")
Back to Journals » International Journal of Nanomedicine » Volume 19
Research Progress on Cyclic-Peptide Functionalized Nanoparticles for Tumor-Penetrating Delivery
Authors Wang C, Shen Z, Chen Y, Wang Y, Zhou X, Chen X , Li Y, Zhang P, Zhang Q
Received 12 September 2024
Accepted for publication 14 November 2024
Published 26 November 2024 Volume 2024:19 Pages 12633—12652
DOI https://doi.org/10.2147/IJN.S487303
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Chenkai Wang,1,2 Zefan Shen,1,2 Yiyang Chen,1,2 Yifan Wang,2 Xuanyi Zhou,2 Xinyi Chen,1 Yuhang Li,2 Pu Zhang,2 Qi Zhang2
1The Second Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 2Urology & Nephrology Center, Department of Urology, Zhejiang Provincial People’s Hospital, Affiliated People’s Hospital, Hangzhou Medical College, Hangzhou, Zhejiang, 310014, People’s Republic of China
Correspondence: Qi Zhang; Pu Zhang, Email [email protected]; [email protected]
Abstract: A key challenge in cancer treatment is the effective delivery of drugs into deep regions of tumor tissues, which are impermeable due to abnormal vascular network, increased interstitial fluid pressure (IFP), abundant extra cellular matrix (ECM), and heterogeneity of tumor cells. Cyclic peptides have been used for the surface engineering of nanoparticles to enhance the tumor-penetrating efficacy of drugs. Compared with other surface ligands, cyclic peptides are more easily produced by automated chemical synthesis, and they are featured by their higher binding affinity with their targets, tumor selectivity, stability against degradation, and low toxicity. In this review, different types of cyclic peptides, their physicochemical properties and their in vivo pharmacokinetics are introduced. Next, the progress of cyclic peptide-functionalized drug delivery nanodevices is updated, and the mechanism underlying the tumor-penetrating properties of cyclic peptide-functionalized drug delivery nanodevices is discussed.
Keywords: cyclic peptide, tumor penetration, drug delivery, nanomaterial functionalization
Introduction
Cancerous tissues are characterized by several distinctive features that impede the delivery of therapeutic agents: an abnormal vascular network, elevated interstitial fluid pressure (IFP), a dense extracellular matrix (ECM), and tumor cell heterogeneity. The ECM is composed of collagen, elastin fibers, proteoglycans, and glycosaminoglycans, which form a complex, cross-linked gel-like structure. This structure significantly hinders the diffusion of drugs within the tumor interstitium. Additionally, IFP increases from the tumor periphery to the center, creating a barrier that prevents nanocarriers and drug molecules from penetrating deep into the tumor after extravasation from peripheral blood microvessels. Moreover, the tumor’s vascular network is highly heterogeneous, resulting in poor perfusion and necrosis. This heterogeneity increases the diffusion distance that nanomedicines must cover to reach target cells. Consequently, most clinically approved drugs, including small molecules, antibodies, hormones, peptides, oncolytic viruses, and nanoparticles, face challenges in penetrating solid tumors.1–3 To overcome these barriers, there is an urgent need to develop vehicles capable of deep tumor penetration to effectively deliver drugs to targeted lesions.
In recent years, many researchers have proposed the potential of cyclic peptides as surface engineering ligands to functionalize drugs or drug delivery systems. The unique structures of cyclic peptides are organized as amino acids connection in a cyclic manner, and they have many advantages in drug delivery into tumors. Cyclic peptides have been engineered to precisely target tumor cells by binding to receptors that are excessively present on the surface of these cells.4,5 These cyclic structures offer greater stability compared to their linear counterparts, as they are less likely to be broken down by proteases due to their rigid, closed-ring shape which restricts conformational changes. Additional stability can be achieved through modifications such as N-methylation of the peptide backbone or by forming covalent bonds.6 These alterations can enhance cyclic peptides’ bioavailability, specificity, and ability to penetrate cell membranes, making them potentially more effective in certain therapeutic applications. Most importantly, the cyclic peptide showed enhanced tumor-penetrating ability. The underlying mechanism is the enhanced transmembrane transport and the enhanced transcellular transport of the cyclic peptide. Thanks to the excellent bioactivity of the cyclic peptide, cyclic-peptide-functionalized drugs or drug delivery systems can more effectively translocate across cancerous tissue barriers, and changing their physical and chemical properties, such as size, charge and hydrophobicity, can regulate their tumor-penetrating efficiency. However, as far as we can know, a comprehensive understanding of the tumor-penetration of the cyclic peptides and their conjugates have not been given in any reviews. In this review, different types of cyclic peptides, their physicochemical properties and their in vivo pharmacokinetics are introduced. Next, the progress of cyclic peptide-functionalized drug delivery nanodevices is updated, and the mechanism underlying the tumor-penetrating properties of cyclic peptide-functionalized drug delivery nanodevices is discussed.
Types and Advantages of Cyclic Peptides
Types of Cyclic Peptides
Cyclic peptides are categorized into two primary groups based on the chemical composition of their backbone.7 Homodetic cyclic peptides are characterized by a backbone solely composed of amide linkages between their constituent amino acid residues. In contrast, heterodetic cyclic peptides feature additional types of linkages with disulfide bonds being the most prevalent non-amide connections. These disulfide bonds are notably present in hormones of the posterior pituitary, such as oxytocin and vasopressin. Furthermore, the presence of ester linkages within the main chain of a peptide classifies it as a depsipeptide. It is noteworthy that cyclic depsipeptides, in particular, represent a significant class of biologically active compounds that are often derived from microbial sources.
The Advantage of Cyclic Peptides
Cyclic peptides offer several advantages over their linear counterparts (Table 1), including enhanced binding affinity to targets, improved membrane permeability, greater structural stability, and reduced cytotoxicity.8 1) A key advantage is their heightened resistance to proteolysis.9 Cyclic peptides tend to form secondary structures such as β-turns, which augment their rigidity. This rigidity confers increased resistance to degradation by endogenous proteases, thus enhancing their in vivo stability.9 Additionally, the covalently closed structure of cyclic peptides shields them from the external environment and eliminates terminal ends, making them particularly resistant to exopeptidase activity.9–11 Empirical evidence supports these advantages; for instance, Bogdanowich-Knipp et al demonstrated that cyclic RGD exhibited superior stability across a pH range of 3 to 7, with a remarkable 30-fold increase in stability at pH 7 compared to its linear counterpart.12 2) Cyclic peptides exhibit an enhanced affinity for their target receptors compared to their linear counterparts. This increased binding specificity is attributed to the reduced conformational flexibility of cyclic peptides, which minimizes entropic contributions to binding.8 For instance, while the linear RGD motif can interact with various integrin subtypes, cyclic RGD peptides demonstrate a more potent and selective binding profile for specific integrin receptors.11,13 3) Cyclic peptides are considered safer for in vivo applications due to their high target specificity, which limits cellular binding to the cells expressing the intended receptors. This precision reduces off-target effects compared to the broader interactions of linear peptides.10 Moreover, 4) cyclic peptides demonstrate enhanced cell-penetrating capabilities. The conjugation of cyclic peptides to anticancer drugs significantly boosts their cell permeability. For instance, Lättig-Tünnemann et al observed that cyclic TAT peptides penetrate cells more rapidly and induce more robust cellular endocytosis than their linear counterparts.14 Sugahara et al showed that drug conjugates with iRGD exhibit higher tumor uptake than the unconjugated drugs, underscoring the superior targeting ability of cyclic peptides.4 Cyclic peptides also exhibit favorable pharmacokinetic properties, including lower surface charge, reduced polarity, decreased hydrodynamic volume, and a smaller topological polar surface area, which, coupled with increased molecular rigidity and stability, augment peptide-cell interactions. Furthermore, 5) cyclic peptides can activate transcellular transport mechanisms, enabling deep tumor penetration. For example, cyclic peptides containing the CendR motif can bind to tumor-specific primary receptors, undergo proteolytic cleavage to expose the CendR motif, and subsequently bind to secondary receptors. This triggers an endocytosis transport pathway known as the C-end Rule (CendR) pathway, facilitating the deep penetration of cyclic peptides into tumor tissues. Sugahara et al found that both the iRGD phage and free iRGD peptide diffuse more extensively into extravascular tumor tissue compared to linear RGD peptides, which are more likely to accumulate around tumor vasculature.4 Additionally, comparative studies on the penetration of iRGD-PPCD and RGD-PPCD couplers in an in vitro avascular C6 glioma spheroid model have revealed that iRGD-mediated couplers penetrate deeper into tumors. The iRGD-PPCD coupler demonstrated a penetration depth of 144 μm, surpassing the 115 μm achieved by RGD-PPCD, indicating a more potent tumor penetration capability for the iRGD-mediated delivery system.11
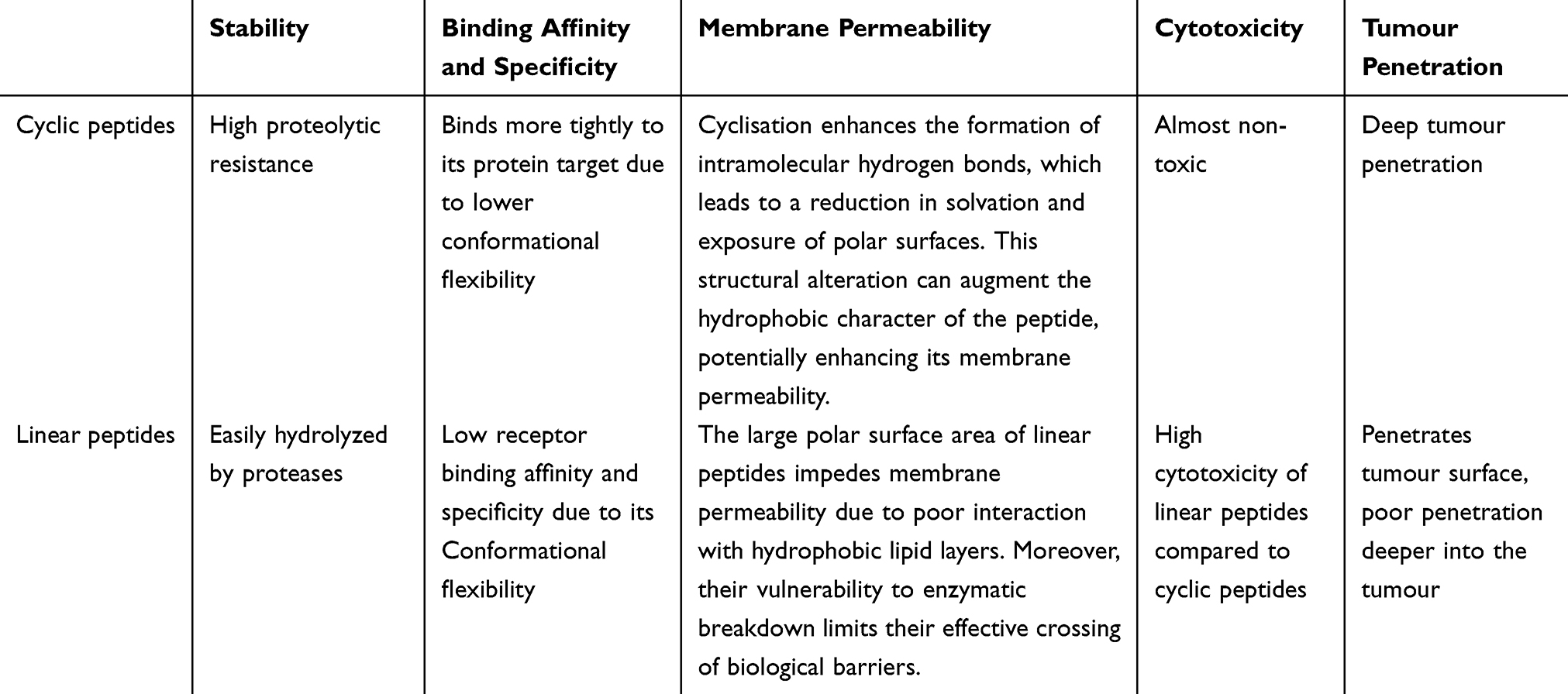 |
Table 1 The Advantage of Cyclic Peptides |
In the realm of targeted cancer therapeutics, drug couplers have emerged as a pivotal area of research. These couplers are sophisticated constructs that attach cytotoxic drugs to tumor-specific carriers via connectors, facilitating the direct delivery of drug payloads to cancer cells. The category of drug couplers encompasses a range of targeted drug conjugates, including antibody-drug conjugates (ADCs), peptide-drug conjugates (PDCs), small molecule-drug conjugates (SMDCs), nucleic acid aptamer-drug conjugates (ApDCs), and viral drug conjugates (VDCs), each demonstrating significant therapeutic potential in clinical settings.15 The evolution of drug coupler development is propelled by several key factors, including enhanced stability to ensure the drug remains intact in the circulation, improved tumor penetration to ensure effective drug reach within the tumor mass, the refinement of targeting vectors for precise cancer cell recognition, the identification of novel targets that are specific to cancer cells, advances in coupling technologies that allow for stable and controlled drug release, the creation of innovative couplers that offer new mechanisms for drug delivery and the diversification of cytotoxic payloads to expand treatment options. However, each type of drug coupler presents unique challenges: For example, ADCs, for instance, often face issues with low permeability, which can limit their effectiveness in reaching all cancer cells within a tumor.16,17 PDCs, are particularly susceptible to degradation by proteases, a challenge that requires strategies to enhance their stability.18,19 SMDCs are limited by the availability of suitable target ligands, which can restrict their targeting specificity. ApDCs, while offering high affinity for targets, are vulnerable to degradation by nucleases, necessitating the development of more resistant nucleic acid structures.
Despite these challenges, the field is actively pursuing solutions to enhance the stability and efficacy of drug couplers. For PDCs, in particular, cyclic peptides and their functionalized systems hold promise for enhancing drug targeting and minimizing non-specific binding. However, several challenges must be addressed regarding their synthesis, functionalization, and in vivo performance. The synthesis of cyclic peptides often requires complex chemical procedures. Traditional methods, such as amide formation, can be challenging due to the need for a predefined precyclised conformation before the intramolecular reaction can take place, which is entropically unfavorable.20 This is especially true for head-to-tail cyclization, where the C-terminus and N-terminus of the peptide must cyclize, a process that typically extends the peptide precursor due to the all-trans confirmation preference of the amide bond.20 Moreover, cyclizations are often conducted in dilute solutions to favor intramolecular reactions over intermolecular oligomerization.21,22
Furthermore, the surface functionalization of nanodrug carriers can impact their interactions with tumor cells, potentially affecting endocytosis and targeted transport. For instance, RGD peptide-modified nanocarriers may face issues with non-specific binding and immune recognition when engaging with tumor cells.22 The metabolic stability of cyclic peptides is crucial, particularly when considering their use as drug candidates. Studies have shown that certain cyclic peptides have limited stability upon contact with hepatocytes, highlighting the need to improve their metabolic stability, such as by replacing thioether bonds.23
In summary, while cyclic peptides offer substantial potential for drug delivery and targeted therapies, their synthesis challenges, stability issues, and the practical aspects of their application in drug delivery are areas that require further optimization. Future research should focus on developing innovative synthetic strategies, enhancing the stability and cellular penetration of cyclic peptides, and optimizing their functionalization methods to boost their efficacy and safety in clinical settings.
Cellular and Tumor Penetration of Cyclic Peptides
Cell Penetration of Cyclic Peptides
Cyclic peptides employ various mechanisms to enter cells, primarily through three pathways: passive diffusion, receptor-mediated endocytosis, and direct translocation.
Passive Diffusion
Passive diffusion is the spontaneous process by which substances move from regions of high extracellular concentration to areas of lower intracellular concentration, without the expenditure of energy (Figure 1A). To predict passive membrane permeability, Lipinski et al24 proposed the “Rule of Five” (Ro5), an empirical guideline. According to Ro5, a molecule with a molecular weight (MW) below 500, no more than 5 hydrogen bond donors (HBDs), no more than 10 hydrogen bond acceptors (HBAs), and a CLogP value less than 5 is likely to be permeable across cell membranes. Romidepsin, a bicyclic peptide consisting of five residues, adheres to these criteria and is known to traverse cell membranes via passive diffusion. Moreover, molecular conformation plays a critical role in passive membrane permeability. N-methylation in the backbone of certain natural cyclic peptides is linked to enhanced membrane permeability. Ovadia et al25 have shown that both the quantity and the spatial arrangement of N-methyl groups within a cyclic peptide significantly affect its ability to penetrate cells. For instance, cyclosporin A, a cyclic peptide with a conformation that accommodates Ro5, has polar groups that are sequestered due to the presence of N-methyl groups, which may contribute to a favorable desolventization energy during the transition from an aqueous phase to the lipid membrane phase, thereby facilitating its passive diffusion.26 Furthermore, cyclosporin A’s conformation is adaptive to its environment. It adopts an “open” conformation in aqueous media, exposing polar groups, but upon encountering a low dielectric medium such as a lipid bilayer, it transitions to a “closed” conformation. This transition involves the elongation of internal hydrogen bonds and the concealment of hydrogen bond donor and acceptor groups, reducing the peptide’s effective polar surface area. These conformational changes enable cyclosporin A to efficiently diffuse across the plasma membrane.
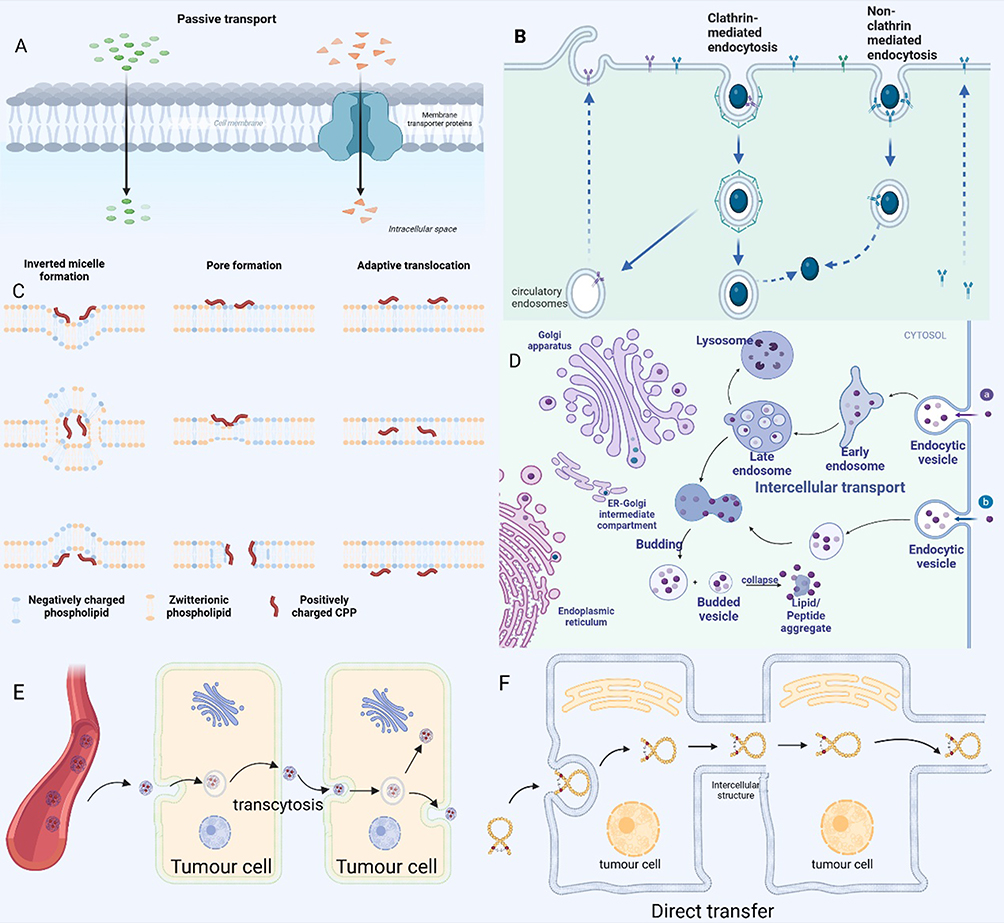 |
Figure 1 Cell penetration of cyclic peptides. (A) Cyclic peptides can enter cells by passive diffusion without consuming energy. (B) Endocytosis can occur through various pathways, including caveolin-mediated, clathrin-mediated, micropinocytosis. (C) The proposed internalization mechanisms of direct translocation for positively charged CPPs. (D) Biological cargo endocytosed through various endocytosis pathways initially enters early endosomes. Early endosomes subsequently mature into late endosomes, which eventually fuse with lysosomes. Within the lysosome, the luminal contents are degraded by hydrolases. However, some of these contents may be recycled back to the cell surface via recycling endosomes. Pei et al recently proposed a novel mechanism involving vesicle growth and collapse to elucidate the mechanism of endocytosis of cyclic cell-penetrating peptides (CPPs). (E)Transcytosis is a complex biological process involving sequential endocytosis and exocytosis across different cell layers. (F) In addition to passive diffusion and endocytosis, a third, well-documented mechanism for the cellular internalization of cyclic peptides is direct translocation across the plasma membrane. This process results in the immediate availability of cyclic cell-penetrating peptides (CPPs) or their conjugates within the cytosol. |
Receptor-Mediated Endocytosis
Macromolecular cyclic peptides, which include cell-penetrating peptides (CPPs) and their nanoparticle conjugates, are known to induce endocytosis—a process critical for cellular uptake27 (Figure 1B). Endocytosis can occur through various pathways, including caveolin-mediated, clathrin-mediated, micropinocytosis, and microtubule-mediated mechanisms. Specifically, Oh et al28 identified that the bicyclic peptide F’-[KW4E]-(β-Ala)-[KR5] is predominantly internalized via clathrin- and caveolin-dependent endocytosis. This finding underscores the role of these pathways in the cellular uptake of certain cyclic peptides. Similarly, Camarero et al29 demonstrated that the cyclic peptide MCoTI-I utilizes multiple endocytic routes for cellular entry, encompassing fluid-phase endocytosis, lipid-dependent endocytosis, and clathrin-mediated endocytosis. This suggests a complex interplay of pathways in the uptake process. Cyclic CPPs, in particular, interact directly with plasma membrane phospholipids and potentially other membrane components, facilitating their internalization through diverse endocytic pathways. These include clathrin-mediated endocytosis, exosome pathway, and cytosolic drinking (also known as macropinocytosis).
Direct Translocation
In addition to passive diffusion and endocytosis, a third, well-documented mechanism for the cellular internalization of cyclic peptides is direct translocation across the plasma membrane (Figure 1C). This process results in the immediate availability of cyclic cell-penetrating peptides (CPPs) or their conjugates within the cytosol. Once inside the cell, cyclic peptides can be selectively distributed to specific organelles, such as the endoplasmic reticulum, Golgi apparatus, or lysosomes, or they may diffuse freely within the cytoplasm to reach their targets. Notably, direct translocation is an energy-independent process that can occur even at temperatures as low as 4°C. Direct translocation offers a more rapid mode of entry compared to endocytosis, often completing the transport within minutes after cell exposure to cyclic CPPs.27 The efficiency of direct translocation can be concentration-dependent for certain types of cyclic CPPs. For instance, the attachment of cyclic Tat to green fluorescent protein (GFP) facilitated the direct translocation of the CPP-protein conjugate into HeLa cells, albeit at relatively high concentrations of 150 μM.27 Similarly, cyclic (Trp-Arg)4 has been shown to enter cells via direct translocation at low temperatures (4°C) or in the presence of metabolic inhibitors like sodium azide.30 Pei et al observed that as little as 25 μM of FITC-labeled cyclic CPP12 could diffuse throughout cells within minutes,27 highlighting the potential of cyclic peptides for rapid cellular uptake.
Intracellular Trafficking
Biological cargo internalized by various endocytic pathways is initially routed to the early endosome. The early endosome then matures into the late endosome, which ultimately fuses with the lysosome. Within the lysosome, the intraluminal contents are degraded by hydrolytic enzymes. However, a portion of these contents may be recycled back to the cell surface via the recycling endosome (Figure 1D). For most non-viral delivery systems, the endosomal escape process is highly inefficient, leading to the entrapment of the vector and/or cargo within the endosome or lysosome.31 Cyclic peptides also follow the endosomal-lysosomal transport pathway for intracellular transport. Yet, a subset of cyclic peptides can evade the endosome and proceed to their target sites or return to the cell surface. Several hypotheses have been proposed to explain the endosomal escape mechanisms of cyclic peptides, including the proton sponge effect,32 pore formation,33 and membrane destabilization.34 Pei et al have recently introduced a novel mechanism involving vesicle outgrowth and collapse to elucidate the endosomal escape of cyclic cell-penetrating peptides (CPPs)27,35(Figure 1D). In this process, cyclic CPPs bind to endosomal membranes, leading to the formation of a CPP-enriched lipid domain that buds from the endosome. These vesicles collapse, releasing their contents into the cytoplasm, a process that is accelerated by the acidic pH within the endosome. Multiple budding and collapse events may occur in a single endosome until the vesicles are depleted of CPPs. Furthermore, cyclic peptides can circumvent lysosomal degradation by engaging the transcytotic pathway. This involves sorting from endosomes and secretion at the opposing membrane, thereby enhancing their tumor-penetration capabilities. Qian et al demonstrated that iRGD binds to cell surface receptors, initiating receptor-mediated transcytosis, which avoids lysosomal trapping and facilitates drug entry into tumor tissue.36
Intercellular Transport
Intercellular transport mechanisms, which are crucial for tissue penetration, primarily consist of paracellular diffusion and transcellular transport. Paracellular diffusion involves the movement of substances through the intercellular spaces, prevented by the tight junctions that connect cells. In the context of tumor biology, the unique structure and function of tumor blood vessels, characterized by increased vascular leakage, provide an avenue for cyclic peptides to penetrate the tumor tissue via the paracellular pathway. For example, cyclic ADT peptides (ADTC1, ADTC5, and ADTC6) have been shown to disrupt calcineurin function at the adhesive junctions of vascular endothelial cells, thereby enhancing their tissue-penetrating properties through paracellular diffusion.37 Transcellular drug delivery, on the other hand, refers to the process by which drugs are transported across cells, rather than through the extracellular matrix. This mode of transport can occur via two primary mechanisms: transcytosis and direct intercellular transfer. Transcytosis is a cellular transport mechanism where substances are carried across the cell, typically from one side to the other, and is particularly relevant for the delivery of drugs to tissues behind biological barriers. Direct intercellular transfer, meanwhile, involves the movement of substances directly from the donor cell to the recipient cell, bypassing the extracellular space.
Transcytosis is a complex biological process involving sequential endocytosis and exocytosis across different cell layers (Figure 1E). Cyclic peptides initiate this process by binding to specific targeting ligands on the cell membrane surface, which then triggers intracellular internalization through interactions with cell surface receptors or transporter proteins. Following endocytosis, the cyclic peptide is encapsulated within an endocytic vesicle. This vesicle traverses the cell, potentially interacting with organelles such as the endoplasmic reticulum or Golgi apparatus, before moving to the cell’s opposite membrane where it fuses and releases the peptide into the extracellular environment. Jiang et al explored the penetration of cyclic RGD peptide-functionalized pol(ethylene glycol) polycarbonate trimethylene nanoparticles (c(RGDyK)-NP) into solid tumors. They conducted in vitro studies and utilized a subcutaneous xenograft mouse model to investigate integrin-mediated transcellular interactions. Their findings indicated that cyclic RGD peptides first target integrin receptors on tumor cells, then bind to neuropilin-1 (NRP-1) receptors for rapid internalization, and subsequently undergo transcytosis to enhance deep penetration into the tumor parenchyma. This process resulted in improved drug penetration and accumulation in tumor tissue compared to conventional nanoparticles and paclitaxel treatment.38 Additionally, Liu et al demonstrated that iRGD (a cyclized RGD peptide) could circumvent the vasculature system barrier in pancreatic ductal adenocarcinoma (PDAC) through activated transcytosis, achieving 3–4 fold enhanced tumor uptake. The iRGD peptide binds to integrins, revealing the CendR sequence, and subsequently binds to NRP-1, initiating a bulk transcytosis pathway that involves vesicular translocation.39,40 This intercellular transport pathway, facilitated by the CendR motif, may also occur via intra-secretory vesicles such as exosomes or microvesicles.
Direct transfer is a distinct class of transcellular drug delivery mechanisms. Unlike traditional transcellular transport, which involves crossing the extracellular matrix, direct transfer is confined to intracellular regions or intercellular connections following endocytosis, and can include direct cell-to-cell contact (Figure 1F). The formation of tubular conduits between tumor cells and endothelial cells, as observed by these researchers, further enhances the efficient intercellular transport of nanomedicines.41,42 Direct transfer can be further categorized based on the type of intercellular connection involved: cell membrane-mediated, gap junction-mediated, and tunneling nanotube (TNT)-mediated. TNTs are long-range intercellular cytoplasmic channels that facilitate direct cell-to-cell communication independent of soluble factors. These dynamic nanoscale membrane structures enable the rapid exchange of cellular components, including organelles, vesicles, molecules, ions, and pathogens, between connected, non-adjacent cells.43–45 Cyclic peptides, once internalized, are transported along the cytoskeleton, often utilizing microtubules as tracks. They can reach TNTs, which are conduits for the direct transport of cellular contents, including cyclic peptides, between neighboring cells.46 This mode of transport is particularly significant for enhancing tumor penetration, as it allows cyclic peptides to bypass the extracellular matrix and reach deeper tumor regions.47
How to Evaluate the Tissue-Penetrating Efficacy?
Transwell Diffusion Assay
Transwell assays are a widely utilized method for evaluating the transcellular permeability of solutes across endothelial monolayers (Figure 2A). These in vitro models offer several advantages, including the simplicity of execution and the elimination of the need for live animal testing. Bhardwaj et al conducted a study to assess the transcellular penetration of a designed macrocyclic peptide using the Transwell Caco-2 assay system. This system employed colorectal epithelial cells to form a barrier between the donor and acceptor compartments, simulating the efflux transport process in the reverse direction. In this assay, an apparent permeability coefficient (Papp) value greater than 1 × 10^-6 cm/s is considered indicative of adequate transcellular penetration for a drug candidate. The study yielded promising results, with high membrane penetration observed for the cyclic peptides in the Caco-2 assays. Specifically, six out of the eight macrocyclic peptides containing eight residues tested demonstrated a Papp value exceeding the threshold of 1 × 10^-6 cm/s, highlighting their potential as drug candidates.48
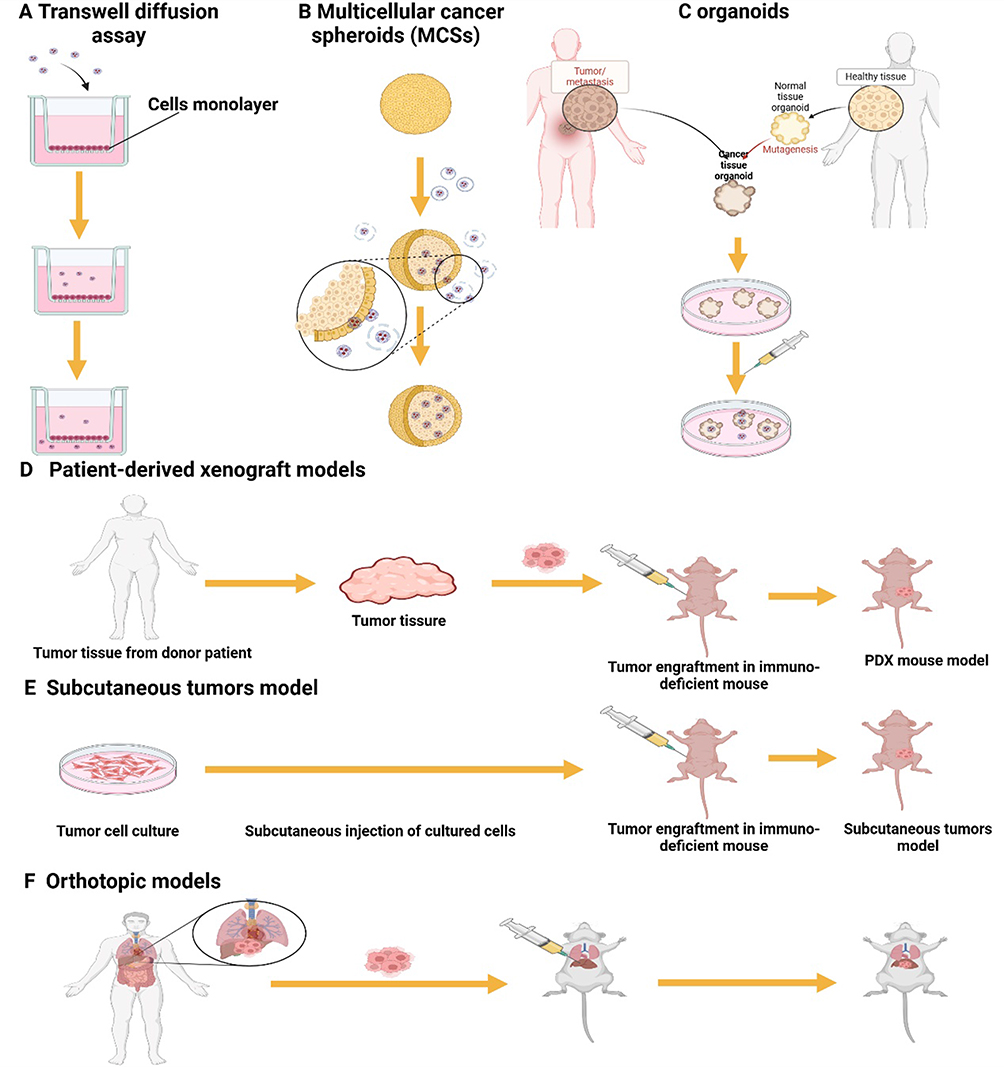 |
Figure 2 Permeation modelling of cyclic peptides. (A) Transwell assays are a widely utilized method for evaluating the transcellular permeability of solutes across endothelial monolayers.(B) Multicellular cancer spheroids (MCSs) are a prevalent in vitro model for assessing the tissue-penetrating efficiency of drugs and nanoparticles. (C) Organoids are three-dimensional (3D) microcell clusters cultured in vitro that closely emulate the structure and function of their corresponding organs in vivo. (D) The Patient-Derived Xenograft (PDX) model, which involves transplanting tumor tissue directly from patients, maintains the genotypic and phenotypic diversity of the original tumor. This model can more realistically reflect the characteristics of the patient’s tumor. (E) Subcutaneous tumours have become a common in vivo model for preclinical oncology research due to their practical advantages, such as short establishment period, high tumourigenicity, ease of handling and low cost. (F) Orthotopic models maintain the natural anatomical context of the tumor, which is important for studying tumor-stroma interactions, metastasis, and responses to treatments. |
Multicellular Spheroids
Multicellular cancer spheroids (MCSs) are a prevalent in vitro model for assessing the tissue-penetrating efficiency of drugs and nanoparticles. MCSs offer a more accurate representation of the cellular microenvironment, cellular interactions, and physiological and biochemical reactions than traditional 2D cell culture models49 (Figure 2B). Jiang et al developed an in vitro MCS simulation to emulate solid tumors. Utilizing a lipid-based system, they generated 3D tumor spheroids with structural and compositional similarities to authentic tumors. These spheroids featured densely packed cells and an organized extracellular matrix enriched with proteins such as fibronectin, laminin, and collagen, closely replicating the natural tumor microenvironment.38 Upon treatment with nanoparticles functionalized with cyclic peptides, confocal microscopy revealed fluorescence throughout the entire tumor sphere, extending to a depth of 90 µm. This observation suggests that cyclic peptides significantly enhance the penetration of nanoparticles into the solid tumor tissue.
Organoids
Organoids are three-dimensional (3D) microcell clusters cultured in vitro that closely emulate the structure and function of their corresponding organs in vivo50 (Figure 2C). These micro-organs not only exhibit self-renewal and self-organization capabilities but also replicate the spatial organization and rudimentary functions of authentic organs. Organoids comprise a diverse array of cell types that engage in complex and tight interactions, extending beyond simple physical contacts to include dynamic relationships with the surrounding extracellular matrix. However, the in vitro cultivation of organoids can be resource-intensive and time-consuming. Additionally, there is often an absence of connective tissues, vasculature, and immune cells within the organoid microenvironment. In a study assessing the tissue penetration of iRGD TPN, a cyclic peptide-based nanocomplex, researchers employed 3D organoids derived from pancreatic ductal adenocarcinoma (PDAC) tumors in mice and humans. The distribution and depth of iRGD TPN penetration within the organoids were tracked, simulating the tumor’s exposure to the nanocomplex in a 3D multicellular setting. The findings demonstrated that iRGD TPNs were capable of effectively penetrating hundreds of micrometers through cell layers in both human and mouse 3D organoids.51
In vivo Tumor Model
Subcutaneous tumors are a prevalent in vivo model for preclinical cancer research due to their practical advantages. They are straightforward to establish and monitor, and their rapid and reliable growth makes them an ideal model for evaluating the penetration capacity of cyclic peptides in tumors52 (Figure 2E). For instance, Jiang’s team assessed the permeability of solid tumors in a subcutaneous xenograft mouse model. They found that c(RGDyK)-binding nanoparticles (c(RGDyK)-NP) demonstrated enhanced penetration and accumulation in solid tumor tissues compared to conventional nanoparticles.38 The Patient-Derived Xenograft (PDX) model, which involves transplanting tumor tissue directly from patients, maintains the genotypic and phenotypic diversity of the original tumor. This model can more realistically reflect the characteristics of the patient’s tumor (Figure 2D). Cyclic peptides, as drug carriers, can be assessed in PDX models for their distribution and penetration within tumor tissues. Although specific examples of cyclic peptide penetration in PDX models are not provided, it is reasonable to speculate that their stable cyclic structure may confer superior tumor penetration.52 The promising efficacy of cyclic peptides in treating subcutaneous and PDX models has encouraged further evaluation of their penetration efficiency in in situ tumor models. Orthotopic models maintain the natural anatomical context of the tumor, which is important for studying tumor-stroma interactions, metastasis, and responses to treatments (Figure 2F). Sugahara’s team4 explored the delivery of anticancer drugs using iRGD-mediated Abraxane®, a 130 nm nanoparticle comprising paclitaxel embedded in albumin, in mice with in situ 22Rv1 tumors. The study showed that the accumulation of Abraxane in the tumor was 8 times higher with iRGD-targeted Abraxane compared to non-targeted Abraxane, highlighting the enhancing effect of iRGD on tissue penetration.4 Furthermore, Justin et al confirmed the tumor penetration ability of iRGD in in situ pancreatic cancer.51 Additional studies have demonstrated that iRGD coupling significantly improves nanoparticle penetration at the tumor site in animal models of prostate cancer and breast cancer.53
Modulating the Tumor-Penetrating Property of Cyclic Peptides
Surface Charge
The surface charge of nanomedicines plays a pivotal role in regulating adsorption-mediated transcytosis, a process crucial for cellular uptake and drug delivery. Cationization, or the introduction of positive charges, can effectively induce transcytosis and enhance the penetration of nanocarriers across multiple cell layers.54,55 For instance, Lättig-Tünnemann et al observed a higher cellular uptake rate, accelerated uptake time, increased accumulation, and improved transduction kinetics for arginine-rich cyclic TAT peptides.14,56 In a follow-up study, Shirazi et al investigated the cell permeability of cationic cyclic cell-penetrating peptides (CPPs) where arginine was substituted with lysine and histidine.56–58 While cationic nanomedicines can interact with the negatively charged cell membrane through direct transmembrane pathways, their highly positive charge may also lead to cytotoxicity by disrupting cellular and mitochondrial membranes across various cell types.59 Furthermore, an overly positive surface charge might interfere with specific endocytosis pathways, such as pinocytosis or receptor-mediated endocytosis, potentially destabilizing cell membranes or obstructing essential membrane bending processes. Positively charged nanomedicines are also at risk of rapid clearance by the reticuloendothelial system, whereas neutral or negatively charged nanomedicines, although having a prolonged circulation time, may face challenges in penetrating deep tumor tissues and being uptaken by cells.60 Cell membranes typically exhibit a slight negative charge, and neutral or negatively charged drug carriers might not interact efficiently with cell membranes via electrostatic interactions, possibly reducing their endocytosis efficiency and subsequent cellular uptake.61 Interestingly, some studies have shown that positively charged peptides or drug carriers can enhance tumor tissue penetration by facilitating Donnan distribution through weak, reversible electrostatic interactions with the tumor extracellular matrix (ECM). In contrast, neutral or negatively charged drug carriers may lack this mechanism, resulting in diminished tumor penetration.61 To achieve a balance between long-term circulation and optimal tumor penetration, nanomedicines should have an adaptable surface charge—neutral or negative in circulating blood and positive at the tumor site. The integration of cyclic peptides into nanoparticle surface functionalization allows for precise, guided delivery of nanoparticles. The net charge of the cyclic peptide ligand, which may alter in the acidic tumor microenvironment, is critical for the surface charge of cyclic peptide-mediated nanopolymers or couplers and directly impacts tumor penetration.61,62
Size
The molecular weight (Mw) of cyclic peptides, which serve as homing peptides to enhance nanoparticle targeting, is a critical factor influencing their tumor penetration ability. Low Mw cyclic peptides (<10 kDa) offer a fundamental advantage over their high Mw counterparts due to their smaller size, which correlates with enhanced penetration.63 Bicyclic peptides, for instance, with molecular weights ranging from 1 to 3 kDa, can effectively diffuse across biological barriers in tissues, a feat that large proteins struggle with. This property holds promise for achieving effective tumor penetration and drug delivery through various local modalities. However, the low Mw of these peptides also results in a shorter plasma half-life and rapid clearance by the kidney. To address this, the blood half-life of peptides can be extended by conjugating them with high Mw proteins or peptides.63 Pollaro’s team demonstrated this approach by injecting bicyclic peptides combined with albumin-binding peptides into human MDA-MB-231 tumor-bearing nude mice. The concentration of bicyclic peptides conjugated to albumin-binding peptides was found to be 54 times higher than that of free bicyclic peptides.64 The ring size of cyclic peptides also impacts their tumor penetration ability. As the ring size increases, cyclic peptides become more flexible and susceptible to proteolytic enzymes. In contrast, bicyclic peptides, with their reduced ring size, exhibit enhanced stability against protein hydrolysis and improved target binding affinity and selectivity due to increased conformational rigidity. Indeed, bicyclic peptide-drug conjugates (BDCs) offer a promising approach for the targeted delivery of chemotherapeutic agents and other therapeutics to specific cell populations. The compact nature of BDCs facilitates their deeper penetration into solid tumors, capitalizing on their small size to traverse the complex tumor microenvironment. Additionally, BDCs demonstrate high renal clearance, which contributes to a reduction in systemic drug toxicity, thereby improving the therapeutic index.65 Moreover, the particle size of cyclic peptide conjugates significantly affects their penetration in tumors. Smaller particle sizes facilitate diffusion within tumor tissues by enabling easier navigation through the complex extracellular matrix (ECM) network. Studies have shown that nanoparticles with sizes greater than 62 nm, yet not exceeding 110 nm, can rapidly diffuse in in vitro ECM models, such as Matrigel, and breast xenograft sections.66 Particle size also influences the cellular uptake efficiency of peptide-drug conjugates (PDCs). Smaller particles are generally more effectively internalized by cells via endocytosis, as they can be more readily encapsulated by cell membranes. However, excessively small particles risk being excreted by renal filtration, necessitating a balanced design approach.67
In conclusion, for optimal tumor penetration, the design of cyclic peptides should aim to minimize both ring size and nanoparticle size, ensuring that the cyclic peptide and its conjugates achieve the strongest tumor penetration ability.
Rigidity
Cyclization of peptides promotes the formation of secondary structures such as β-turns, which increases the rigidity of cyclic peptides.13 This enhanced rigidity boosts lipophilicity and enables the formation of intermolecular hydrogen bonds, allowing for tighter binding to target proteins with minimal entropy loss, and ultimately enhancing cell penetration. Lättig-Tünnemann24 demonstrated that arginine-rich cell-penetrating peptides with greater structural rigidity had increased cellular transduction rates, and rigid cyclic peptides showed superior cellular uptake kinetics compared to their linear counterparts. The increased rigidity, achieved through cyclization that positions the guanidinium moiety in a distal configuration, enhances membrane contacts and improves cell penetration. Zhang68 introduced rigid fragments into cyclic peptides, which induced apoptosis in tumor cells, underscoring the potential of rigidity to augment biological activity and protein-protein interactions. Additionally, enhanced conformational rigidity may improve metabolic stability by slowing down protease cleavage kinetics.69 Bicyclic peptides, in particular, offer higher conformational rigidity, target affinity, selectivity, metabolic stability, and membrane permeability compared to monocyclic counterparts.70 These attributes expand their application potential and enhance tissue permeability, making bicyclic peptides valuable tools in drug delivery and therapeutic development.
Multivalency or Surface Density
Multivalency, or the presentation of multiple binding sites on a surface, is a key factor influencing the efficacy and function of cyclic peptides. Cell surface proteins, such as integrins and selectins, engage in multivalent interactions with small structural motifs like RGD, which are found on neighboring cells or extracellular matrices and play a role in cell migration regulation. By incorporating multiple targeting ligands, cyclic peptides can significantly enhance their affinity and binding strength to cell surface receptors or extracellular matrix components.71,72 This increased binding avidity improves the penetration and internalization efficiency of cyclic peptides within tissues. For example, Wang et al developed a cyclic TMTP1 peptide dimer connected by two PEG4 junctions, which resulted in enhanced tumor uptake and more sensitive tumor imaging compared to free ICG or TMTP1-PEG4-ICG.73 In another study, Gao74 investigated the multivalent effect on blood-brain barrier penetration by labeling nanoparticles with varying amounts of angiopep-2 peptides. The research demonstrated that multivalency significantly increased the intracerebral delivery of nanoparticles, thereby enhancing their ability to cross the blood-brain barrier. These findings underscore the potential of cyclic peptides to improve tumor penetration through multivalent interactions.
Anti-Cancer Application of Cyclic Peptides
Nanoscale drug delivery systems offer distinct advantages for advanced therapeutic strategies, including the co-administration of multiple drugs, extended circulation times, controlled release mechanisms, and the potential for multifaceted interactions within the biological milieu. However, the success of these systems is not solely dependent on the choice of drug carriers; the surface modification and functionalization of nanoparticles are equally crucial for effective drug delivery.
To enhance the accumulation of nanoparticles in tumor tissues through active targeting, the surfaces of these nanoparticles are often decorated with specific targeting molecules. These include antibodies, aptamers, and various ligands that can recognize and bind to cancer cell surface markers with high affinity. By incorporating such targeting elements, nanoparticles can actively recognize and bind to their homologous receptors present in the circulatory system. This targeted interaction facilitates the highly efficient internalization of nanoparticles into tumor cells via receptor-mediated endocytosis, thereby improving the therapeutic index of the encapsulated drugs.75–77
The use of cyclic peptides in this context is particularly noteworthy. Their inherent stability, precise targeting capabilities, and the ability to be functionalized with various chemical groups make them ideal candidates for surface modification of nanoparticles. Cyclic peptides can be engineered to selectively bind to specific receptors overexpressed on tumor cells, enhancing the active targeting and internalization of drug-loaded nanoparticles. This precision targeting not only improves drug delivery efficiency but also minimizes the side effects associated with nonspecific drug distribution.
Furthermore, cyclic peptides can be designed to respond to specific stimuli within the tumor microenvironment, such as changes in pH or the presence of certain enzymes, allowing for triggered drug release at the site of action. This adds an additional layer of control to the drug delivery system, ensuring that drug release is localized to the tumor and occurs under the appropriate conditions. The integration of cyclic peptides into the design of nanoscale drug delivery systems offers a powerful approach to enhance tumor targeting, improve drug release profiles, and ultimately, provide more effective cancer treatments. It has been shown indicates that surface functionalization of carriers and drugs with cyclic peptides can significantly enhance active targeting and cellular penetration. For instance, Wang et al demonstrated that solid lipid nanoparticles loaded with paclitaxel and naringenin, with surfaces modified with cyclic peptides, showed improved tumor targeting in glioblastoma multiforme78 (Table 2). Similarly, Larmandie’s team conjugated cyclic peptides to the biocompatible polymer poly(2-hydroxypropylmethacrylamide) (pHPMA), creating an efficient drug delivery system for organometallic anticancer complexes79,80 (Table 2).
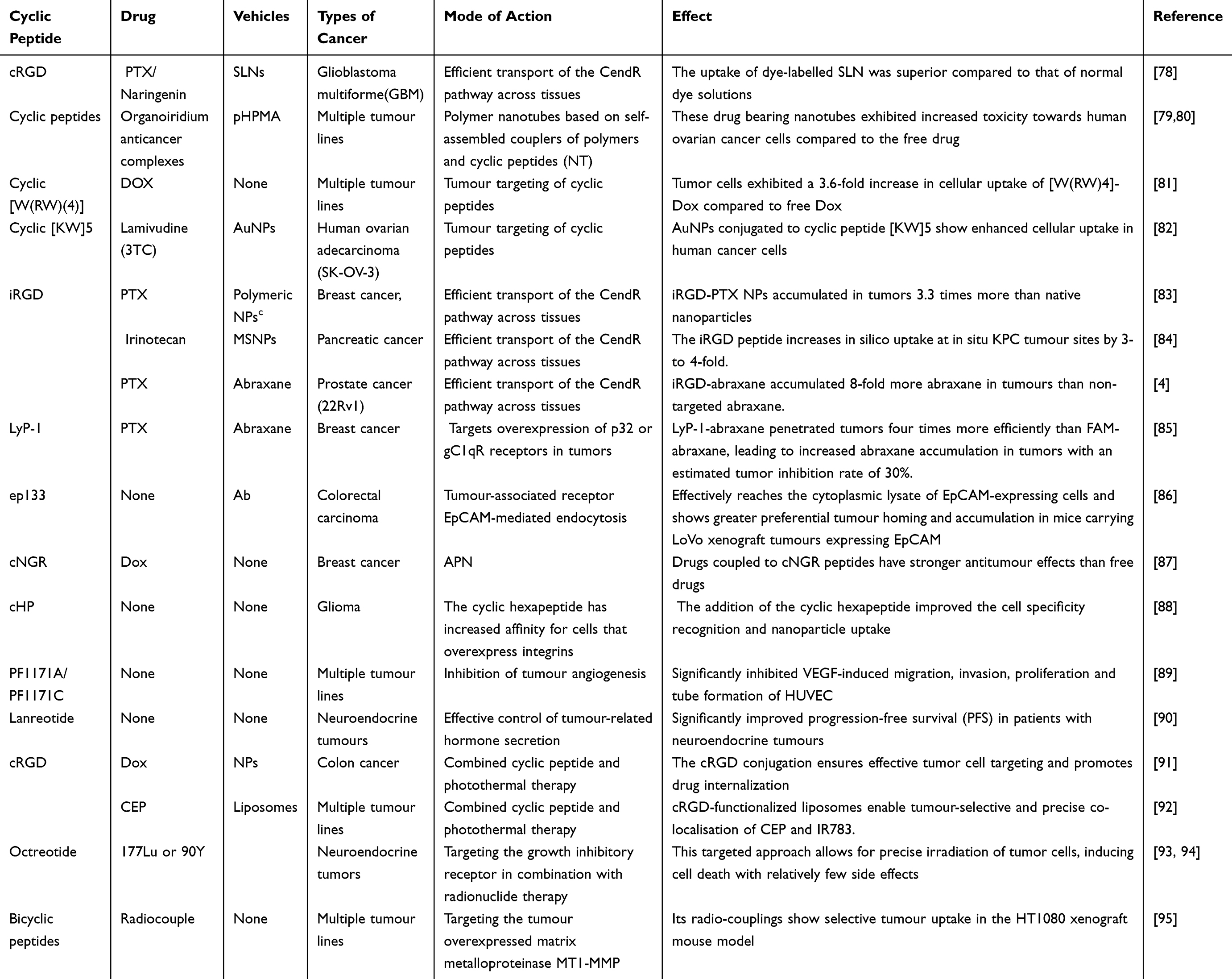 |
Table 2 Anti-Cancer Application of Cyclic Peptides |
Doxorubicin (Dox) is a hydrophilic anticancer agent that faces rapid efflux in certain cancer cells, leading to a short retention time. To address this challenge and enhance the drug’s sustained anticancer activity, Dox was conjugated to the cyclic peptide [W(RW)4] and its linear counterpart (RW)4 using appropriate linkers. The cyclic peptide conjugate [W(RW)4]-Dox demonstrated the ability to inhibit cell proliferation in various cancer cell lines, including ovarian adenocarcinoma (SK-OV-3), colorectal cancer (HCT-116), and breast cancer (MDA-MB-468). Notably, SK-OV-3 cells exhibited a 3.6-fold increase in cellular uptake of [W(RW)4]-Dox compared to free Dox. These results indicate that the cyclic peptide conjugate [W(RW)4]-Dox holds promise as a prodrug candidate for improving the cellular delivery and retention of Dox 81 (Table 2).
Shirazi et al developed an environmentally friendly and efficient method to synthesize gold nanoparticles (AuNPs) functionalized with both linear (KW)5 and cyclic [KW]5 peptides 82 (Table 2). These peptide-decorated AuNPs were assessed for their potential as nanodrug delivery systems. The study revealed that AuNPs conjugated with the cyclic peptide [KW]5 demonstrated superior cellular uptake of fluorescently labeled drugs in human cancer cells when compared to those conjugated with the linear (KW)5 peptide. Additionally, the research mentioned the potential use of cyclic peptide-functionalized Gadolinium nanoparticles to enhance intracellular drug delivery96.
“Irgd” is a cyclic RGD peptide with the sequence CRGDKGPDC (Cys-Arg-Gly-Asp-Lys-Gly-Pro-Asp-Cys). It demonstrates a tropism for various solid tumors, including breast, pancreatic, and prostate cancers, by selectively targeting tumor-associated biomarkers such as integrin αvβ3 and αvβ5 on the cell membrane surface4,97,98. The binding of iRGD to these integrins induces conformational changes, activating downstream signaling pathways and exposing the CendR sequence (C-end Rule motif). This motif then interacts with the NRP-1 receptor on tumor cells, initiating NRP-1-mediated endocytosis and facilitating iRGD internalization. This process promotes trans-cellular transport, thereby enhancing tumor penetration (Figure 3). Numerous studies have confirmed that iRGD can significantly improve the tumor targeting and penetration capabilities of therapeutic agents or nanocarriers (Table 2). For example, Hu’s team synthesized iRGD-PTX conjugates by coupling paclitaxel (PTX) to MCA through esterification, followed by attachment to iRGD-SH via a Michael addition reaction. These conjugates self-assembled into nanoparticles that, when administered systemically in a 4T1 breast cancer mouse model, achieved 3.3-fold higher tumor accumulation compared to native nanoparticles83 (Table 2). This strategy effectively enhanced the anticancer activity of the delivered drugs. Cyclic hexapeptide c(RGDf(N-me)VK)-C (cHP) exhibits an increased affinity for cells that overexpress integrins, a characteristic that renders it a promising agent for tumor targeting. To evaluate the in vitro penetration capability of cHP into tumor tissues, Zhang et al constructed a tumor cell spheroid model. Their findings indicated that cHP/Cou-NPs (cyclic hexapeptide-modified curcumin nanoparticles), penetrated deeper into C6 cell spheroids over time, in contrast to the unmodified nanoparticles that displayed limited penetration. Further in vitro cellular assays revealed that cHP/Cur-NPs (cyclic hexapeptide-modified curcumin nanoparticles) achieved a higher cellular uptake efficiency compared to their unmodified counterparts. Specifically, in C6 cells, which highly express integrin αvβ3, cHP/Cur-NPs demonstrated a cellular uptake efficiency of 99.5% within 90 minutes, significantly surpassing the 19.4% efficiency of unmodified Cur-NPs (Table 2). These results underscore the enhanced targeting ability of cHP-modified nanoparticles towards tumor cells. The broad-spectrum anticancer activities of cyclic peptides have been corroborated in various cancer types, such as colon cancer, pancreatic cancer, and lung cancer, underscoring their potential utility in oncology5,88,99. The asparagine-glycine-arginine (NGR) motif, similar to the RGD sequence, specifically targets tumors by engaging with the aminopeptidase N (APN) receptor, which is prevalent in the tumor vasculature (Table 2). The cyclic NGR peptide CNGRC demonstrates superior tumor targeting efficiency over linear NGR-containing peptides. For instance, Giorgio et al reported that the antitumor activity of the cyclic peptide CNGRC-TNF was over 10-fold higher than that of the linear peptide GNGRG-TNF, indicating the enhanced effectiveness of the cyclic structure in tumor targeting87,100.
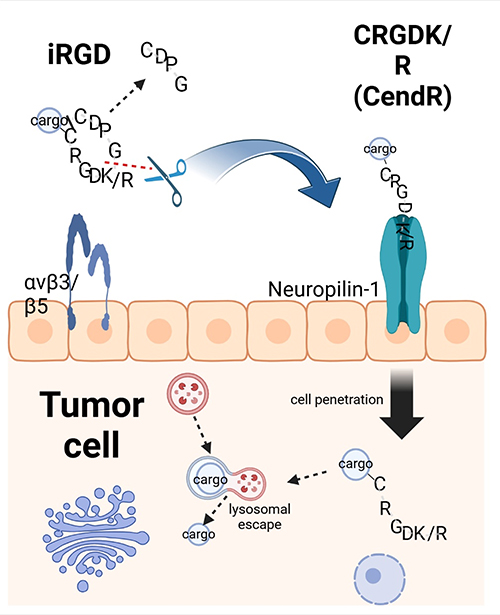 |
Figure 3 Cell penetration of iRGD. The iRGD peptide selectively accumulates on the surface of endothelial cells and other cell types within tumors that express αv integrins. It utilizes its RGD motif to mediate binding to these integrins. Subsequently, the peptide is cleaved by cell surface-associated proteases, which exposes the CendR element, characterized by the motif RXXK/R, at the C-terminus (indicated by a red dotted line). This CendR element then facilitates binding to neuropilin-1 (NRP-1), enabling the peptide to penetrate cells and tissues. The iRGD peptide can deliver cargo, such as simple chemicals or nanoparticles, into tumor cells and tissues, provided that the cargo is conjugated to the N-terminus of the iRGD peptide. It is noteworthy that the disulfide bond within the peptide is cleaved, allowing for internalization of the peptide and its cargo (represented by a black line). |
In vitro tumor penetration assays revealed that phages containing CendR motifs, such as iNGR and iRGD, could penetrate deeper into tumor tissues, following a similar pattern. In contrast, non-CendR phages, like CNGRC and controls, remained primarily in the outer tumor layers. This finding suggests that iNGR and iRGD utilize a similar CendR-mediated transport mechanism for tumor penetration. Notably, iNGR can induce tumor-specific penetration when co-administered with drugs, such as adriamycin (doxorubicin), leading to increased local drug accumulation in tumor tissues. The combination of doxorubicin with iNGR has exhibited significantly enhanced efficacy in tumor therapy compared to doxorubicin monotherapy101.
LyP-1 is another cyclic peptide, consisting of 9 amino acids with the sequence Cys-Gly-Gln-Lys-Arg-Thr-Arg-Gly-Cys, which exhibits antitumor effects by targeting p32 or gC1qR receptors on tumor lymphatics or cells102 (Table 2). Karmali et al demonstrated the superior in vivo homing efficacy of LyP-1-abraxane in a MDA-MB-435 xenograft tumor model using mice. The conjugation of the LyP-1 peptide to abraxane particles via a cysteine sulfhydryl linkage resulted in FAM-LyP-1-abraxane exhibiting four times the fluorescence intensity in tumors compared to FAM-abraxane, signifying a substantial enhancement in targeting efficiency. Furthermore, LyP-1-abraxane displayed a markedly higher accumulation in extravascular tumor tissues. This accumulation was particularly evident in clusters of tumor cells expressing the p32 receptor, which are often found in lymphatic-rich, less vascularized areas that are typically challenging for chemotherapeutic agents to penetrate. These findings suggest that LyP-1-abraxane has the potential to access deeper, less vascularized regions of tumor tissue, where conventional drugs may not effectively reach.85 This resulted in increased accumulation of abraxane in tumors, with an estimated tumor inhibition rate of 30%.
Ep133, a 12-amino acid cyclic peptide, is internalized specifically by tumor cells via endocytosis that is mediated by the tumor-associated receptor EpCAM (Table 2). Building upon this mechanism, Park et al employed yeast surface-display technology to develop a high-affinity cyclic peptide-fusion antibody named ep6Ras37. This construct exhibits a significantly enhanced binding affinity for EpCAM, approximately 10-fold greater than the native peptide. In mouse tumor models, ep6Ras37 demonstrated a pronounced preference for tumor cells, achieving peak accumulation within 48 hours post-injection. Confocal immunofluorescence microscopy revealed that ep6Ras37 localizes more efficiently in the cytoplasm than the parental antibody, indicating improved endocytosis and lysosomal escape capabilities. Furthermore, the intensified fluorescence of ep6Ras37 in tumor tissues suggests a higher accumulation level, indicative of its potential for enhanced tumor targeting and therapeutic efficacy.86
Cyclic peptides have emerged as potent inhibitors of tumor angiogenesis by targeting key signaling molecules such as ERK, FAK, and Akt, which are essential for the process. This inhibition impedes the formation of new blood vessels necessary for tumor growth. Specifically, the cyclic peptides PF1171A and PF1171C, derived from microbial soil fungi, have been shown to significantly suppress VEGF-induced activities in human umbilical vein endothelial cells (HUVECs), including cell migration, invasion, proliferation, and tube formation—critical steps in the angiogenic cascade89 (Table 2).
Lanreotide, a cyclic octapeptide with a well-established role in oncology, is utilized to manage clinical syndromes associated with functional neuroendocrine tumors (NETs) and to mitigate tumor growth in patients with advanced low- or intermediate-grade NETs (Table 2). Clinical studies have demonstrated that treatment with lanreotide significantly enhances progression-free survival (PFS) in individuals with neuroendocrine tumors. Specifically, patients administered lanreotide experienced a notable prolongation in PFS compared to those receiving a placebo, underscoring the therapeutic potential of this cyclic peptide in extending disease control and improving patient outcomes90.
In the realm of oncology, combination therapies have emerged as a superior approach to monotherapies, offering enhanced therapeutic benefits. Photothermal therapy (PTT) is a promising modality within this context, particularly for its potential in the clinical treatment of tumors. The synergy between cyclic peptides and photothermal agents, such as indocyanine green (ICG), has led to the development of tumor-targeting nanoparticles. These nanoparticles, when exposed to near-infrared light, generate heat that induces thermal damage, selectively destroying tumor cells while minimizing harm to surrounding healthy tissues. The precision of circulating peptides in targeting tumor tissues increases the concentration of photothermal agents, thereby improving treatment efficacy73. Fan et al have contributed to this field by developing cRGD-conjugated nanoparticles (Table 2). These adriamycin (DOX)-loaded Fe3O4@polydopamine (PDA) nanoparticles are designed for both MRI imaging and antitumor chemo-photothermal therapy. Their research findings indicate that PTT not only enhances cell membrane permeability and drug uptake but also expedites the release of nanoparticle-encapsulated drugs. The cRGD conjugation ensures effective tumor cell targeting and promotes drug internalization, demonstrating an excellent ability to enhance therapeutic outcomes91. Building on these advancements, Wu’s team in 2024 introduced a novel therapeutic approach using cRGD-encapsulated liposomes. These liposomes encapsulate a synergistic combination of ceftazidime (CEP), a chemotherapeutic agent, and IR783, a phototherapeutic agent, for combined chemotherapy and PTT. The cRGD-functionalized liposomes serve as a multifunctional platform, achieving tumor selectivity and precise co-localization of CEP and IR783 (Table 2). This strategic integration of chemotherapy and phototherapy has significantly improved the efficacy of in vivo antitumor therapy, showcasing the potential of cyclic peptides in enhancing multimodal treatment strategies92.
Cyclic peptides possess the unique ability to self-assemble into peptide-dye nanostructures, which have shown promise in the field of in vivo tumor imaging and photodynamic therapy (PDT). The self-assembly process relies on non-covalent interactions, such as electrostatic forces and hydrophobic stacking, which endow these nanostructures with the versatility needed for precise tumor targeting, effective therapy, and high-resolution imaging. A prime example of this approach involves the non-covalent assembly of iRGD peptides with methylene blue dye. The resulting nanomaterials harness the tumor-homing properties of iRGD to selectively bind to cancer cells. Once bound, these nanostructures can be utilized for photoacoustic imaging, which provides deep tissue penetration and high spatial resolution. Following imaging, the same nanostructures can initiate photodynamic therapy, where the methylene blue dye, upon activation by light, generates reactive oxygen species that induce cell death in the targeted cancer cells. This multifunctional approach not only enhances the specificity of cancer treatment but also streamlines the diagnostic and therapeutic process, offering a potent strategy for combating tumors103.
Cyclic peptides represent a promising therapeutic vehicle in Radionuclide Therapy (RT) due to their unique biological properties and targeting capabilities. These peptides can bind to radionuclides, forming Peptide Receptor Radionuclide Therapy (PRRT) agents that specifically target receptors on tumor cells, such as somatostatin receptors (SSTRs). Upon binding, these agents release radioactivity that effectively destroys tumor cells, achieving the desired therapeutic outcome104. The primary advantage of cyclic peptides in PRRT lies in their selectivity and targeting ability, which enable efficient delivery of radionuclides directly to the tumor site while minimizing damage to surrounding healthy tissues. For instance, growth-inhibitory analogs, such as Octreotide, labeled with radionuclides like 177Lu or 90Y, have been widely utilized for treating neuroendocrine tumors that express SSTR293,94 (Table 2). This targeted approach allows for precise irradiation of tumor cells, inducing cell death with relatively few side effects. Recent studies have further demonstrated the potential of cyclic peptides in this field. For example, Matthias et al identified a bis-cyclic peptide with sub-nanomolar affinity for membrane-type 1 matrix metalloproteinase (MT1-MMP), which exhibited selective tumor uptake in the HT1080 xenograft mouse model following radio-coupling95 (Table 2).
In terms of clinical application, the integration of cyclic peptides with radionuclide therapy is becoming increasingly refined. Notably, cyclic peptide-labeled radiopharmaceuticals, such as 177Lu-FAP-2286, have shown promising tumor uptake and retention across various malignant tumors, with favorable tolerance profiles105. This suggests that the application of cyclic peptides in radionuclide therapy holds significant promise, particularly when combined with other therapeutic modalities, such as immunotherapy and chemotherapy, which may enhance overall treatment efficacy.
Conclusion
Cyclic peptide-functionalized nanoparticles show great potential for enhancing tumor penetration and improving drug delivery efficiency. These cyclic peptides enhance drug cellular uptake and tumour tissue penetration through multiple mechanisms, including passive diffusion, receptor-mediated endocytosis and direct transmembrane transport. In particular, the rigid structure, multivalency and high affinity for specific receptors of cyclic peptides make them excellent for tumour targeting and intracellular drug delivery. We highlight the potential of cyclic peptides in the areas of enhanced tumour penetration, improved delivery efficiency, multimodal treatment strategies, and radionuclide therapy applications. Cyclic peptides offer unique advantages, including the potential for reduced immunogenicity compared to larger proteins such as antibodies and excellent stability, making them a preferred choice over other surface modification technologies. In addition, the physical and chemical properties of certain nanoparticles can be tailored to further enhance tumour penetration. Factors such as particle size, surface charge and shape can greatly affect the ability of nanoparticles to penetrate tumour tissue.
However, the further development of cyclic peptides in drug delivery systems still faces many challenges, including the synthesis efficiency, stability and in vivo pharmacokinetic properties of cyclic peptides. Future research needs to go to focus on how to optimise synthetic strategies and develop new synthetic methods to increase the synthetic efficiency and yield of cyclic peptides while reducing costs. To improve the stability of cyclic peptides in vivo through structural modification and chemical modification, and to prolong the circulation time of cyclic peptides. In addition, we are also focusing on pharmacokinetic studies, in-depth study of the distribution, metabolism and excretion of cyclic peptides and their conjugates in vivo, in order to optimise the dosage design and frequency of administration. Preclinical and clinical studies will be conducted to evaluate the efficacy and safety of cyclic peptide-functionalized nanoparticles in animal models and to advance clinical trials.
In conclusion, cyclic peptide-functionalized nanoparticles show great potential and application prospects in the field of tumor penetration drug delivery. Through continuous research and optimisation, we believe that these systems will provide more effective and precise therapeutic solutions for cancer treatment.
Funding
This research was funded by the National Natural Science Foundation of China (82300868), Medical Technology Plan of Zhejiang Province (grant number: 2022497314), the Natural Science Foundation of Zhejiang Province (grant number: LQ21H160041), the Natural Science Foundation of Zhejiang Province (grant number: LBQ20H050001).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Miao L, Lin CM, Huang L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J Control Release. 2015;219:192–204.
2. Souri M, Soltani M, Moradi Kashkooli F, Kiani Shahvandi M. Engineered strategies to enhance tumor penetration of drug-loaded nanoparticles. J Control Release. 2022;341:227–246. doi:10.1016/j.jconrel.2021.11.024
3. Zhang YR, Lin R, Li HJ, He WL, Du JZ, Wang J. Strategies to improve tumor penetration of nanomedicines through nanoparticle design. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2019;11(1):e1519. doi:10.1002/wnan.1519
4. Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520.
5. Ramadhani D, Maharani R, Gazzali AM, Muchtaridi M. Cyclic peptides for the treatment of cancers: a review. Molecules. 2022;27:4428. doi:10.3390/molecules27144428
6. Remesic M, Lee YS, Hruby VJ. Cyclic opioid peptides. Curr Med Chem. 2016;23:1288–1303. doi:10.2174/0929867323666160427123005
7. Kopple KD. Synthesis of cyclic peptides. J Pharmaceut Sci. 1972;61:1345–1356. doi:10.1002/jps.2600610902
8. Tyler TJ, Durek T, Craik DJ. Native and engineered cyclic disulfide-rich peptides as drug leads. Molecules. 2023;28:3189. doi:10.3390/molecules28073189
9. Qian Z, Rhodes CA, McCroskey LC, et al. Enhancing the cell permeability and metabolic stability of peptidyl drugs by reversible bicyclization. Angew Chem Int Ed Engl. 2017;56:1525–1529. doi:10.1002/anie.201610888
10. Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: past, present and future. Curr Opin Chem Biol. 2017;38:24–29. doi:10.1016/j.cbpa.2017.02.006
11. Wang K, Zhang X, Liu Y, Liu C, Jiang B, Jiang Y. Tumor penetrability and anti-angiogenesis using iRGD-mediated delivery of doxorubicin-polymer conjugates. Biomaterials. 2014;35(30):8735–8747. doi:10.1016/j.biomaterials.2014.06.042
12. Bogdanowich-Knipp SJ, Chakrabarti S, Williams TD, Dillman RK, Siahaan TJ. Solution stability of linear vs. cyclic RGD peptides. J Pept Res. 1999;53:530–541. doi:10.1034/j.1399-3011.1999.00052.x
13. Pfaff M, Tangemann K, Müller B, et al. Selective recognition of cyclic RGD peptides of NMR defined conformation by alpha IIb beta 3, alpha V beta 3, and alpha 5 beta 1 integrins. J Biol Chem. 1994;269:20233–20238.
14. Lättig-Tünnemann G, Prinz M, Hoffmann D, et al. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat Commun. 2011;2:453.
15. Li C, Shi K, Zhao S, et al. Natural-source payloads used in the conjugated drugs architecture for cancer therapy: recent advances and future directions. Pharmacol Res. 2024;207:107341. doi:10.1016/j.phrs.2024.107341
16. Yang Y, Wang S, Ma P, et al. Drug conjugate-based anticancer therapy - Current status and perspectives. Cancer Lett. 2023;552:215969. doi:10.1016/j.canlet.2022.215969
17. Li WQ, Guo HF, Li LY, Zhang YF, Cui JW. The promising role of antibody drug conjugate in cancer therapy: combining targeting ability with cytotoxicity effectively. Cancer Med. 2021;10(14):4677–4696. doi:10.1002/cam4.4052
18. Zhu YS, Tang K, Lv J. Peptide-drug conjugate-based novel molecular drug delivery system in cancer. Trends Pharmacol Sci. 2021;42(10):857–869. doi:10.1016/j.tips.2021.07.001
19. Heh E, Allen J, Ramirez F, et al. Peptide drug conjugates and their role in cancer therapy. Int J Mol Sci. 2023;24(1):829. doi:10.3390/ijms24010829
20. Bechtler C, Lamers C. Macrocyclization strategies for cyclic peptides and peptidomimetics. RSC Med Chem. 2021;12(8):1325–1351. doi:10.1039/d1md00083g
21. White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nat Chem. 2011;3(7):509–524. doi:10.1038/nchem.1062
22. Yin L, Li X, Wang R, et al. Recent research progress of RGD peptide–modified nanodrug delivery systems in tumor therapy. Int J Pept Res Ther. 2023;29:53. doi:10.1007/s10989-023-10523-4
23. Merz ML, Habeshian S, Li B, et al. De novo development of small cyclic peptides that are orally bioavailable. Nat Chem Biol. 2024;20(5):624–633. doi:10.1038/s41589-023-01496-y
24. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi:10.1016/S0169-409X(00)00129-0
25. Ovadia O, Greenberg S, Chatterjee J, et al. The effect of multiple N-methylation on intestinal permeability of cyclic hexapeptides. Mol Pharm. 2011;8:479–487. doi:10.1021/mp1003306
26. Wang CK, Swedberg JE, Harvey PJ, Kaas Q, Craik DJ. Conformational flexibility is a determinant of permeability for cyclosporin. J Phys Chem B. 2018;122:2261–2276.
27. Dougherty PG, Sahni A, Pei D. Understanding cell penetration of cyclic peptides. ChemRev. 2019;119:10241–10287.
28. Oh D, Darwish SA, Shirazi AN, Tiwari RK, Parang K. Amphiphilic bicyclic peptides as cellular delivery agents. ChemMedChem. 2014;9:2449–2453. doi:10.1002/cmdc.201402230
29. Contreras J, Elnagar AY, Hamm-Alvarez SF, Camarero JA. Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J Control Release. 2011;155:134–143. doi:10.1016/j.jconrel.2011.08.030
30. Mandal D, Nasrolahi Shirazi A, Parang K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew Chem Int Ed Engl. 2011;50(41):9633–9637. doi:10.1002/anie.201102572
31. Pei D, Buyanova M. Overcoming endosomal entrapment in drug delivery. Bioconjug Chem. 2019;30:273–283. doi:10.1021/acs.bioconjchem.8b00778
32. Moreira C, Oliveira H, Pires LR, Simões. S, Barbosa MA, Pêgo AP. Improving chitosan-mediated gene transfer by the introduction of intracellular buffering moieties into the chitosan backbone. Acta Biomater. 2009;5:2995–3006.
33. Bus T, Traeger A, Schubert US. The great escape: how cationic polyplexes overcome the endosomal barrier. J Mater Chem B. 2018;6:6904–6918. doi:10.1039/C8TB00967H
34. Herce HD, Garcia AE, Litt J, et al. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys J. 2009;97:1917–1925.
35. Sahni A, Qian Z, Pei D. Cell-penetrating peptides escape the endosome by inducing vesicle budding and collapse. ACS Chem Biol. 2020;15:2485–2492. doi:10.1021/acschembio.0c00478
36. Qian J, Zhou S, Lin P, et al. Recent advances in the tumor-penetrating peptide internalizing RGD for cancer treatment and diagnosis. Drug Dev Res. 2023;84(4):654–670. doi:10.1002/ddr.22056
37. Laksitorini MD, Kiptoo PK, On NH, Thliveris JA, Miller DW, Siahaan TJ. Modulation of intercellular junctions by cyclic-ADT peptides as a method to reversibly increase blood-brain barrier permeability. J Pharm Sci. 2015;104:1065–1075.
38. Jiang X, Xin H, Gu J, et al. Solid tumor penetration by integrin-mediated pegylated poly(trimethylene carbonate) nanoparticles loaded with paclitaxel. Biomaterials. 2013;34:1739–1746.
39. Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A. 2009;106:16157–16162. doi:10.1073/pnas.0908201106
40. Liu X, Lin P, Perrett I, et al. Tumor-penetrating peptide enhances transcytosis of silicasome-based chemotherapy for pancreatic cancer. J Clin Invest. 2017;127:2007–2018. doi:10.1172/JCI92284
41. Pang HB, Braun GB, Friman T, et al. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat Commun. 2014;5:4904. doi:10.1038/ncomms5904
42. Connor Y, Tekleab S, Nandakumar S, et al. Physical nanoscale conduit-mediated communication between tumour cells and the endothelium modulates endothelial phenotype. Nat Commun. 2015;6:8671.
43. Ariazi J, Benowitz A, De Biasi V, et al. Tunneling nanotubes and gap junctions-their role in long-range intercellular communication during development, health, and disease conditions. Front Mol Neurosci. 2017;10:333. doi:10.3389/fnmol.2017.00333
44. Gerdes HH, Bukoreshtliev NV, Barroso JF. Tunneling nanotubes: a new route for the exchange of components between animal cells [published correction appears in FEBS lett. FEBS Lett. 2007;581(11):2194–2201. doi:10.1016/j.febslet.2007.03.071
45. Gousset K, Schiff E, Langevin C, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11(3):328–336. doi:10.1038/ncb1841
46. Turos-Korgul L, Kolba MD, Chroscicki P, Zieminska A, Piwocka K. Tunneling nanotubes facilitate intercellular protein transfer and cell networks function. Front Cell Dev Biol. 2022;10:915117. doi:10.3389/fcell.2022.915117
47. Wang Y, Yi S, Sun L, Huang Y, Lenaghan SC, Zhang M. Doxorubicin-loaded cyclic peptide nanotube bundles overcome chemoresistance in breast cancer cells. J Biomed Nanotechnol. 2014;10(3):445–454. doi:10.1166/jbn.2014.1724
48. Bhardwaj G, O’Connor J, Rettie S, et al. Accurate de novo design of membrane-traversing macrocycles. Cell. 2022;185(19):3520–3532. doi:10.1016/j.cell.2022.07.019
49. Kapałczyńska M, Kolenda T, Przybyła W, et al. 2D and 3D cell cultures - a comparison of different types of cancer cell cultures. Arch Med Sci. 2018;14(4):910–919. doi:10.5114/aoms.2016.63743
50. Clevers H. Modeling Development and Disease with Organoids. Cell. 2016;165(7):1586–1597. doi:10.1016/j.cell.2016.05.082
51. Lo JH, Hao L, Muzumdar MD, et al. iRGD-guided tumor-penetrating nanocomplexes for therapeutic siRNA delivery to pancreatic cancer. Mol Cancer Ther. 2018;17:2377–2388. doi:10.1158/1535-7163.MCT-17-1090
52. Gengenbacher N, Singhal M, Augustin HG. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat Rev Cancer. 2017;17(12):751–765. doi:10.1038/nrc.2017.92
53. Liu X, Jiang J, Ji Y, Lu J, Chan R, Meng H. Targeted drug delivery using iRGD peptide for solid cancer treatment. Mol Syst Des Eng. 2017;2:370–379. doi:10.1039/C7ME00050B
54. Syvänen S, Edén D, Sehlin D. Cationization increases brain distribution of an amyloid-beta protofibril selective F(ab’)2 fragment. Biochem Biophys Res Commun. 2017;493(1):120–125. doi:10.1016/j.bbrc.2017.09.065
55. Miura S, Suzuki H, Bae YH. A multilayered cell culture model for transport study in solid tumors: evaluation of tissue penetration of polyethyleneimine based cationic micelles. Nano Today. 2014;9(6):695–704. doi:10.1016/j.nantod.2014.10.003
56. Sajid MI, Moazzam M, Stueber R, et al. Applications of amphipathic and cationic cyclic cell-penetrating peptides: significant therapeutic delivery tool. Peptides. 2021;141:170542. doi:10.1016/j.peptides.2021.170542
57. Shirazi AN, El-Sayed NS, Mandal D, et al. Cysteine and arginine-rich peptides as molecular carriers. Bioorg Med Chem Lett. 2016;26(2):656–661. doi:10.1016/j.bmcl.2015.11.052
58. Shirazi AN, Mozaffari S, Sherpa RT, Tiwari R, Parang K. Efficient intracellular delivery of cell-impermeable cargo molecules by peptides containing tryptophan and histidine. Molecules. 2018;23(7):1536. doi:10.3390/molecules23071536
59. Mott L, Akers C, Pack DW. Effect of polyplex surface charge on cellular internalization and intracellular trafficking. J Drug Delivery Sci Technol. 2023;84:104465. doi:10.1016/j.jddst.2023.104465
60. Zhou Q, Shao S, Wang J, et al. Enzyme-activatable polymer-drug conjugate augments tumour penetration and treatment efficacy. Nat Nanotechnol. 2019;14:799–809. doi:10.1038/s41565-019-0485-z
61. Mohanty RP, Liu X, Ghosh D. Electrostatic driven transport enhances penetration of positively charged peptide surfaces through tumor extracellular matrix. Acta Biomater. 2020;113:240–251. doi:10.1016/j.actbio.2020.04.051
62. Boehnke N, Dolph KJ, Juarez VM, Lanoha JM, Hammond PT. Electrostatic conjugation of nanoparticle surfaces with functional peptide motifs. BioconjugChem. 2020;31:2211–2219.
63. Scodeller P, Asciutto EK. Targeting Tumors Using Peptides. Molecules. 2020;25:808.
64. Pollaro L, Raghunathan S, Morales-Sanfrutos J, Angelini A, Kontos S, Heinis C. Bicyclic peptides conjugated to an albumin-binding tag diffuse efficiently into solid tumors. Mol Cancer Ther. 2015;14:151–161. doi:10.1158/1535-7163.MCT-14-0534
65. Rhodes CA, Pei D. Bicyclic peptides as next-generation therapeutics. Chemistry. 2017;23:12690–12703. doi:10.1002/chem.201702117
66. Dancy JG, Wadajkar AS, Schneider CS, et al. Non-specific binding and steric hindrance thresholds for penetration of particulate drug carriers within tumor tissue. J Control Release. 2016;238:139–148. doi:10.1016/j.jconrel.2016.07.034
67. Ding J, Chen J, Gao L, et al. Engineered nanomedicines with enhanced tumor penetration. Nano Today. 2019;29:100800. doi:10.1016/j.nantod.2019.100800
68. Zhang H, Wu J, Wang J, et al. Novel isoindolinone-based analogs of the natural cyclic peptide fenestin a: synthesis and antitumor activity. ACS Med Chem Lett. 2022;13:1118–1124.
69. Khatri B, Nuthakki VR, Chatterjee J. Strategies to enhance metabolic stabilities. Methods Mol Biol. 2019;2001:17–40.
70. Dahiya R, Dahiya S, Kumar P, et al. Structural and biological aspects of natural bridged macrobicyclic peptides from marine resources. Arch Pharm (Weinheim). 2021;354:e2100034.
71. Elias DR, Cheng Z, Tsourkas A. An intein-mediated site-specific click conjugation strategy for improved tumor targeting of nanoparticle systems. Small. 2010;6:2460–2468. doi:10.1002/smll.201001095
72. Goyard D, Ortiz AM, Boturyn D, Renaudet O. Multivalent glycocyclopeptides: conjugation methods and biological applications. Chem Soc Rev. 2022;51:8756–8783. doi:10.1039/D2CS00640E
73. Wang L, Zhang D, Li J, et al. A novel ICG-labeled cyclic TMTP1 peptide dimer for sensitive tumor imaging and enhanced photothermal therapy in vivo. Eur J Med Chem. 2022;227:113935. doi:10.1016/j.ejmech.2021.113935
74. Gao X, Qian J, Zheng S, et al. Up-regulating blood brain barrier permeability of nanoparticles via multivalent effect. Pharm Res. 2013;30:2538–2548. doi:10.1007/s11095-013-1004-9
75. Cheng Q, Liu Y. Multifunctional platinum-based nanoparticles for biomedical applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(2). doi:10.1002/wnan.1410
76. Shi S, Zhou M, Li X, et al. Synergistic active targeting of dually integrin αvβ3/CD44-targeted nanoparticles to B16F10 tumors located at different sites of mouse bodies. J Control Release. 2016;235:1–13. doi:10.1016/j.jconrel.2016.05.050
77. Qin SY, Zhang AQ, Cheng SX, Rong L, Zhang XZ. Drug self-delivery systems for cancer therapy. Biomaterials. 2017;112:234–247. doi:10.1016/j.biomaterials.2016.10.016
78. Wang L, Wang X, Shen L, et al. Paclitaxel and naringenin-loaded solid lipid nanoparticles surface modified with cyclic peptides with improved tumor targeting ability in glioblastoma multiforme. Biomed Pharmacother. 2021;138:111461.
79. Larnaudie SC, Brendel JC, Romero-Canelón I, et al. Cyclic peptide-polymer nanotubes as efficient and highly potent drug delivery systems for organometallic anticancer complexes. Biomacromolecules. 2018;19(1):239–247. doi:10.1021/acs.biomac.7b01491
80. Blunden BM, Chapman R, Danial M, et al. Drug conjugation to cyclic peptide-polymer self-assembling nanotubes. Chemistry. 2014;20(40):12745–12749. doi:10.1002/chem.201403130
81. Nasrolahi Shirazi A, Tiwari R, Chhikara BS, Mandal D, Parang K. Design and biological evaluation of cell-penetrating peptide-doxorubicin conjugates as prodrugs. Mol Pharm. 2013;10(2):488–499. doi:10.1021/mp3004034
82. Nasrolahi Shirazi A, Tiwari RK, Oh D, et al. Surface decorated gold nanoparticles by linear and cyclic peptides as molecular transporters. Mol Pharm. 2013;10(8):3137–3151. doi:10.1021/mp400199e
83. Hu H, Wang B, Lai C, et al. iRGD-paclitaxel conjugate nanoparticles for targeted paclitaxel delivery. Drug Dev Res. 2019;80:1080–1088. doi:10.1002/ddr.21589
84. Liu X, Jiang J, Nel AE, Meng H. Major effect of transcytosis on nano drug delivery to pancreatic cancer. Mol Cell Oncol. 2017;4:e1335273.
85. Karmali PP, Kotamraju VR, Kastantin M, et al. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine. 2009;5:73–82. doi:10.1016/j.nano.2008.07.007
86. Park SW, Jun SY, Kim JS, Kim YS. Engineering of an EpCAM-targeting cyclic peptide to improve the EpCAM-mediated cellular internalization and tumor accumulation of a peptide-fused antibody. Biochem Biophys Res Commun. 2021;573:35–41. doi:10.1016/j.bbrc.2021.08.021
87. Pasqualini R, Koivunen E, Kain R, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–727.
88. Zhang X, Li X, Hua H, et al. Cyclic hexapeptide-conjugated nanoparticles enhance curcumin delivery to glioma tumor cells and tissue. Int J Nanomed. 2017;12:5717–5732. doi:10.2147/IJN.S138501
89. Jang JP, Jung HJ, Han JM, et al. Two cyclic hexapeptides from Penicillium sp. FN070315 with antiangiogenic activities. PLoS One. 2017;12(9):e0184339. doi:10.1371/journal.pone.0184339
90. La Salvia A, Modica R, Rossi RE, et al. Targeting neuroendocrine tumors with octreotide and lanreotide: key points for clinical practice from NET specialists. Cancer Treat Rev. 2023;117:102560. doi:10.1016/j.ctrv.2023.102560
91. Fan X, Yuan Z, Shou C, et al. cRGD-conjugated Fe3O4@PDA-DOX multifunctional nanocomposites for MRI and antitumor chemo-photothermal therapy. Int J Nanomed. 2019;14:9631–9645. doi:10.2147/IJN.S222797
92. Wu Y, Zeng C, Lv J, et al. Tumor-targeted cRGD-coated liposomes encapsulating optimized synergistic cepharanthine and IR783 for chemotherapy and photothermal therapy. Int J Nanomed. 2024;19:6145–6160. doi:10.2147/IJN.S457008
93. Bergsma H, van Vliet EI, Teunissen JJ, et al. Peptide receptor radionuclide therapy (PRRT) for GEP-NETs. Best Pract Res Clin Gastroenterol. 2012;26(6):867–881. doi:10.1016/j.bpg.2013.01.004
94. Eychenne R, Bouvry C, Bourgeois M, Loyer P, Benoist E, Lepareur N. Overview of radiolabeled somatostatin analogs for cancer imaging and therapy. Molecules. 2020;25(17):4012. doi:10.3390/molecules25174012
95. Eder M, Pavan S, Bauder-Wüst U, et al. Bicyclic peptides as a new modality for imaging and targeting of proteins overexpressed by tumors. Cancer Res. 2019;79(4):841–852. doi:10.1158/0008-5472.CAN-18-0238
96. Shirazi AN, Park SE, Rad S, et al. Cyclic peptide-gadolinium nanoparticles for enhanced intracellular delivery. Pharmaceutics. 2020;12(9):792. doi:10.3390/pharmaceutics12090792
97. Yin H, Yang J, Zhang Q, et al. iRGD as a tumor-penetrating peptide for cancer therapy (review). Mol Med Rep. 2017;15:2925–2930.
98. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi:10.1038/nrc2748
99. Xiao W, Ma W, Wei S, et al. High-affinity peptide ligand LXY30 for targeting α3β1 integrin in non-small cell lung cancer. J Hematol Oncol. 2019;12:56.
100. Colombo G, Curnis F, De Mori GM, et al. Structure-activity relationships of linear and cyclic peptides containing the NGR tumor-homing motif. J Biol Chem. 2002;277(49):47891–47897. doi:10.1074/jbc.M207500200
101. Alberici L, Roth L, Sugahara KN, et al. De novo design of a tumor-penetrating peptide. Cancer Res. 2013;73(2):804–812. doi:10.1158/0008-5472.CAN-12-1668
102. Fogal V, Zhang L, Krajewski S, Ruoslahti E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008;68:7210–7218. doi:10.1158/0008-5472.CAN-07-6752
103. Borum RM, Retout M, Creyer MN, et al. Self-assembled peptide-dye nanostructures for in vivo tumor imaging and photodynamic toxicity. Npj Imaging. 2024;2(4). doi:10.1038/s44303-024-00008-4.
104. Zhang T, Lei H, Chen X, et al. Carrier systems of radiopharmaceuticals and the application in cancer therapy. Cell Death Discov. 2024;10(1):16. doi:10.1038/s41420-023-01778-3
105. Chakravarty R, Song W, Chakraborty S, Cai W. Fibroblast activation protein (FAP)-targeted radionuclide therapy: which ligand is the best? Eur J Nucl Med Mol Imaging. 2023;50(10):2935–2939. doi:10.1007/s00259-023-06338-6
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.