observed appears to be different. We therefore performed a small-scale ex vivo experiment in which we incubated mouse erythrocytes with DiO-labeled DCC-EMVs to detect any membrane fusion and payload transfer under physiological conditions. After 24 and 48 hours of coincubation, several erythrocytes acquired DiO fluorescence (Figure S11c), indicating membrane fusion, as the diffusion rate of such a lipophilic tracker in lipid bilayers is low 80 In the absence or presence of EMVs, many erythrocytes take the form of echinocytes 81 while the addition of RPMI medium caused the aggregation of DCC-EMVs, which was confirmed by DLS analysis (Figure S11b and); agglomerates of 2–5 µm were indeed absent in the DCC-EMVs dispersed in PBS (Figure S11a).
 |
Figure 14 Distribution of Cy-5/DiO DCC-siRNA-EMVs at different time points in frozen sections of the mouse brain via siRNA-Cy-5 (red) and DiO (green) signals. Nuclei were labeled with Hoechst 33342 (blue). The scale bar is 100 µm. |
Successful in Vivo Delivery of siRNA-EMVs to Tumors and Knockdown of tdTomato Expression After Systemic Administration of DCC-siRNA-EMVs
Having confirmed the strong in vitro knockdown ability and successful delivery of EMVs to B16F10-tdTomato tumors, we aimed to ascertain whether the DCC-siRNA-EMVs had the ability to elicit gene knockdown in vivo. For this purpose, we again subcutaneously induced B16F10-tdTomato tumors in C57BL/6 mice, and when the tumors reached 50–60 mm3, we intravenously injected 100 µL of naked siRNA (2.5 mg/kg in 0.9% NaCl, n = 4), unlabeled DCC-siRNA-EMVs (2.5 mg/kg in 0.9% NaCl, corresponding to 50 µg for each mouse, n = 5), and, finally, 100 µL of saline (0.9% NaCl, n=4, due to the passing out of one mouse during the experiment). At this dose, the DCC-siRNA-EMVs caused 79% ± 4.38% (SEM, n=5) tdTomato gene knockdown at 48 hours post injection (Figure 12a). The relative expression of the tdTomato gene in the naked siRNA-injected group (n=5) did not significantly differ from that in the control group because of the previously reported13 quick clearance and rapid degradation of dsRNAs. The mean fluorescence intensity of whole tumors (Figures 12b and S12), on the other hand, suggested that tdTomato protein elimination is a much slower process: indeed, 48 hours post-administration, the reduction in MFI was 48 ± 10% SEM, whereas the variation from 24--48 hours was, compared with the control group, 14 ± 7% (Table 6). Unsurprisingly, the MFI of the B16F10-tdTomato tumors continued to increase throughout the experiment in both the naked siRNA and control groups. We also assessed whether there was any knockdown in the biodistribution mouse group injected with a 1.5 mg/kg dose (30 µg/mouse). We monitored tdTomato gene expression at 24 and 48 hours via qRT‒PCR and then measured the MFI of its protein product in thin sections. The presence of Cy-5 on the 5′ end of the passenger strand did not jeopardize the RNA interference process. The knockdown effect on tdTomato gene expression appears to be a relatively early phenomenon because most of the gene silencing occurred within the first 24 hours post-injection (Table 6), with 47% ± 10 (SEM, n = 5) total tdTomato gene silencing, reaching 69% ± 5.4% (SEM, n=5) at 48 hours. In frozen tissue sections, after 48 hours, the fluorescent signal of the target gene, tdTomato, was indeed lower or absent in the proximity of capillaries (Figures 12c and 13), indicating successful internalization of EMVs and gene knockdown. Logically, the Cy-5 and tdTomato signals hardly ever superimpose at 48 hours within 5–6 cells from the closest capillary. Overall, as shown in Table 6, the total MFI of tdTomato was 48 ± 15% greater than that of the control at 48 hours after injection (Figure S13).
 |
Table 6 TdTomato Gene Knockdown and Relative MFI Decrease at Two Different Doses of Anti-tdTomato siRNA |
Discussion
The RBC membrane has already been used as a coating material for polycationic nanoparticles in siRNA delivery37 to take advantage of endocytosis and, subsequently, endosomal escape; Wang et al failed to address the stability of their co-assemblies. Our platform is RBC-based as well, but we consciously decided to avoid the involvement of any polyplexes in the development of a siRNA carrier and to rely, for cell uptake, only on membrane properties to minimize toxicity and ensure biocompatibility. When small nucleic acids are loaded into neutral, lipid-based, and naturally occurring nanocarriers, such as extracellular vesicles and exosomes,78,82 available techniques—electroporation, sonication and lipofection—are laborious and work intensive79 or can lead to RNA degradation.56,82 Furthermore, the removal of unloaded cargo can pose another problem: naturally occurring EVs are already tightly packed with miRNAs and other small RNAs, and the systemic administration of such contents may lead to unwanted off-target effects and hinder the loading efficiency of exogenous nucleic acids.80
In contrast, the whole process of production and purification of DCC-siRNA-EMVs requires approximately four hours. We tested different methods for the removal of unloaded siRNA and cell debris: simple ultracentrifugation, with pelletization of loaded EMVs, sucrose gradient ultracentrifugation, density-changing centrifugation, and size exclusion chromatography with G25‒PD10 Sephadex packed columns, which are commonly used for liposome purification.45,83 Density-changing centrifugation was proven to be the technically least demanding, most repeatable separation method among those tested: a solution of 10% trisodium citrate could select EMVs according to their RNA content in precise and well-defined layers (Figure 1).
While successful loading of siRNAs into a single EV seems to be the limiting step for their ability as silencing carriers,84 we confirmed that loading of siRNAs in our EMVs via freeze-fracture TEM (Figure 2) and a STED laser equipped with a confocal microscope (Figure 4) effectively visualized the siRNAs within the lumen of the vesicles. Loading resulted in a final encapsulation efficiency of 67%. Freeze fracturing of EMVs has already been successfully used in,54 and despite being a widely exploited technique in liposome and nanocapsule characterization,85,86 our results are the first, to our knowledge, to show RNA aggregates in the lumen of lipid/protein objects. Notably, despite appearing as amorph aggregates, the integrity and ability of the loaded siRNA to elicit RNA interference were maintained both in vitro and in vivo. Moreover, as it is a valuable tool for the in-depth characterization of such elusive objects, an ever-growing number of studies have investigated the characterization of extracellular vesicles/exosomes/liposomes via fluorescence microscopy.87–90 We used this technique to characterize the density of packing of DCC-siRNA-EMVs, and we confirmed the efficient removal of unencapsulated siRNA and of membrane debris not shaped into EMVs. Furthermore, owing to the high STED resolution, we confirmed that this technique is reliable for size characterization. The rare superimposition of Cy-5 and CM-DiI signals hints at a high density of packing of siRNA inside the EMVs, enough for Cy-5 to self-quench, as already reported for a similar system.91
Membrane-derived vesicles have already been exploited for nucleic acid delivery,92,93 but red blood cell-derived vesicles have never been fully characterized as TNA delivery systems. The Lejeune group extensively described these objects – termed them “nanoerythrosomes” – in a series of follow-up papers.94–96 When P3881 cells were loaded with daunorubicin, the internalization pathway was remarkably similar to that we observed (Figure 6), with initial accumulation of DCC-siRNA-EMVs around the plasma membrane followed by robust internalization of the payload. The migration of the CM-DiI signal inside the cytoplasm indicates EMV–cell membrane fusion and subsequent translocation of the dye in the internal membrane system, as reported elsewhere.97 Naturally occurring EVs are highly likely to follow some endosomal escape pathways to expose their RNA payload to the cytosol without rupturing the endosomal membrane, as happens in cationic gene delivery, but through a simple fusion event,98 whose minute molecular mechanism has yet to be elucidated. RBCs express flotillin 1 and 2 on their lipid rafts,18 a protein pair known to elicit a caveolae/clathrin-independent endocytosis process.99,100 Therefore, siRNAs are either directly translocated into the cytoplasm by direct membrane fusion or released from endosomes and are ready to be complexed with Ago2. BCAM1 might also play a role in membrane fusion, as it is present both in the RBC proteome and in naturally released EVs.5,18 Similarly, even if they are of minor importance in the native RBC membrane,5 typical exosome markers such as TSG101 and ALIX have also been reported. This nonspecific uptake mechanism seems to be shared among the cell lines we evaluated (CT26, B16F10 and NHLF) with no preferred target. Finally, the in vitro ability of DCC-siRNA-EMVs to silence B16F10 expression on tdTomato mRNA was found to be approximately 80% at a final well concentration of 30 nM, a result comparable to that reported for RBC-derived EVs.17 Notably, compared with HEK293-derived EVs, DCC-siRNA-EMVs elicited RNA interference at a markedly lower dose,56 Sk-hep1-derived DPPC hybrid EVs,87 and Neuro2a-EVs loaded with lipidic siRNA.101
Our DCC-siRNA-EMVs showed promising results for future clinical translation. In vivo, in melanoma-bearing mice, at 2.5 mg/kg body weight, upon systemic administration via the caudal vein, DCC-siRNA-EMVs were capable of causing 80% tdTomato gene silencing, whereas intact EMVs containing the anti-tdTomato siRNA were still present in the blood after 48 h of administration. The early occurrence of gene knockdown is consistent with siRNA activity models,102 which favor early mRNA degradation rather than GW182 translation blockage. In similar experiments (ie single injection, systemic administration of siRNA-loaded nanoparticles towards B16F10 derived tumors and metastasis), DCC-siRNA-EMVs outperformed untargeted PEGylated cationic lipid nanoparticles in terms of knock-down103 and transferrin-based nanoparticles.104 Remarkably, similar knock-down levels are reached after multiple administrations.105,106
The accumulation of EMVs in the tumor directly confirms the enhanced permeation and retention effect (EPR effect), as established for rodents,107,108 for the passive accumulation of 100–200 nm nanoparticles in tumor tissues. B16F10 tumors are known to have a leaky vasculature and lower recruitment of CD8+ cells,109 suggesting that melanoma-related metastasis is a possible target of EMVs. Furthermore, the lack of uptake by white pulp macrophages in the spleen (Figure 11) confirms the masking activity of the CD47 marker on EMVs. EMVs also managed to elude clearance by Kupffer cells in the liver, with the siRNA concentration in tumors being twice that in the liver after 48 hours. The high apparent volume of distribution Vd of DCC-siRNA-EMVs allowed precocious delivery of siRNA to tumors, leading to 50% gene knockdown (at 1.5 mg/kg weight) as early as 24 hours after administration. As the EPR effect is known to fail in vivo in humans due to the higher blood flow rate,108,110 increased washout to achieve a longer circulation time and lower clearance by RES is of utmost importance, together with active targeting of the tissue of interest.
Interestingly, with respect to the tissue penetration of our carriers, 48 hours postinjection, DCC-siRNA-EMVs exhibited a knockdown ability within a layer of 5--6 cells from the closest blood vessel, thereby successfully crossing the endothelium but, unfortunately, extremely slow diffusion within the extracellular matrix (ECM) of B16F10 tumors. Nevertheless, we observed increased homogenization of the Cy-5 signal, which was conjugated with the siRNA passenger strand in EMVs, in tumor tissue over the course of the experiment. This finding indicated that despite being extremely hindered by collagen and hyaluronic acids and interstitial fluid pressure,111 our carriers diffuse from the endothelium within the interstitial space. Liposomes and other spherical nanoparticles larger than 60 nm have been shown not to be able to diffuse into the ECM and interstitial tissue,112 but such a phenomenon appears to be heavily dependent on the physical characteristics of the nanoparticle,113,114 such as its size. The ECM indeed behaves like a hydrogel matrix: the penetration of nanoparticles is more efficient when collagen fibers are aligned in an ordered, isotropic architecture. As elucidated by cryo-EM, EMVs are flat objects overall, with a pronounced discoidal/ellipsoidal shape. In the literature, a vast majority of papers report differential diffusion efficiency within solid tumors for hard nanoparticles, such as Au nanorods, nanodisks or nanospheres,114,115 but many fewer reports are available for soft matter, such as polymeric nanoparticles,116 or plant-derived virus-like particles,117 and to our knowledge, soft-matter, hydrophilic nanodisk solid–tumor diffusion depths have never been reported.
The fusion or attachment/hitchhiking event between EMVs and circulating erythrocytes, which has already occurred 2 hours post-administration, is highly important for the advancement and optimization of biodistribution techniques. A common method used to determine the presence of fluorescently labeled siRNA in mice is monitoring the remaining fluorescence of the labeled carrier in the mouse whole body at fixed time points via equipment such as the IVIS.118,119 This technique, even if fast, does not focus on the tissue level. Murine and human red blood cells are prone to membrane fusion with synthetic liposomes,120,121 but fusion with EVs, exosomes, or membrane-derived artificial vesicles has never been reported. Moreover, red blood cells are known to adsorb, on their outer layers, a variety of artificial nanoparticles in the absence of plasma proteins;122,123 unfortunately, investigations of putative hitchhiking or membrane fusion of EMVs with red blood cells are outside the scope of this paper.
We also characterized the best storage option of our DCC-siRNA-EMVs to evaluate the most cost/effective storage option for long-term potential treatment. We concluded that disaccharides—sucrose and trehalose—are our system’s best cryolyoprotectant, leading to minimal payload loss upon rehydration. Sucrose FD-DCC-siRNA-EMVs retained the ability to induce RNA interference (Figure 6). Upon storage at 4°C, the EMVs caused phospholipid loss over time and a significant increase in size. Such storage lesions have already been reported for whole red blood cells and are connected to oxidative stress and subsequent EVs release.124 As reported in,125 larger extracellular vesicles may arise from the fusion of smaller protein/lipid complexes released during the early stages of storage. These data, taken together with the findings of siRNA leakage upon storage, lead us to speculate that a budding mechanism, similar to that observed for whole RBCs, is maintained in EMVs. An inner membrane-bound proteasome has been previously reported59,126 and confirmed in5 and,127 and despite being the main target of hemoglobin, it can also digest Band 3 and glycophorin A proteins, thus explaining the colloidal instability of stored EMVs and the presence of storage lesions. However, whether this protease activity might play a role in transfection efficiency and tumor ECM diffusion has yet to be elucidated.
Conclusion
Red blood cells are the ideal starting material for a biomimetic and biocompatible nanodelivery system with low immunogenicity and prolonged circulation time. Additionally, the intriguing mechanical properties of their cytoskeleton lead to interesting soft-matter stiff nanoparticles. We developed an easy and repeatable procedure that allowed us to obtain a high encapsulation efficiency of a therapeutic nucleic acid, siRNA, in a relatively short amount of time. Despite relying only on passive targeting, our erythrocyte membrane vesicles, in vivo, had a remarkable circulation time and safely delivered their payload to tumors. The passive high accumulation of these objects in the lungs may lead to the development of new therapies for lung cancer. We were therefore able to develop a simple, cost-effective, therapeutic nucleic acid carrier that is also prone to easy scaling-up. Most importantly, the patient’s own blood might be used, avoiding immune reactions, for actual personalized medicine. As of 2024, many nanoparticle-siRNA formulations (such as CALAA-01, ARC-520) fail in clinical translation due to off-site accumulation and knock-down of off-target mRNAs, insufficient gene knock-down in target tissue, and hepatic or kidney toxicity. Therefore, for a successful drug approval, not only the TNA sequence must be carefully tailored and chemically modified (incorporation of locked-nucleic acids, methylation of the guide strand in position 2)128 to avoid unwanted knockdown, but also carrier tropism needs careful tuning to ensure the lowest possible effective dose without significant side effects. Future developments for these carriers will include active targeting tumor sites, to avoid healthy cells accumulation, and simultaneous loading to a reporter agent to obtain an all-in-one theranostic agent. Furthermore, we aim to quantitatively investigate the correlation between soft-matter nanoparticle shapes and their tissue penetration.
Ethical Approvals
Red blood cells were obtained from the Blood Transfusion Centre of Slovenia (Ethical approval number 0120-592/2020/7), which belong to two young anonymous males, A RhD+ blood type. The blood donors provided informed consent, in accordance with the Declaration of Helsinki. All in vivo experiments were approved by the Ministry of Agriculture, Forestry and Food of the Republic of Slovenia (permission no. U34401-35/2020/8). All the procedures involving animals were conducted in concordance with the EU directive 2010/63/EU, the NC3Rs ARRIVE guidelines and complied with Slovenian legislation.
Acknowledgments
We thank Rok Zupanc for his contribution to thin section preparation. We also thank Dr Simona Kranjc Brezar for her precious help during in vivo experiments.
Funding
The study was supported by the Slovenian Research and Innovation Agency ARIS (program numbers P1-0207, P2-0084, P3-0003, P3-0108, project numbers PR-10482, J3-14531 and J4-50150). CryoTEM was performed at the National Institute of Chemistry CryoEM Facility, supported by the Slovenian Research Agency Infrastructure Programme IO-0003.
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Della Pelle G, Kostevšek N. Nucleic acid delivery with red-blood-cell-based carriers. Int J mol Sci. 2021;22(10):5264. doi:10.3390/ijms22105264
2. Rossi L, Pierigè F, Aliano MP, Magnani M. Ongoing developments and clinical progress in drug-loaded red blood cell technologies. BioDrugs. 2020;34(3):265–272. doi:10.1007/s40259-020-00415-0
3. Fondaj D, Arduino I, Lopedota AA, Denora N, Iacobazzi RM. Exploring the microfluidic production of biomimetic hybrid nanoparticles and their pharmaceutical applications. Pharmaceutics. 2023;15(7):1953. doi:10.3390/pharmaceutics15071953
4. Fang RH, Jiang Y, Fang JC, Zhang L. Cell membrane-derived nanomaterials for biomedical applications. Biomaterials. 2017;128:69–83. doi:10.1016/j.biomaterials.2017.02.041
5. Bryk AH, Wiśniewski JR. Quantitative analysis of human red blood cell proteome. J Proteome Res. 2017;16(8):2752–2761. doi:10.1021/acs.jproteome.7b00025
6. Bosman GJCGM, Lasonder E, Groenen-Döpp YAM, Willekens FLA, Werre JM. The proteome of erythrocyte-derived microparticles from plasma: new clues for erythrocyte aging and vesiculation. J Proteomics. 2012;76(286):203–210. doi:10.1016/j.jprot.2012.05.031
7. Jain V, Yang WH, Wu J, Roback JD, Gregory SG, Chi JT. Single cell RNA-seq analysis of human red cells. Front Physiol. 2022;13:1–9. doi:10.3389/fphys.2022.828700
8. Shiga T, Maeda N, Kon K. Erythrocyte rheology. Crit Rev Oncol Hematol. 1990;10(1):9–48. doi:10.1016/1040-8428(90)90020-S
9. Dao M, Lim CT, Suresh S. Mechanics of the human red blood cell deformed by optical tweezers. J Mech Phys Solids. 2003;51(11–12):2259–2280. doi:10.1016/j.jmps.2003.09.019
10. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051–2054. doi:10.1126/science.288.5473.2051
11. Vahedi A, Bigdelou P, Farnoud AM. Quantitative analysis of red blood cell membrane phospholipids and modulation of cell-macrophage interactions using cyclodextrins. Sci Rep. 2020;10(1):1–13. doi:10.1038/s41598-020-72176-3
12. Sioud M. RNA interference: challenges and therapeutic opportunities. RNA Interf Challenges Ther Oppor. 2014;1218:1–489. doi:10.1007/978-1-4939-1538-5
13. Friedrich M, Aigner A. Therapeutic siRNA: state-of-the-art and future perspectives. BioDrugs. 2022;36(5):549–571. doi:10.1007/S40259-022-00549-3
14. Digital Science. Dimensions [Software]. 2018. Available from: https://app.dimensions.ai.
15. Harisa GI, Badran MM, Alanazi FK. Erythrocyte nanovesicles: biogenesis, biological roles and therapeutic approach: erythrocyte nanovesicles. Saudi Pharm J. 2017;25(1):8–17. doi:10.1016/j.jsps.2015.06.010
16. Song J, Song B, Yuan L, Yang G. Multiplexed strategies toward clinical translation of extracellular vesicles. Theranostics. 2022;12(15):6741–6761. doi:10.7150/thno.75899
17. Usman WM, Pham TC, Kwok YY, et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat Commun. 2018;9(1):1–15. doi:10.1038/s41467-018-04791-8
18. Prudent M, Delobel J, Hübner A, Benay C, Lion N, Tissot J-D-D. Proteomics of stored red blood cell membrane and storage-induced microvesicles reveals the association of flotillin-2 with band 3 complexes. Front Physiol. 2018;9:1–11. doi:10.3389/fphys.2018.00421
19. Yona S, Gordon S, Van Nostrand EL, et al. Co-development of diagnostic vectors to support targeted therapies and theranostics: essential tools in personalized cancer therapy. Front Oncol. 2017;11(1):1–12. doi:10.1002/marc.200500777
20. Moenner M, Vosoghi M, Ryazantsev S, Glitz DG. Ribonuclease inhibitor protein of human erythrocytes: characterization, loss of activity in response to oxidative stress, and association with Heinz bodies. Blood Cells, mol Dis. 1998;24(2):149–164. doi:10.1006/bcmd.1998.0182
21. Ohashi A, Murata A, Cho Y, et al. The expression and localization of RNase and RNase inhibitor in blood cells and vascular endothelial cells in homeostasis of the vascular system. PLoS One. 2017;12(3):e0174237. doi:10.1371/journal.pone.0174237
22. Blanco E, Shen H, Ferrari M, Blanco HSMF E. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature Biotechnology. 2015;33(9):941–951. doi:10.1038/nbt.3330
23. Partikel K, Korte R, Stein NC, et al. Effect of nanoparticle size and PEGylation on the protein corona of PLGA nanoparticles. Eur J Pharm Biopharm. 2019;141:70–80. doi:10.1016/j.ejpb.2019.05.006
24. Chen M, Leng Y, He C, et al. Red blood cells: a potential delivery system. J Nanobiotechnology. 2023;21(1):1–19. doi:10.1186/s12951-023-02060-5
25. Mao S, Neu M, Germershaus O, et al. Influence of polyethylene glycol chain length on the physicochemical and biological properties of poly(ethylene imine)-graft-poly(ethylene glycol) block copolymer/SiRNA polyplexes. Bioconjug Chem. 2006;17(5):1209–1218. doi:10.1021/bc060129j
26. Janaszewska A, Lazniewska J, Trzepiński P, Marcinkowska M, Klajnert-Maculewicz B. Cytotoxicity of dendrimers. Biomolecules. 2019;9(8):330. doi:10.3390/biom9080330
27. Abedi-Gaballu F, Dehghan G, Ghaffari M, et al. PAMAM dendrimers as efficient drug and gene delivery nanosystems for cancer therapy. Appl Mater Today. 2018;12(3):177–190. doi:10.1016/j.apmt.2018.05.002
28. Correia JS, Mirón-Barroso S, Hutchings C, et al. How does the polymer architecture and position of cationic charges affect cell viability? Polym Chem. 2022;14(3):303–317. doi:10.1039/d2py01012g
29. Della Pelle G, Bozic T, Vukomanović M, Sersa G, Markelc B, Kostevšek N. Efficient siRNA delivery to murine melanoma cells via a novel genipin-based nano-polymer. Nanoscale Adv. 2024;6(18):4704–4723. doi:10.1039/d4na00363b
30. Beddoes CM, Case CP, Briscoe WH. Understanding nanoparticle cellular entry: a physicochemical perspective. Adv Colloid Interface Sci. 2015;218:48–68. doi:10.1016/j.cis.2015.01.007
31. Franchi L, Eigenbrod T, Muñoz-Planillo R, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ Efflux. J Immunol. 2014;193(8):4214–4222. doi:10.4049/jimmunol.1400582
32. Wang W, Wang WH, Azadzoi KM, et al. Activation of innate antiviral immune response via double-stranded RNA-dependent RLR receptor-mediated necroptosis. Sci Rep. 2016;6:1–14. doi:10.1038/srep22550
33. Gao S, Dagnaes-Hansen F, Nielsen EJB, et al. The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol Ther. 2009;17(7):1225–1233. doi:10.1038/mt.2009.91
34. Pérez-Carrión MD, Posadas I, Ceña V. Nanoparticles and siRNA: a new era in therapeutics? Pharmacol Res. 2024;201:107102. doi:10.1016/j.phrs.2024.107102
35. Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22(12):1567–1572. doi:10.1038/nbt1037
36. Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108(27):10980–10985. doi:10.1073/pnas.1106634108
37. Wang Y, Ji X, Ruan M, et al. Worm-like biomimetic nanoerythrocyte carrying siRNA for melanoma gene therapy. Small. 2018;14(47):1–13. doi:10.1002/smll.201803002
38. Yang L, Wang T, Zhang D, et al. Black phosphorus nanosheets assist nanoerythrosomes for efficient mRNA vaccine delivery and immune activation. Adv Healthc Mater. 2023;12(26):1–12. doi:10.1002/adhm.202300935
39. Huang Y, Hong J, Zheng S, et al. Elimination pathways of systemically delivered siRNA. Mol Ther. 2011;19(2):381–385. doi:10.1038/mt.2010.266
40. Perepelyuk M, Thangavel C, Liu Y, et al. Biodistribution and pharmacokinetics study of siRNA-loaded anti-NTSR1-mAb-functionalized novel hybrid nanoparticles in a metastatic orthotopic murine lung cancer model. mol Ther - Nucleic Acids. 2016;5:e282. doi:10.1038/mtna.2015.56
41. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(1–2):248–254. doi:10.1006/ABIO.1976.9999
42. Della Pelle G, Delgado López A, Salord Fiol M, Kostevšek N. Cyanine dyes for photo-thermal therapy: a comparison of synthetic liposomes and natural erythrocyte-based carriers. Int J mol Sci. 2021;22(13):6914. doi:10.3390/ijms22136914
43. Steck TL. Preparation of impermeable inside-out and right-side-out vesicles from erythrocyte membranes. In: Methods in Membrane Biology. Vol 255. Springer US; 1974:245–281. doi:10.1007/978-1-4615-8960-0_4
44. Stewart JCM, JC S. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104(1):10–14. doi:10.1016/0003-2697(80)90269-9
45. Resnier P, Lepeltier E, Emina AL, et al. Model affitin and PEG modifications onto siRNA lipid nanocapsules: cell uptake and in vivo biodistribution improvements. RSC Adv. 2019;9(47):27264–27278. doi:10.1039/C9RA03668G
46. Han L, Zhan H, Sun X, Zhang Z-R-R, Deng L. A density-changing centrifugation method for efficient separation of free drugs from drug-loaded particulate delivery systems. AAPS J. 2019;21(3):1–11. doi:10.1208/s12248-019-0306-1
47. Sezgin E, Schneider F, Galiani S, et al. Measuring nanoscale diffusion dynamics in cellular membranes with super-resolution STED–FCS. Nat Protoc. 2019;44:1–51. doi:10.1038/s41596-019-0127-9
48. Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. doi:10.1016/j.cmpb.2010.01.007
49. Yen J-C, Chang F-J, Chang S. A new criterion for automatic multilevel thresholding. IEEE Trans Image Process. 1995;4(3):370–378. doi:10.1109/83.366472
50. Kabanova S, Kleinbongard P, Volkmer J, Andrée B, Kelm M, Jax TW. Gene expression analysis of human red blood cells. Int J Med Sci. 2009;6(4):156–159. doi:10.7150/ijms.6.156
51. Sun L, Yu Y, Niu B, Wang D, Lamont SJ. Red blood cells as potential repositories of microRNAs in the circulatory system. Front Genet. 2020;11:11. doi:10.3389/FGENE.2020.00442
52. Deák R, Mihály J, Szigyártó IC, Wacha A, Lelkes G, Bóta A. Physicochemical characterization of artificial nanoerythrosomes derived from erythrocyte ghost membranes. Colloids Surfaces B Biointerfaces. 2015;135:225–234. doi:10.1016/j.colsurfb.2015.07.066
53. Kuo YC, Wu HC, Hoang D, Bentley WE, D’Souza WD, Raghavan SR. Colloidal properties of nanoerythrosomes derived from bovine red blood cells. Langmuir. 2016;32(1):171–179. doi:10.1021/acs.langmuir.5b03014
54. Deák R, Mihály J, Szigyártó IC, et al. Nanoerythrosomes tailoring: lipid induced protein scaffolding in ghost membrane derived vesicles. Mater Sci Eng C. 2020;109:110428. doi:10.1016/j.msec.2019.110428
55. Singh P, Szigyártó IC, Ricci M, et al. Membrane active peptides remove surface adsorbed protein corona from extracellular vesicles of red blood cells. Front Chem. 2020:8(August):1–10. doi:10.3389/fchem.2020.00703
56. Lamichhane TN, Jeyaram A, Patel DB, et al. Oncogene knockdown via active loading of small RNAS into extracellular vesicles by sonication. Cell Mol Bioeng. 2016;9(3):315–324. doi:10.1007/s12195-016-0457-4
57. Kang J, Kaczmarek O, Liebscher J, Dähne L. Prevention of H-aggregates formation in Cy5 labeled macromolecules. Int J Polym Sci. 2010;2010:1–7. doi:10.1155/2010/264781
58. Fagan JM, Waxman L. Purification of a protease in red blood cells that degrades oxidatively damaged haemoglobin. Biochem J. 1991;277(3):779–786. doi:10.1042/bj2770779
59. Murakami T, Suzuki Y, Murachi T. An acid protease in human erythrocytes and its localization in the inner membrane. Eur J Biochem. 1979;96(2):221–227. doi:10.1111/j.1432-1033.1979.tb13032.x
60. Bosman GJCGM, Stappers M, Novotný VRMJ. Changes in band 3 structure as determinants of erythrocyte integrity during storage and survival after transfusion. Blood Transfus. 2010;8(SUPPL. 3):48–52. doi:10.2450/2010.008S
61. Tarone G, Hamasaki N, Fukuda M, Marchesi VT. Proteolytic degradation of human erythrocyte band 3 by membrane-associated protease activity. J Membr Biol. 1979;48(1):1–12. doi:10.1007/BF01869253
62. El Baradie KBY, Nouh M, O’Brien F, et al. Freeze-dried extracellular vesicles from adipose-derived stem cells prevent hypoxia-induced muscle cell injury. Front Cell Dev Biol. 2020;8:181. doi:10.3389/FCELL.2020.00181/BIBTEX
63. Trenkenschuh E, Richter M, Heinrich E, Koch M, Fuhrmann G, Friess W. Enhancing the stabilization potential of lyophilization for extracellular vesicles. Adv Healthc Mater. 2021;11:2100538. doi:10.1002/ADHM.202100538
64. Takahashi T, Hirsh A, Erbe E, Williams RJ. Mechanism of cryoprotection by extracellular polymeric solutes. Biophys J. 1988;54(3):509–518. doi:10.1016/S0006-3495(88)82983-7
65. Abdelwahed W, Degobert G, Fessi H. Investigation of nanocapsules stabilization by amorphous excipients during freeze-drying and storage. Eur J Pharm Biopharm. 2006;63(2):87–94. doi:10.1016/j.ejpb.2006.01.015
66. Buera MDP, Levi G, Karel M. Glass transition in poly(vinylpyrrolidone): effect of molecular weight and diluents. Biotechnol Prog. 1992;8(2):144–148. doi:10.1021/bp00014a008
67. Berezhkovskiy LM. Determination of mean residence time of drug in plasma and the influence of the initial drug elimination and distribution on the calculation of pharmacokinetic parameters. J Pharm Sci. 2009;98(2):748–762. doi:10.1002/jps.21440
68. Sun CY, Shen S, Xu CF, et al. Tumor acidity-sensitive polymeric vector for active targeted siRNA delivery. J Am Chem Soc. 2015;137(48):15217–15224. doi:10.1021/jacs.5b09602
69. Khaja F, Jayawardena D, Kuzmis A, Önyüksel H. Targeted sterically stabilized phospholipid siRNA nanomedicine for hepatic and renal fibrosis. Nanomaterials. 2016;6(1):8. doi:10.3390/nano6010008
70. Li SD, Chen YC, Hackett MJ, Huang L. Tumor-targeted delivery of siRNA by self-assembled nanoparticles. Mol Ther. 2008;16(1):163–169. doi:10.1038/sj.mt.6300323
71. Liang X, Ye X, Wang C, et al. Photothermal cancer immunotherapy by erythrocyte membrane-coated black phosphorus formulation. J Control Release. 2019;296:150–161. doi:10.1016/J.JCONREL.2019.01.027
72. Sosale NG, Ivanovska II, Tsai RK, et al. “Marker of Self” CD47 on lentiviral vectors decreases macrophage-mediated clearance and increases delivery to SIRPA-expressing lung carcinoma tumors. mol Ther Methods Clin Dev. 2016;3:16080. doi:10.1038/mtm.2016.80
73. Jalil AAR, Tobin MP, Discher DE. Suppressing or enhancing macrophage engulfment through the use of CD47 and related peptides. Bioconjug Chem. 2022;33(11):1989–1995. doi:10.1021/acs.bioconjchem.2c00019
74. Cipolla MJ. The Cerebral Circulation. Colloq Ser Integr Syst Physiol From mol to Funct. 2009;1(1):1–59. doi:10.4199/C00005ED1V01Y200912ISP002
75. Cataldi M, Vigliotti C, Mosca T, Cammarota MR, Capone D. Emerging role of the spleen in the pharmacokinetics of monoclonal antibodies, nanoparticles and exosomes. Int J mol Sci. 2017;18(6):1249. doi:10.3390/ijms18061249
76. Hoffman AS. The origins and evolution of “controlled” drug delivery systems. J Control Release. 2008;132(3):153–163. doi:10.1016/j.jconrel.2008.08.012
77. Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. doi:10.1016/S0002-9440(10)65006-7
78. O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev mol Cell Biol. 2020;21(10):585–606. doi:10.1038/s41580-020-0251-y
79. Roerig J, Mitrach F, Schmid M, et al. Synergistic siRNA loading of extracellular vesicles enables functional delivery into cells. Small Method. 2022;6(12). doi:10.1002/smtd.202201001
80. Faruqu FN, Xu L, Al-Jamal KT. Preparation of exosomes for siRNA delivery to cancer cells. JoVE. 2018;2018(142):e58814. doi:10.3791/58814
81. Reinhart WH, Chien S. Red cell rheology in stomatocyte-echinocyte transformation: roles of cell geometry and cell shape. Blood. 1986;67(4):1110–1118. doi:10.1182/blood.v67.4.1110.1110
82. Kooijmans SAA, Stremersch S, Braeckmans K, et al. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release. 2013;172(1):229–238. doi:10.1016/j.jconrel.2013.08.014
83. Li C, Wang J, Wang Y, et al. Recent progress in drug delivery. Acta Pharm Sin B. 2019;9(6):1145–1162. doi:10.1016/j.apsb.2019.08.003
84. Murphy DE, de Jong OG, Evers MJW, Nurazizah M, Schiffelers RM, Vader P. Natural or synthetic RNA delivery: a stoichiometric comparison of extracellular vesicles and synthetic nanoparticles. Nano Lett. 2021;21(4):1888–1895. doi:10.1021/acs.nanolett.1c00094
85. Kuvichkin VV. Investigation of ternary complexes: DNA-phosphatidylcholine liposomes-mg2+ by freeze-fracture method and their role in the formation of some cell structures. J Membr Biol. 2009;231(1):29–34. doi:10.1007/S00232-009-9201-5/FIGURES/5
86. Toub N, Maksimenko A, Maccario J, Malvy C, Fattal E, Couvreur P. Efficacy of siRNA nanocapsules targeted against the EWS-Fli1 oncogene in Ewing sarcoma efficacy of siRNA nanocapsules targeted against the EWS – fli1 oncogene in Ewing sarcoma. Pharm Res. 2006;23:892–900. doi:10.1007/s11095-006-9901-9
87. Zhou X, Miao Y, Wang Y, et al. Tumour‐derived extracellular vesicle membrane hybrid lipid nanovesicles enhance siRNA delivery by tumour‐homing and intracellular freeway transportation. J Extracell Vesicles. 2022;11(3). doi:10.1002/jev2.12198
88. Szatanek R, Baj-Krzyworzeka M, Zimoch J, Lekka M, Siedlar M, Baran J. The methods of choice for extracellular vesicles (EVs) characterization. Int J mol Sci. 2017;18(6):1153. doi:10.3390/ijms18061153
89. Chen C, Zong S, Wang Z, et al. Imaging and intracellular tracking of cancer-derived exosomes using single-molecule localization-based super-resolution microscope. ACS Appl Mater Interfaces. 2016;8(39):25825–25833. doi:10.1021/acsami.6b09442
90. Ter-Ovanesyan D, Kowal EJK, Regev A, Church GM, Cocucci E. Imaging of isolated extracellular vesicles using fluorescence microscopy. In: Buchwald’s Atlas of Metabolic & Bariatric Surgical Techniques and Procedures. Vol 1660. Elsevier; 2017:233–241. doi:10.1007/978-1-4939-7253-1_19
91. Blau R, Epshtein Y, Pisarevsky E, et al. Image-guided surgery using near-infrared Turn-ON fluorescent nanoprobes for precise detection of tumor margins. Theranostics. 2018;8(13):3437–3460. doi:10.7150/thno.23853
92. Phua KKL, Boczkowski D, Dannull J, Pruitt S, Leong KW, Nair SK. Whole blood cells loaded with messenger RNA as an anti-tumor vaccine. Adv Healthc Mater. 2014;3(6):837–842. doi:10.1002/adhm.201300512
93. Xu C, Ju D, Zhang X. Cell membrane-derived vesicle: a novel vehicle for cancer immunotherapy. Front Immunol. 2022;13:3573. doi:10.3389/FIMMU.2022.923598/BIBTEX
94. Lejeune A, Moorjani M, Gicquaud C, Lacroix J, Poyet P, Gaudreault CR. Nanoerythrosome, a new derivative of erythrocyte ghost: preparation and antineoplastic potential as drug carrier for daunorubicin. Anticancer Res. 1994;14(3 A):915–919.
95. Lejeune A, Poyet P, C.-Gaudreault R, Gicquaud C. Nanoerythrosomes, a new derivative of erythrocyte ghost: III. Is phagocytosis involved in the mechanism of action? Anticancer Res. 1997;17(5 A):3599–3603.
96. Moorjani M, Lejeune A, Gicquaud C, Lacroix J, Poyet P, Gaudreault RC. Nanoerythrosomes, a new derivative of erythrocyte ghost II: identification of the mechanism of action. Anticancer Res. 1996;16(5A):2831–2836.
97. Lassailly F, Griessinger E, Bonnet D. “Microenvironmental contaminations” induced by fluorescent lipophilic dyes used for noninvasive in vitro and in vivo cell tracking. Blood. 2010;115(26):5347–5354. doi:10.1182/BLOOD-2009-05-224030
98. Joshi BS, de Beer MA, Giepmans BNG, Zuhorn IS. Endocytosis of extracellular vesicles and release of their cargo from endosomes. ACS Nano. 2020;14(4):4444–4455. doi:10.1021/acsnano.9b10033
99. Ciana A, Achilli C, Gaur A, Minetti G. Membrane remodelling and vesicle formation during ageing of human red blood cells. Cell Physiol Biochem. 2017;42(3):1127–1138. doi:10.1159/000478768
100. Frick M, Bright NA, Riento K, Bray A, Merrified C, Nichols BJ. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr Biol. 2007;17(13):1151–1156. doi:10.1016/j.cub.2007.05.078
101. O’Loughlin AJ, Mäger I, de Jong OG, et al. Functional delivery of lipid-conjugated siRNA by extracellular vesicles. Mol Ther. 2017;25(7):1580–1587. doi:10.1016/j.ymthe.2017.03.021
102. Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci U S A. 2003;100(17):9779–9784. doi:10.1073/pnas.1630797100
103. Li S-D, Chono S, Huang L. Efficient gene silencing in metastatic tumor by siRNA formulated in surface-modified nanoparticles. J Control Release. 2008;126(1):77–84. doi:10.1016/j.jconrel.2007.11.002
104. Yhee JY, Lee SJ, Lee S, et al. Tumor-targeting transferrin nanoparticles for systemic polymerized siRNA delivery in tumor-bearing mice. Bioconjug Chem. 2013;24(11):1850–1860. doi:10.1021/bc400226b
105. Reddy TL, Garikapati KR, Reddy SG, et al. Simultaneous delivery of Paclitaxel and Bcl-2 siRNA via pH-Sensitive liposomal nanocarrier for the synergistic treatment of melanoma. Sci Rep. 2016;6(1):35223. doi:10.1038/srep35223
106. Ehexige E, Bao M, Bazarjav P, et al. Silencing of STAT3 via peptidomimetic LNP-mediated systemic delivery of RNAi downregulates PD-L1 and inhibits melanoma growth. Biomolecules. 2020;10(2):1–13. doi:10.3390/biom10020285
107. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6. doi:10.1016/j.addr.2015.01.002
108. Danhier F. To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244:108–121. doi:10.1016/j.jconrel.2016.11.015
109. Zang G, Gustafsson K, Jamalpour M, Hong JW, Genové G, Welsh M. Vascular dysfunction and increased metastasis of B16F10 melanomas in Shb deficient mice as compared with their wild type counterparts. BMC Cancer. 2015;15(1):1–11. doi:10.1186/s12885-015-1269-y
110. Leporatti S. Thinking about Enhanced Permeability and Retention Effect (EPR). J Pers Med. 2022;12(8):12–14. doi:10.3390/jpm12081259
111. Hofmann M, Guschel M, Bernd A, et al. Lowering of tumor interstitial fluid pressure reduces tumor cell proliferation in a xenograft tumor model. Neoplasia. 2006;8(2):89–95. doi:10.1593/neo.05469
112. Alexandrakis G, Brown EB, Tong RT, et al. Two-photon fluorescence correlation microscopy reveals the two-phase nature of transport in tumors. Nat Med. 2004;10(2):203–207. doi:10.1038/nm981
113. Munir MU. Nanomedicine penetration to tumor: challenges, and advanced strategies to tackle this issue. Cancers. 2022;14(12):2904. doi:10.3390/cancers14122904
114. Black KCL, Wang Y, Luehmann HP, et al. Radioactive 198 au-doped nanostructures with different shapes for in vivo analyses of their biodistribution, tumor uptake, and intratumoral distribution. ACS Nano. 2014;8(5):4385–4394. doi:10.1021/nn406258m
115. Zhang YR, Lin R, Li HJ, ling HW, Du JZ, Wang J. Strategies to improve tumor penetration of nanomedicines through nanoparticle design. Wiley Interdiscip Rev Nanomed Nanobiotech. 2019;11(1):1–12. doi:10.1002/wnan.1519
116. Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. doi:10.1038/nnano.2011.166
117. Shukla S, Eber FJ, Nagarajan AS, et al. The impact of aspect ratio on the biodistribution and tumor homing of rigid soft‐matter nanorods. Adv Healthc Mater. 2015;4(6):874–882. doi:10.1002/adhm.201400641
118. Wen Y, Bai H, Zhu J, Song X, Tang G, Li J. A supramolecular platform for controlling and optimizing molecular architectures of siRNA targeted delivery vehicles. Sci Adv. 2020;6(31). doi:10.1126/sciadv.abc2148
119. Jiang T, Qiao Y, Ruan W, et al. Cation-free siRNA micelles as effective drug delivery platform and potent RNAi nanomedicines for glioblastoma therapy. Adv Mater. 2021;33(45):1–10. doi:10.1002/adma.202104779
120. Constantinescu I, Levin E, Gyongyossy‐Issa M. Liposomes and blood cells: a flow cytometric study. Artif Cells, Blood Substitutes, Biotechnol. 2003;31(4):395–424. doi:10.1081/BIO-120025410
121. Zhu K, Xu Y, Zhong R, et al. Hybrid liposome–erythrocyte drug delivery system for tumor therapy with enhanced targeting and blood circulation. Regen Biomater. 2023:10. doi:10.1093/rb/rbad045
122. Pan DC, Myerson JW, Brenner JS, et al. nanoparticle properties modulate their attachment and effect on carrier red blood cells. Sci Rep. 2018;8(1):1–12. doi:10.1038/s41598-018-19897-8
123. Lenders V, Escudero R, Koutsoumpou X, et al. Modularity of RBC hitchhiking with polymeric nanoparticles: testing the limits of non-covalent adsorption. J Nanobiotechnology. 2022;20(1):1–12. doi:10.1186/s12951-022-01544-0
124. Chaudhary R, Katharia R. Oxidative injury as contributory factor for red cells storage lesion during twenty eight days of storage. Blood Transfus. 2012;10(1):59–62. doi:10.2450/2011.0107-10
125. Bebesi T, Kitka D, Gaál A, et al. Storage conditions determine the characteristics of red blood cell derived extracellular vesicles. Sci Rep. 2022;12(1):1–13. doi:10.1038/s41598-022-04915-7
126. Ballas S, Burka E. Protease activity in the human erythrocyte: localization to the cell membrane. Blood. 1979;53(5):875–882. doi:10.1182/blood.V53.5.875.875
127. Sae-Lee W, McCafferty CL, Verbeke EJ, et al. The protein organization of a red blood cell. Cell Rep. 2022;40(3):111103. doi:10.1016/J.CELREP.2022.111103
128. Ranjbar S, Zhong X, Manautou J, Lu X. A holistic analysis of the intrinsic and delivery-mediated toxicity of siRNA therapeutics. Adv Drug Deliv Rev. 2023;201:1–38. doi:10.1016/j.addr.2023.115052
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.