")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Screening of Covalent Kinase Inhibitors Yields Hits for Cysteine Protease USP7 / HAUSP
Authors Ernst LN , Jaag SJ , Wydra VR , Masberg B , Knappe C , Gerstenecker S, Serafim RAM, Liang XJ, Seidler NJ, Lämmerhofer M , Gehringer M , Boeckler FM
Received 21 December 2024
Accepted for publication 12 March 2025
Published 25 March 2025 Volume 2025:19 Pages 2253—2284
DOI https://doi.org/10.2147/DDDT.S513591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Solomon Tadesse Zeleke
Larissa N Ernst,1 Simon J Jaag,2 Valentin R Wydra,3 Benedikt Masberg,2 Cornelius Knappe,2 Stefan Gerstenecker,3 Ricardo AM Serafim,3 Xiaojun Julia Liang,4,5 Nico J Seidler,3,5 Michael Lämmerhofer,2 Matthias Gehringer,3– 5 Frank M Boeckler1,6
1Department of Pharmacy and Biochemistry, Eberhard Karls Universität Tübingen, Laboratory for Molecular Design and Pharmaceutical Biophysics, Institute of Pharmaceutical Sciences, Tübingen, 72076, Germany; 2Department of Pharmacy and Biochemistry, Eberhard Karls Universität, Pharmaceutical (Bio-)Analysis, Institute of Pharmaceutical Sciences, Tübingen, 72076, Germany; 3Department of Pharmacy and Biochemistry, Eberhard Karls Universität, Pharmaceutical Chemistry, Institute of Pharmaceutical Sciences, Tübingen, 72076, Germany; 4Department of Medicinal Chemistry, Eberhard Karls Universität Tübingen, Faculty of Medicine, Institute for Biomedical Engineering, Tübingen, 72076, Germany; 5Cluster of Excellence iFIT (EXC 2180) ‘Image-Guided and Functionally Instructed Tumor Therapies’, Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany; 6Interfaculty Institute for Biomedical Informatics (IBMI), Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany
Correspondence: Frank M Boeckler, Lab for Molecular Design and Pharm. Biophysics, Institute for Pharmaceutical Sciences, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 8, Tübingen, 72076, Germany, Tel +49 7071 29 74567, Email [email protected]
Purpose: The ubiquitin-specific protease 7 (USP7), also known as herpes-associated ubiquitin-specific protease (HAUSP) is an interesting target due to its role in the tumor suppressor p53 pathway. In recent years targeted covalent inhibitors have gained significant importance in pharmaceutical research. Thus, we have investigated a small library of 129 ligands bearing different types of covalent reactive groups (“warheads”) from various kinase drug discovery projects for their reactivity towards the catalytic cysteine of USP7, as well as their influence on its melting temperature. These compounds mainly encompassed α,β-unsaturated amides specifically acrylamides, SNAr reacting compounds, aryl fluorosulfates and sulfonyl fluorides.
Methods: We analyzed an array of 129 electrophilic compounds which had been designed as covalent kinase inhibitors in a DSF-based (differential scanning fluorimetry) screen against USP7. The hits were evaluated for their ability to cause similar thermal shifts for a CYS-deficient USP7 control mutant (USP7asoc), where only the catalytic Cys223 was retained. Additionally, covalent binding was evaluated by intact protein mass spectrometry (MS).
Results: The DSF screen revealed that, predominantly 18 of the 129 tested compounds decreased the melting temperature of USP7 and its mutant USP7asoc. For 8 of these, the hypothesized covalent binding mode was corroborated with native and mutant USP7 by intact protein MS. Nearly all identified hits have a covalent warhead that reacts via nucleophilic aromatic substitution (SNAr).
Conclusion: The screening and evaluation of the kinase library revealed several initial hits of interest. Seven SNAr warheads and one acrylamide warhead compound covalently modified the target protein (USP7) and showed clear shifts in the melting temperatures ranging from − 6.0 °C to +1.7 °C.
Keywords: covalent cysteine modification, differential scanning fluorimetry (DSF), intact protein mass spectrometry, nucleophilic aromatic substitution (SNAr), repurposing of kinase inhibitors
Graphical Abstract:

Introduction
The cysteine protease ubiquitin-specific protease 7 (USP7) is one of over 100 deubiquitinases, which catalyzes the removal of ubiquitin and protects the substrate protein from being degraded.1 Deubiquitinating enzymes (DUB) can be categorized into seven families, among which the USPs make up the largest subfamily.2–5 Over the last few years USP7, also known as herpes-associated ubiquitin-specific protease (HAUSP), gained much interest due to its role in the tumor suppressor protein p53 pathway.6–8 It deubiquitinates and subsequently stabilizes mouse double minute 2 homolog (MDM2), thus causing ubiquitination and degradation of p53.9 Furthermore, USP7 also showed influence on the regulation and stabilization of additional proteins associated with tumorigenesis,10–13 DNA damage response, cell cycle regulation and apoptosis.14–16 Thus, inhibiting USP7 represents a promising strategy for various cancer types for example by stabilizing p53 through the promotion of MDM2 degradation.17 In recent years, small molecules have been shown to stabilize p53.18–23 Furthermore, one candidate for p53 is even in Phase 1/2 clinical trials.24–26 Moreover, a number of USP7 inhibitors have been identified.27–30 For instance 4-hydroxypiperidine FT827 with a vinyl sulfonamide warhead reacts covalently with the catalytic cysteine 223 (Cys223).31 In addition, proteolysis targeting chimera (PROTAC) degraders targeting USP7 such as U7D-132 and CST967 represent a novel approach for abrogating USP7 function.3 However, to date, no USP7-targeted agents have entered clinical trials. Thus, improving the diversity of chemotypes, implementing different ways how to address the binding site including the use of covalent mechanisms could foster the potential of the portfolio of available hits, leads and, eventually, candidates for clinical development.33
USP7 is a cysteine protease with a nucleophilic catalytic cysteine which makes it particularly amenable to covalent targeting approaches. However, USP7 is known for its structural changes upon ubiquitin binding where the catalytic Cys223 moves from a conserved apoenzyme form to a catalytically competent conformation in complex with ubiquitin where the catalytic triad comes closer together.34 In order to target this cysteine covalently, we recently designed and screened a library of covalently reactive fragments with different electrophilic warheads (CovLib) against USP7 and two other proteins. The identified hit fragments all caused destabilization of USP7 by reducing its melting temperature (Tm).18 In the present study, the focus was on screening more lead-like electrophilic compounds containing different warhead classes, which were initially designed as covalent kinase inhibitors. With this approach, we aimed for the identification of hits with a stronger non-covalent binding contribution and a more tempered reactivity and, thus, a higher selectivity for USP7. Using DSF and intact protein MS, the screening was further facilitated with the help of two control mutants of the catalytic domain of USP7 (USP7CD): USP7asoc (active site only cysteine), retaining only the catalytic cysteine 223 and USP7nc (no cysteine), where all cysteine residues are changed into serine residues.
Materials and Methods
Materials
The compounds studied were all synthesized at purity levels of 95 % or higher according to the peak areas at the two different wavelengths (254 nm and 230 nm) except compound 27 (92.17% at 230 nm) (Figures S1.1–S1.18). High performance liquid chromatography (HPLC) purity analysis was conducted on an Agilent 1100 series HPLC system including injection module, column compartment, degasser and binary pump (Agilent Technologies Inc., Santa Clara, CA, USA) coupled to a 1260 DAD detector module. The system was either equipped with a Phenomenex Kinetex® 2.6 µm C8 100 Å 150×4.6 mm column (Phenomenex Inc., Torrance, CA, USA) or with a Phenomenex Luna® 5 µm (150 x 4.6 mm, 5 µm) reversed phase C8 separation column (Phenomenex Inc., Torrance, CA, USA). A flow rate of either 0.5 mL/min or 1.5 mL/min at 23 °C was used with an injection volume of 5 µL or 10 µL. Elution was performed with the following gradients: 0.01 M KH2PO4 pH 2.3 (solvent A) and MeOH (solvent B).
Method A = 0 min: 40% B / 60% A, 9 min: 95% B / 5% A, 10 min: 95% B / 5% A, 11 min: 40% B / 60% A, 16 min: 40% B / 60% A.
Method B = 0 min: 40% B / 60% A, 15 min: 85% B / 15% A, 20 min: 85% B / 15% A, 22 min: 40% B / 60% A, 28 min: 40% B / 60% A.
Method C = 0 min: 40% B / 60% A, 8 min: 85% B / 15% A, 13 min: 85% B / 15% A, 14 min: 40% B / 60% A, 16 min: 40% B / 60% A.
Molecular Biology
The expression and purification of USP7 catalytic domain (USP7CD) (208–560) using a pET24a(+)_HLT_USP7 construct, were carried out as previously described.18 The purity of USP7 was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and the correct protein mass was confirmed by ultra-high performance liquid chromatography electrospray ionization mass spectrometry (Supporting Information) (Table S2.1 and Figure S2.1A).
The pET24a(+)_HLT_USP7asoc and pET24a(+)_HLT_USP7nc plasmid were transformed into E. coli BL 21 (DE3) pLysS cells (Novagen, Merck, Darmstadt, Germany). For the USP7asoc (active site only cysteine) six of the seven cysteines are mutated to serines (208–560, C300/315/334/448/478/510S). The USP7nc (no cysteine) has no cysteine residues left in the protein. Here all cysteines are mutated to serines (208–560, C223/300/315/334/448/478/510S). Expression and purification were performed as for USP7CD and as previously described.18 Wherever USP7 is written, it refers to the wild type USP7 catalytic domain. The purity of USP7asoc and USP7nc were checked by SDS-PAGE and the correct protein mass was confirmed by UHPLC-ESI-MS (Table S3.1, Figure S3.1B and C). All used protein sequences are depicted in Table 1.
 |
Table 1 All Used Protein Sequences |
Differential Scanning Fluorimetry (DSF)
The melting temperatures of USP7 and USP7asoc in presence or absence of fragments were determined by DSF. Experiments were conducted on a Qiagen Rotor-Q Model-5-Plex HRM real-time PCR instrument (Qiagen, Hilden, Germany). SYPRO Orange (Life Technologies Corporation, Eugene, OR, USA) served as a fluorescent dye at a final concentration of 5x. Most experiments were performed with 8 µM protein and a final compound concentration of 250 µM corresponding to a protein-to-compound ratio of 1:31.25. Due to solubility issues, compound concentrations were lower for compounds 7 and 8 (90 µM), as well as compounds 9, 10, and 11 (225 µM). Moreover, compounds 7–10 were dissolved in 1,4-dioxane instead of dimethyl sulfoxide (DMSO). For concentration-dependent measurements, the compound concentration was varied. A Tris buffer (25 mM Tris pH 8.0, 150 mM NaCl, 5 mM TCEP, 5% (v/v) DMSO) was used for all proteins.
All measurements of USP7 were performed after 30 min, 4 h and 24 h of incubation. Only the hits of the DSF screen with USP7 were measured with USP7asoc after 30 min, 4 h and 24 h incubation time. DSF experiments were performed with a constant heating rate of 270 °C/h.22 The temperature was ramped from 28 °C to 60–70 °C and the excitation and emission filters were set to 470 nm and 610 nm, respectively. The melting temperatures of USP7 and USP7asoc were determined from the maxima of the first derivatives of the melting curves in OriginPro2020 (OriginLab, Northampton, MA, USA). Tm was calculated by subtracting the Tm of the protein sample with pure DMSO or 1,4-dioxane from the Tm of the protein sample containing the compound. All samples were measured at least in triplicate. Multiple runs for a compound were averaged using error propagation. Compounds were considered as a hit, if the shift in ∆Tm was at least 0.50 °C.35
Intact Protein Mass Spectrometry
USP7 was prepared in Tris puffer (25 mM Tris pH 8.0, 150 mM NaCl, 5 mM TCEP) with a protein-to-compound ratio of 1:31.25 and 5% (v/v) DMSO. The mixtures of USP7, USP7asoc and USP7nc were all incubated at 20 °C for 24 h on a rotating shaker. Intact protein mass analysis was performed using UHPLC-ESI-MS measurements, data acquisition and data analysis as previously described.20
Results
Characteristics and Evaluation of the Compounds Against USP7
The rationale behind our screening approach, encompassing compounds synthesized for various kinase projects (see ref36–41 plus some unpublished analogues from the same compound series), was to scout for covalent mechanism targeting the catalytic Cys223 of USP7, by employing more drug-like compounds with tempered reactivity in comparison to our previous covalent fragment screening. In addition, their thermal shift behavior in a DSF experiment was of interest: Are these compounds with more elaborate non-covalent recognition motifs likewise inducing a decrease of the melting temperature of USP7 upon covalent binding to the catalytic Cys223, as we have evidenced with the fragments of our covalent library (CovLib)18. The search for such larger covalent inhibitors was enabled by using two control mutants of USP7. The first one has only the catalytic Cys223 left while all other six cysteines of USP7CD are mutated to serines. Thus, it was named: “USP7 active site only cysteine” (USP7asoc). In the other control mutant termed “USP7 no cysteine” (USP7nc), all cysteine residues are mutated to serines. Usage of these two control mutants enables the identification of compounds which are likely to bind to catalytic Cys223. The compounds contained in the screening library mainly encompassed α,β-unsaturated amides specifically acrylamides and SNAr electrophiles, while a few other warheads such as ketones, aryl fluorosulfates and sulfonyl fluorides were also included despite being known to form instable cysteine adducts.42,43
DSF Measurements
The influence of the compounds on the melting temperature of USP7 and its mutant USP7asoc was investigated by DSF. DSF is an efficient and fast primary screening method for ligand identification which relies on the detection of a shift in the stability of the protein and thus melting temperature upon compound binding.44–46
Incubation times of 30 min, 4 h and 24 h were chosen for the primary DSF screen (Table 2), to also allow for the detection of compounds with slow inactivation kinetics. Measurements using the USP7asoc mutant were only performed for compounds, which influenced the melting temperature of native USP7 (Table 3). For both proteins, the employed protein concentration was 8 µM. DSF screening results of USP7 and USP7asoc, as well as the corresponding melting curves of compounds where an influence on the proteins was observed, are shown in Tables 2 and 3, as well as in Figures 1–6, respectively. Analogue results and corresponding melting curves of all compounds can be found in Table S3.1 and Figures S3.1–S3.6 in the Supporting Information.
 |
Table 2 Overview of ∆Tm ± SD of USP7 (8 µM Protein) Incubated With 250 µM (Protein-to-Compound Ratio 1:31.25) for 30 min, 4 h and 24 h at 20 °C. Measurements Were Performed at Least in Triplicate. Hits With a ∆Tm of >0.5 °C are Highlighted as Bold Text |
 |
Table 3 Overview of ∆Tm ± SD of USP7asoc (8 µM Protein) Incubated With 250 µM (Protein-to-Compound Ratio 1:31.25) for 30 min, 4 h and 24 h at 20 °C. Measurements Were Performed at Least in Triplicate. Hits With a ∆Tm of >0.5 °C are Highlighted as Bold Text |
 |
Figure 1 First derivatives of the melting curves of compound 5 (A), compound 6 (B), compound 7 (C), compound 10 (D), compound 17 (E) and compound 18 (F) with USP7 (8 µM protein) after 30 min, 4 h and 24 h of incubation. |
 |
Figure 2 First derivatives of the melting curves of compound 20 (A), compound 21 (B), compound 27 (C), compound 28 (D), compound 29 (E) and compound 30 (F) with USP7 (8 µM protein) after 30 min, 4 h and 24 h of incubation. |
 |
Figure 3 First derivatives of the melting curves of compound 31 (A), compound 34 (B), compound 52 (C), compound 90 (D), compound 98 (E) and compound 121 (F) with USP7 (8 µM protein) after 30 min, 4 h and 24 h of incubation. |
 |
Figure 4 First derivatives of the melting curves of compound 5 (A), compound 6 (B), compound 7 (C), compound 10 (D), compound 17 (E) and compound 18 (F) with USP7asoc (8 µM protein) after 30 min, 4 h and 24 h of incubation. |
 |
Figure 5 First derivatives of the melting curves of compound 20 (A), compound 21 (B), compound 27 (C), compound 28 (D), compound 29 (E) and compound 30 (F) with USP7asoc (8 µM protein) after 30 min, 4 h and 24 h of incubation. |
 |
Figure 6 First derivatives of the melting curves of compound 31 (A), compound 34 (B), compound 52 (C), compound 90 (D), compound 98 (E) and compound 121 (F) with USP7asoc (8 µM protein) after 30 min, 4 h and 24 h of incubation. |
Of all compounds tested three induced a Tm change of USP7 only after 24 h of incubation at 20 °C. Compound 6 with an isoquinoline linked to a fluoronitropyridine decreased the Tm by about −4.20 °C (Figure 1B), compound 31 with a pyrimidine linked to a chloronitropyridine about −3.75 °C (Figure 3A) and compound 98 containing a quinazoline with a fluorophenol about −0.90 °C after 24 h incubation (Figure 1E). Notably, they all possessed an electron-deficient aryl ring bearing either a fluorine (6 and 31) or chlorine (98) as leaving group reacted engaging the target cysteine via an SNAr reaction. Compound 5, the chlorine analogue of 6, did not influence the melting temperature of USP7 (Figure 1A) whereas compound 7, the bromine analogue of 6, decreased it by more than 1 °C after all three incubation times (Figure 1C). Of note, this compound was measured at lower concentration and solubilized in 1,4-dioxane because of its reactivity with DMSO causing stability issues upon storage. Compound 18 and 30 both showed a decrease in the melting temperature of USP7 after 4 h (both about −2 °C) and 24 h (between −3 °C and −4 °C) of incubation (Figures 1F and 2F). Compound 10 was the only compound stabilizing USP7 after 4 h of incubation namely with a ΔTm of 0.80 °C (Figure 1D). All three last mentioned compounds react in a SNAr reaction with chlorine as leaving group.
Compound 52 has an acrylamide warhead and increased the Tm of USP7 already after 30 min of incubation. The extent of the temperature increase, indicative of the level of USP7 stabilization was practically constant over the three incubation times (i.e. about 0.65 °C to 0.70 °C) (Figure 3C). Inducing a consistent Tm increase of about 1°C, the ketone compound 90, potentially acting in a reversible covalent manner, stabilized USP7 across all three incubation times (Figure 3D). A slightly stabilizing effect on USP7 was observed for compound 34, which is structurally related to compound 31. Here, Tm increased by less than 1 °C across all three incubation times (Figure 3B). This stabilization behavior is rather unusual, as only destabilizing compounds are known to date.18,30,47
Compounds 17 and 27 are structurally similar (isoquninolines with difluorophenyls) only differing in their fluorination pattern as two fluorine atoms are located either in the para (17) and meta (27) positions, respectively. They both displayed a similar decrease in the melting temperature of around 1 °C for all incubation times (Figures 1E and 2C), suggesting a non-covalent mechanism as their relatively electron-rich SNAr warheads are unlikely to undergo full covalent modification already at early time points. Similarly, both the sulfonyl-activated putative SNAr compound 20 (Figure 2A) and its sulfinyl analogue 21 (Figure 2B) consistently induced a time-independent destabilization of USP7 (Tm decrease of around 1 °C). Likewise, compounds 28 and 29 representing ring-opened analogues of 20 and 21, destabilized USP7 equally with a time-independent Tm decrease of slightly more than 1 °C (Figure 2D and E).
Interestingly, compound 121 also with a chloroacetamide warhead, induced a gradual decrease of the Tm with prolonged incubation time (Figure 3F) with temperature shifts of −0.90 °C after 30 min of incubation, −1.90 °C after 4 h of incubation and −2.45 °C after 24 h of incubation.
In conclusion, most of the tested compounds decreased the melting temperature of native USP7, whereas four compounds (10, 34, 52 and 90) stabilized the protein. These first interesting DSF hits required further investigation with USP7asoc (Table 3) to investigate whether they have a similar influence on the Tm of the mutant.
Compound 6 behaved with USP7asoc as it did with USP7 inducing a decrease in the Tm (−6 °C) only after 24 h of incubation (Figure 4B). In contrast to native USP7, where any change in Tm was only detected after 24 h, compound 31 showed a slight decrease in the Tm of USP7asoc exclusively after 4 h (Figure 6A). Interestingly, the structurally similar compound 34 had no effect on the melting temperature of USP7asoc (Figure 6B), but stabilized USP7 after all three incubation times (∆Tm about 1 °C). Compound 98 (2-chloroqinazoline) neither stabilized nor destabilized USP7asoc at any incubation time, whereas it destabilized USP7 after 24 h (Figure 6E). For compound 18 (trifluoromethylpyridine), a shift in USP7asoc Tm was only observed after 24 h while a decrease in the Tm of USP7 was already present at 4 h of incubation (Figure 4F). The chlorine analogue (compound 5) did not affect the melting temperature of either the mutant or USP7 (Figure 4A). In contrast, the bromine analogue (compound 7) showed a decrease in Tm after 30 min of incubation. Unfortunately, the melting curves after 4 h and 24 h could not be evaluated, likely due to fluorescence quenching and low signal intensity.44,48 Similar issues also rendered the curve for compound 17 (after 30 min) non-analyzable. In contrast, Figure 4E shows evaluable melting curves for longer incubation times. The destabilization effect of compound 17 on mutant USP7 was in a same range as for USP7 (less than 1 °C for 4 h and 24 h of incubation). Compound 30 destabilized the Tm of USP7asoc after 4 h (−1.50 °C) and after 24 h (−4.55 °C) of incubation (Figure 5F) with the magnitude of Tm decrease being similar to that of the catalytic domain of USP7. Compound 10 was the only compound stabilizing the mutant only after 30 min incubation (1.10 °C) whereas it stabilized USP7 only after 4 h and 24 h (Figure 4D). The two sulfonyl-activated SNAr compounds 20 and 29 and the sulfinyl analogues 21 and 28 all destabilized USP7asoc equally as USP7 and in the same range of about 1 °C (Figure 5A, B, D and E). In similar fashion, compounds 52 (∆Tm 1 °C) and 90 (∆Tm 2 °C) both stabilized USP7asoc at all timepoints as analogue to USP7 (Figure 6C and D). With a ∆Tm of −2.10 °C, the pyrrolopyridine-derived chloroacetamide 121 already destabilized USP7asoc after 30 min (Figure 6F). This decrease remained almost constant over 24 h hinting towards a complete inactivation already at early time points. In contrast, USP7 was gradually destabilized more over time (from 0.90 °C to almost −2.5 °C) suggesting slower inactivation kinetics on the latter.
In summary, only three compounds were left which also stabilized USP7asoc (10, 52, 90). For two other compounds (34, 98) an effect on the Tm was no longer observed for USP7asoc while they shifted the melting temperature of USP7. All other compounds behaved in the same way in USP7asoc as in USP7.
Intact Protein Mass Spectrometry
Compound hits identified by DSF were validated by UHPLC-ESI-MS to verify covalent binding by corresponding mass shift. Intact protein mass spectrometry is a major screening method in covalent fragment-based drug discovery (FBDD) and for covalent inhibitor screens in general enabling the direct detection of covalent protein-compound adducts.49–55 However, the covalent adducts of reversibly acting covalent ligands such as compound 90 can typically not be observed by protein mass spectrometry due to rapid dissociation under denaturing conditions where the stabilization of the covalent complex by non-covalent interactions is lost.42,56,57
The covalent modifications of USP7, USP7asoc and the mutant with no cysteine left (USP7nc) were confirmed by the deconvoluted protein MS spectra for 8 of the 20 Tm shifting compounds (Figures 7–12). In addition, the spectrum of one compound (7) did only show one wide peak making a clear attribution to individual mass shifts difficult. The deconvoluted MS spectra indicated compounds with multiple or only single modifications. Only compounds that covalently bound to USP7 were also measured against its mutants.
 |
Figure 7 Deconvoluted MS spectra of compound 5 (A), 6 (B), 7 (C), 10 (D), 17 (E), and 18 (F) with USP7 (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7: 41,145.61 Da) after 24 h of incubation at 20 °C. |
 |
Figure 8 Deconvoluted MS spectra of compound 20 (A), 21 (B), 27 (C), 28 (D), 29 (E), and 30 (F) with USP7 (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7: 41,145.61 Da) after 24 h of incubation at 20 °C. |
 |
Figure 9 Deconvoluted MS spectra of compound 31 (A), 34 (B), 52 (C), 90 (D), 98 (E), and 121 (F) with USP7 (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7: 41,145.61 Da) after 24 h of incubation at 20 °C. |
 |
Figure 10 Deconvoluted MS spectra of 5 (A), 6 (C), and 18 (E) with USP7asoc (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7asoc: 41,049.22 Da), as well as 5 (B), 6 (D), and 18 (F) with USP7nc (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7nc: 41,033.16 Da) after 24 h of incubation at 20 °C. |
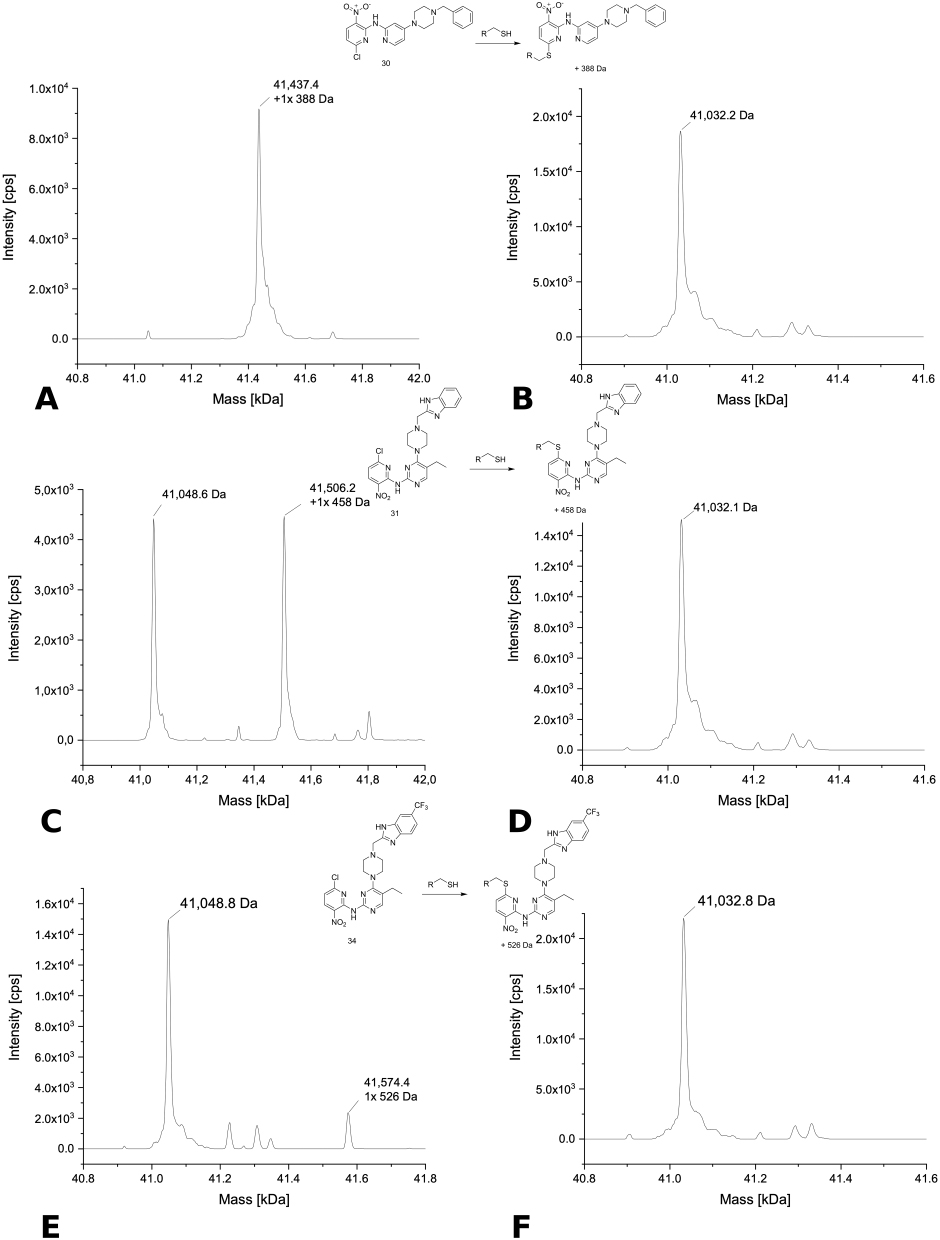 |
Figure 11 Deconvoluted MS spectra of 30 (A), 31 (C) and 34 (E) with USP7asoc (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7asoc: 41,049.22 Da), as well as 30 (B), 31 (D), and 34 (F) with USP7nc (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7nc: 41,033.16 Da) after 24 h of incubation at 20 °C. |
 |
Figure 12 Deconvoluted MS spectra of 121 (A) with USP7asoc (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7asoc: 41,049.22 Da) and 121 (B) with USP7nc (protein-to-compound ratio 1:31.25, theoretical mass of unmodified USP7nc: 41,033.16 Da) after 24 h of incubation at 20 °C. |
The deconvoluted MS spectra of structural analogues 5, 6, and 7 with USP7 are shown in Figure 7A–C. Multiple peaks with an increase in the native protein mass were only detected for compound 6 (Figure 7B) with a fluoride leaving group where the main peak was the single modified protein, while a second smaller peak was assigned to the double-arylated form. The smallest peak in the spectrum of compound 6 was unmodified USP7. The less reactive chloro analog 5 only showed two signals with one representing native protein with a mass of 41,145.1 Da and the other single modified protein accompanied by loss of the chloride thus increasing the mass by 344 Da. The deconvoluted spectrum of the bromine analogue 7 could not be evaluated, because it only showed one wide peak (Figure 7C). This compound was highly reactive, eg with DMSO, for which reason it had to be dissolved in 1,4-dioxane. Maybe this can explain the uncommon behavior. Interestingly, the compound (5) that did not show any change in the melting temperature of either USP7 or USP7asoc did bind to USP7 and USP7asoc (Figure 10A and B) under the conditions employed in the MS experiments. Moreover, it modified both proteins only one time indicating a specific covalent interaction with the catalytic Cys223. However, its more reactive fluoride analog 6 showed in USP7asoc a second small peak corresponding to a double modified protein aside from the main peak (single modified protein) (Figure 10C and D). Consequently, this compound arylated another amino acid than cysteine, a feature that has previously been describe for the more reactive ones among the SNAr electrophiles.42,43 In contrast, none of the compounds showed an arylation of USP7nc. The only detected signal corresponded to the native protein USP7nc with a mass of approximately 41,033 Da.
The MS spectrum of USP7 with compounds 10 and 17 showed no protein modification (Figure 7D and E). Compounds 10 and 17 differ from compound 5 by bearing a quite reactive 2-chloro-5-CF3 pyrimidine (10) ring or a rather unreactive para-difluorophenyl warhead (17), respectively.
In the spectrum of USP7 incubated with compound 18 (Figure 7F), the doubly arylated protein species represented the highest peak with the two smaller signals corresponding to the threefold and the onefold modified protein. With USP7asoc compound 18 reacted only once as there were no additional peaks detectable (Figure 10E and F). Thus, one of the modifications observed for this compound is located at the catalytic Cys223.
None of the deconvoluted MS spectra of sulfonyl SNAr compounds 20 and 29, the sulfinyl analogs 21 and 28, meta di-fluorinated compound 27, (Figure 8A–E) indicate any arylation of USP7, which is likely linked to their comparably low intrinsic reactivity. In all four cases, the only detected signal was unmodified USP7. Importantly, they all decreased the melting temperature of USP7 at all incubation times. This might indicate non-covalent binding.
Covalent modification was detected with the SNAr-reactive chloronitropyridine 30 as the spectra showed single arylated USP7 species as the main peak (Figure 8F) and single arylated USP7asoc as the only visible peak (Figure 11A and B). As mentioned before, only native USP7nc was detectable. Thus, it can be concluded that compound 30 bound to the catalytic Cys223.
Single arylated USP7 species also represented the main signal in the MS spectrum of compound 31 with a smaller visible peak corresponding to unmodified USP7 (Figure 9A). In the spectrum of USP7asoc, two signals of identical intensity were visible representing native protein and a single modification with compound 31 (Figure 11C and D) in line with the expected binding to the catalytic Cys223.
Upon incubation of closely related compound 34 with USP7, the spectra showed unmodified protein as main signal as well as two more peaks representing onefold and doubly arylated protein species, with the latter appearing clearly smaller (Figure 9B). As expected, the spectrum of USP7asoc mainly showed unmodified protein species (Figure 11E and F) along with a small additional signal representing single arylated USP7asoc species. Compound 34 was one of the four compounds, which increased the melting temperature of USP7, which is interesting since structurally related analogue 31 showed the opposite trend.
The acrylamide 52, which stabilized USP7 in the DSF experiment, also mainly showed unmodified USP7 species in its MS spectrum. In addition, there was a small peak representing single modified protein (Figure 9C). However, this signal was not significantly greater than noise and thus should be interpreted with caution.
As expected, compound 90 only showed unmodified protein species (Figure 9D). This compound increased the melting temperature of USP7 and it might react with the target amino acid in a reversible covalent way.
In the MS spectrum of compound 98, likewise only the native protein was detected (Figure 9E). This compound features a quinazoline-linked chlorine atom as a potential leaving group in the SNAr reaction.
The spectrum of chloroacetamide compound 121 with USP7 displayed multiple peaks with a mass shift compared to the native protein mass (Figure 9F). This compound showed the highest number of USP7 modifications as the binding of three times modified protein appeared with almost the same intensity as the double alkylated one. A smaller peak corresponding to mono-alkylated protein was also detected whereas unmodified USP7 was not detected. USP7asoc showed single alkylated protein species as main peak, along with a second small signal corresponding to unmodified USP7asoc indicating chemo-selectivity for cysteine (Figure 12A and B).
In summary, the MS experiment confirmed that 8 out of the 18 compounds which influenced the melting temperature of USP7, reacted in a covalent manner. Notably, all of them reacted in a SNAr reaction with fluorine or chlorine as the leaving group. The only exception is the chloroacetamide compound 121 which reacts in an SN2 alkylation. Only compound 52 had an acrylamide warhead resulting in a small negligible signal. Compounds 5, 30, and 31 only showed mono-arylated USP7, whereas compounds 6, 18, 34, and 121 showed multiple USP7 modifications. In addition, all 7 aforementioned compounds also reacted with USP7asoc indicating binding to the catalytic Cys223. All the other tested compounds did not bind covalently to USP7.
Concentration-Dependent Measurements With DSF
Concentration-dependent DSF measurements with USP7 and its hexa-mutant (USP7asoc) after 24 h of incubation were performed with the four previously identified covalent hits 6, 18, 30, and 121. These compounds all showed a predominant single modification of USP7asoc in the intact protein MS spectra.
All measurements were performed at least in triplicate and with an incubation time of 24 h at 20 °C because otherwise no decrease in the melting temperature could be detected. Table 4 shows the results of the concentration-dependent measurements of compound 6.
 |
Table 4 ∆Tm ± SD of USP7 and USP7asoc (8 µm Protein) Incubated With Different Concentrations of Compound 6 for 24 h at 20 °C |
With increasing concentrations of compound 6, the extent of the negative thermal shift of USP7 and its mutant also increased up to a maximum at a concentration of 125 µM, culminating with a ∆Tm of approximately −5 °C (Table 4 and Figure 13). Thereby, the curves have a hyperbolic shape. Interestingly, the destabilization effect was abruptly lost in the compound concentration range from 15.63 µM (about −3 °C) to 7.81 µM (about 0.20 °C for USP7 and 0.45 °C for USP7asoc, respectively) and remained insignificant at further lower concentrations. Also noticeable are two peaks in the melting curves of USP7 and USP7asoc at a compound concentration of 7.81 µM and 15.63 µM. The first peak at lower temperature should represent the modified protein species, whereas the second peak corresponds to the unmodified protein species. Thus, the amount of compound is no longer sufficient to modify and destabilize the entire amount of protein. Here, the protein-to-compound ratios were approximately 1:2 (15.63 µM) and 1:1 (7.81 µM). This could indicate that the affinity of the ligand is too low to achieve sufficient target occupancy to induce a thermal shift at the lower concentration..
 |
Figure 13 First derivatives of the melting curves and resulting ∆Tm values of USP7 (A and B) and USP7asoc (C and D) (8 µM protein) with various concentrations of compound 6 after 24 h of incubation at 20 °C. |
The results of the concentration-dependent measurements are listed in Table 5 and Figure 14, with 24 h as the standard incubation time.
 |
Table 5 ∆Tm ± SD of USP7 and USP7asoc (8 µm Protein) Incubated With Different Concentrations of Compound 18 for 24 h at 20 °C |
 |
Figure 14 First derivatives of the melting curves and resulting ∆Tm values of USP7 (A and B) and USP7asoc (C and D) (8 µM protein) with various concentrations of compound 18 after 24 h of incubation at 20 °C. |
In similar fashion to compound 6, an increase in compound 18 concentration was in line with a greater degree of destabilization of USP7 with a maximum around −5 °C at 250 µM (Table 5 and Figure 14). In this case, the curve again resembled a hyperbolic shape. At a compound concentration of 15.63 µM (protein-to-compound ratio of approximately 1:2), there was no longer any effect on the melting temperature of USP7. Accordingly, at this point, the affinity is too low to obtain sufficient occupancy to induce USP7 destabilization.
The melting temperature curve of USP7asoc is U-shaped and shows impaired negative shifts in comparison to USP7. Consequently, compound 18 displayed the greatest destabilizing effect not at the maximum compound concentration (250 µM), but rather at a slightly lower one (125 µM). Compared to USP7, the value of the maximum temperature reduction was notably lower with USP7asoc (USP7asoc with −1.90 °C versus −5.35 °C USP7). This compound showed still a minor destabilizing effect (-0.55 °C) toward USP7asoc at a compound concentration of 15.63 µM. At even lower concentrations, the melting temperature was no longer affected.
These concentration-dependent DSF measurements are in line with the results from the MS experiment. Data of the concentration-dependent measurements for compound 30 are shown in Table 6 and Figure 15.
 |
Table 6 ∆Tm ± SD of USP7 and USP7asoc (8 µm Protein) Incubated With Different Concentrations of Compound 30 for 24 h at 20 °C |
 |
Figure 15 First derivatives of the melting curves and resulting ∆Tm values of USP7 (A and B) and USP7asoc (C and D) (8 µM protein) with various concentrations of compound 30 after 24 h of incubation at 20 °C. |
Compound 30 showed the greatest negative thermal shift (−4.50 °C) with USP7 at 125 µM. At 250 µM and lower concentrations than 62.5 µM, the destabilization was reduced, but still more than −3 °C. No further change in the melting temperature (−0.05 °C) was visible at a compound concentration of 7.8 µM. However, a concentration of 15.63 µM still caused a decrease of −3 °C with USP7 corresponding to a protein-to-compound ratio of nearly 1:2 suggesting that protein occupancy becomes almost maximal already at this relatively small excess.
In contrast, this compound destabilized USP7asoc almost constantly from 250 µM to 15.63 µM with about −4.40 °C (Table 6 and Figure 15). This level of destabilization only decreased slightly to −3.60 °C at 7.81 µM. This is still a strong destabilization of the mutant at a protein-to-compound ratio of almost 1:1. This indicates that compound 30 still engages the catalytic cysteine residue 223 at relatively low excess.
Table 7 and Figure 16 show the results of the pyrrolopyridine-derived chloroacetamide 121. It destabilized both USP7 and USP7asoc most at the highest compound concentration (250 µM). For USP7, the corresponding maximal destabilization was −2.40 °C and was reduced by half with half compound concentration to −1.45 °C. Below a concentration of 62.5 µM, the melting temperature of USP7 no longer changed.
 |
Table 7 ∆Tm ± SD of USP7 and USP7asoc (8 µm Protein) Incubated With Different Concentrations of Compound 121 for 24 h at 20 °C |
 |
Figure 16 First derivatives of the melting curves and resulting ∆Tm values of USP7 (A and B) and USP7asoc (C and D) (8 µM protein) with various concentrations of compound 121 after 24 h of incubation at 20 °C. |
In contrast, compound 121 still destabilized USP7asoc at 31.25 µM, namely by −0.55 °C. However, at 15.63 µM or lower decreases in the melting temperature were no longer visible. Overall, the value of the maximum decrease in melting point was slightly lower (−1.98 °C) for USP7asoc. The shape of the temperature shift curves is more linear for both proteins.
This compound caused a decrease in the melting temperatures for both proteins at lower compound concentrations. Based on this behavior, it can be assumed that the compound also binds to the catalytic Cys223 in USP7 which is in line with the observations from intact protein MS.
Take Home Message
In summary (Figure 17), 18 of the 129 tested compounds influenced the melting temperature of USP7 and USP7asoc with most of them decreasing it. These compounds were measured in an intact protein MS experiment to confirm a possible covalent binding with the catalytic Cys223 with the help of the USP7asoc and the USP7nc mutant. The MS experiment confirmed that mainly 7 out of the 18 compounds reacted in a covalent manner with USP7 and USP7asoc, featuring an SNAr warhead with fluoride or chloride as leaving group. Only one of them showed a SN2-based alkylation reaction. Four of these compounds were additionally tested in concentration-dependent DSF measurements with USP7 and USP7asoc to make any estimates about a possible affinity. The only compound which still caused a destabilization of the mutant USP7asoc at a protein-to-compound ratio of almost 1:1, was the chloronitropyridine bearing a 4-(4-benzylpiperazin-1-yl)pyridin-2-amine moiety (30). Due to these interesting results, we will aim at solving the crystal structure of its protein complex for this particular compound and the other three in the future.
 |
Figure 17 Overview and summary of the results of the USP7 compound screen. |
Conclusion
The screening and evaluation of a covalent kinase inhibitor library against USP7 revealed several interesting hits which may serve as valuable starting points for the development of potent and selective USP7 inhibitors. In total, 18 of the 129 tested compounds influenced the melting temperature of USP7 with most of them decreasing it consistently across all three incubation times. Only compounds 10, 34, 52, and 90 stabilized USP7 by about 1 °C. However, among those only compound 34 bound irreversibly to USP7 although it is worth noting that it only showed comparably little modification of USP7asoc in the MS spectrum. Consequently, it probably did not exclusively bind to the catalytic cysteine in USP7. Compound 90 may react in a reversible covalent way, making it difficult to prove the potential covalent reaction mechanism in an intact protein MS experiment.42,56,57 The spectrum of compound 52 revealed one small peak corresponding to modified USP7. The peak did not stand out significantly from the noise. Therefore, this compound was not classified as a covalent hit. However, it was the only acrylamide compound in the set which influenced the melting temperature of USP7, although this was the largest group tested in the library.
Covalent modification of USP7 was confirmed for six of the other compounds causing a decrease in melting temperature. Except for compound 121, they all reacted in a SNAr mechanism and showed clear shifts in the melting temperatures of up to −4.20 °C for USP7 and −6.0 °C for USP7asoc. Compound 5 was the only one which did not influence the melting temperature of either USP7 or USP7asoc but bound covalently to the catalytic cysteine of USP7. Unfortunately, and opposed to the commonly observed order of SNAr reactivity, its bromo analogue 7 was too reactive to obtain a proper MS spectrum.43
Compounds 6, 18, 30, and 121 all consistently induced destabilization of both USP7 and USP7asoc. Only 121 already destabilized USP7 after 30 min of incubation at 20 °C whereas the other three only changed the melting temperature after 4 and 24 hours, respectively, indicating slower on-target reaction kinetics. All four compounds mainly bound one time to USP7asoc, hence, only the catalytic Cys223 was modified. In these compounds, the leaving group was a chlorine atom neighboring the pyridine nitrogen atom in a the 3-nitropyridine ring, while only compound 6 contained a fluorine atom as the leaving group instead.
However, only the spectra of compound 18 and 30 showed one single signal representing the mono-arylated protein without any additional peaks and, hence, no unmodified protein. In addition, compound 18 shifted the melting temperature of USP7 and USP7asoc to a greater extent than compound 30. Moreover, 18 decreased the Tm at a compound concentration of 31.25 µM, whereas compound 30 still showed an effect at a compound concentration of 15.63 µM. Compound 30 was the only compound which still showed a strong destabilization of the mutant USP7asoc at a protein-to-compound ratio of almost 1:1. This fact makes the compound worth mentioning and it should definitely be investigated further for inhibitory effects.
The experiments given above lead to the conclusion that the compounds featuring an SNAr warhead were more likely to react covalently with USP7 than other warheads. Furthermore, taking into account the results of both experiments, a correlation between DSF shifts and a possible covalent binding with USP7 was visible. However, not every compound bound to the catalytic cysteine of USP7, which also influenced the melting temperature of the latter. All in all, this screen also showed that compounds with more elaborate non-covalent recognition motifs decreased the melting temperature of USP7 as well upon covalent binding to the catalytic Cys223 which we have already seen with our CovLib.
These interesting screening results will serve as starting points for early-stage drug discovery, facilitated by hit optimization and structure elucidation to determine the binding mode and the inhibitory effect on USP7.
Abbreviations
CD, catalytic domain; Cys223, cysteine 223; DMSO, dimethyl sulfoxide; DSF, differential scanning fluorimetry; FBDD, fragment-based drug discovery; HPLC, high performance liquid chromatography; MDM2, mouse double minute 2 homolog; n.a., not available; PROTAC, proteolysis targeting chimera; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; SNAr, nucleophilic aromatic substitution; Tm, melting temperature; UHPLC-ESI-MS, ultra-high performance liquid chromatography electrospray ionization mass spectrometry; USP7, ubiquitin-specific protease 7.
Author Contributions
All authors made a significant contribution to the work reported: F.M.B. envisioned the research. L.N.E. and F.M.B. conceptualized the experiments and designed the study. L.N.E. prepared the proteins by heterologous expression and performed the DSF studies. S.J., B.M., C.K., and M.L. performed and analyzed the UHPLC-ESI-MS experiments. V.R.W., S.G., X.J.L., R.A.M.S. and N.J.S. synthesized the compounds. M.G. conceived and compiled the covalent library of kinase inhibitors and supervised the synthesis and characterization of these compounds. L.N.E. conducted data analysis and reprocessing of the experimental data. L.N.E. and F.M.B. wrote the manuscript. All authors have at least substantially revised or critically reviewed the manuscript. All authors have agreed on the journal to which the article has been submitted. All authors agreed on all versions of the article at any stage of the submission, revision, publication and proofing process, particularly including the final version accepted for publication. All authors agree to take responsibility and be accountable for the contents of the article.
Funding
X.J.L., R.A.M.S. and M.G. gratefully acknowledge the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s excellence strategy – EXC 2180 – 390900677 [“Image Guided and Functionally Instructed Tumor Therapies” (iFIT)] for funding. M.G. and S.G. thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 511101075, and the Postdoctoral Fellowship Program of the Baden-Württemberg Stiftung for financial support. Funding by the Deutsche Krebshilfe (German Cancer Aid) – Dr. Mildred Scheel Stiftung für Krebsforschung is gratefully acknowledged by R.A.M.S. and M.G.
Disclosure
Ricardo A M Serafim is currently affiliated with Department of Organic and Pharmaceutical Chemistry, School of Engineering, Institut Químic de Sarrià (IQS), Universitat Ramon Llull (URL), 08017 Barcelona, Spain. The authors report no conflicts of interest in this work.
References
1. Mevissen TET, Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu Rev Biochem. 2017;86:159–192. doi:10.1146/annurev-biochem-061516-044916
2. Davis MI, Simeonov A. Ubiquitin-specific proteases as druggable targets. Drug Target Rev. 2015;2(3):60–64.
3. Murgai A, Sosič I, Gobec M, et al. Targeting the deubiquitinase USP7 for degradation with PROTACs. Chem Commun. 2022;58(63):8858–8861. doi:10.1039/d2cc02094g
4. Abdul Rehman SA, Kristariyanto YA, Choi SY, et al. MINDY-1 is a member of an evolutionarily conserved and structurally distinct new family of deubiquitinating enzymes. Mol Cell. 2016;63(1):146–155. doi:10.1016/j.molcel.2016.05.009
5. Kwasna D, Abdul Rehman SA, Natarajan J, et al. Discovery and characterization of ZUFSP/ZUP1, a distinct deubiquitinase class important for genome stability. Mol Cell. 2018;70(1):150–164.e6. doi:10.1016/j.molcel.2018.02.023
6. Tavana O, Gu W. Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J Mol Cell Biol. 2017;9(1):45–52. doi:10.1093/jmcb/mjw049
7. Leger PR, Hu DX, Biannic B, et al. Discovery of potent, selective, and orally bioavailable inhibitors of USP7 with in vivo antitumor activity. J Med Chem. 2020;63(10):5398–5420. doi:10.1021/acs.jmedchem.0c00245
8. Wu J, Kumar S, Wang F, et al. Chemical approaches to intervening in ubiquitin specific protease 7 (USP7) function for oncology and immune oncology therapies. J Med Chem. 2018;61(2):422–443. doi:10.1021/acs.jmedchem.7b00498
9. Lee JT, Gu W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010;17(1):86–92. doi:10.1038/cdd.2009.77
10. Song MS, Salmena L, Carracedo A, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP–PML network. Nature. 2008;455(7214):813–817. doi:10.1038/nature07290
11. Zhang L, Wang H, Tian L, Li H. Expression of USP7 and MARCH7 is correlated with poor prognosis in epithelial ovarian cancer. Tohoku J Exp Med. 2016;239(3):165–175. doi:10.1620/tjem.239.165
12. Gavory G, O’Dowd CR, Helm MD, et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol. 2018;14(2):118–125. doi:10.1038/nchembio.2528
13. Futran AS, Lu T, Amberg-Johnson K, et al. Ubiquitin-specific protease 7 inhibitors reveal a differentiated mechanism of p53-driven anti-cancer activity. iScience. 2024;27(5):109693. doi:10.1016/j.isci.2024.109693
14. Pfoh R, Lacdao IK, Saridakis V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr Relat Cancer. 2015;22(1):T35–54. doi:10.1530/erc-14-0516
15. Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60(1–2):61–68. doi:10.1007/s12013-011-9185-5
16. Kim RQ, Sixma TK. Regulation of USP7: a high incidence of E3 complexes. J Mol Biol. 2017;429(22):3395–3408. doi:10.1016/j.jmb.2017.05.028
17. Qi SM, Cheng G, Cheng XD, et al. Targeting USP7-mediated deubiquitination of MDM2/MDMX-p53 pathway for cancer therapy: are we there yet? Front Cell Dev Biol. 2020;8:233. doi:10.3389/fcell.2020.00233
18. Klett T, Schwer M, Ernst LN, et al. Evaluation of a covalent library of diverse warheads (CovLib) binding to JNK3, USP7, or p53. Drug Des Devel Ther;2024. 2653–2679. doi:10.2147/DDDT.S466829
19. Klett T, Stahlecker J, Jaag S, et al. Covalent fragments acting as tyrosine mimics for mutant p53-Y220C rescue by nucleophilic aromatic substitution. ACS Pharmacol Transl Sci. 2024;7(12):3984–3999. doi:10.1021/acsptsci.4c00414
20. Stahlecker J, Klett T, Schwer M, et al. Revisiting a challenging p53 binding site: a diversity-optimized HEFLib reveals diverse binding modes in T-p53C-Y220C. RSC Med Chem. 2022;13(12):1575–1586. doi:10.1039/d2md00246a
21. Wilcken R, Wang G, Boeckler FM, Fersht AR. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc Natl Acad Sci U S A. 2012;109(34):13584–13589. doi:10.1073/pnas.1211550109
22. Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A. 2008;105(30):10360–10365. doi:10.1073/pnas.0805326105
23. Wilcken R, Liu X, Zimmermann MO, et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J Am Chem Soc. 2012;134(15):6810–6818. doi:10.1021/ja301056a
24. Dumble M, Xu L, Dominique R, et al. Abstract LB006: PC14586: the first orally bioavailable small molecule reactivator of Y220C mutant p53 in clinical development. Cancer Res. 2021;81(13_Supplement):LB006. doi:10.1158/1538-7445.Am2021-lb006
25. Dumbrava EE, Johnson ML, Tolcher AW, et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J Clin Oncol. 2022;40(16_suppl):3003. doi:10.1200/JCO.2022.40.16_suppl.3003
26. US National Library of Medicinal Clinical Trials.gov. Available from: https://clinicaltrials.gov/study/NCT04585750.
27. Chauhan D, Tian Z, Nicholson B, et al. Deubiquitylating enzyme USP-7, a novel therapeutic target in multiple myeloma. Blood. 2009;114(22):610. doi:10.1182/blood.V114.22.610.610
28. Li X, Kong L, Yang Q, et al. Parthenolide inhibits ubiquitin-specific peptidase 7 (USP7), Wnt signaling, and colorectal cancer cell growth. J Biol Chem. 2020;295(11):3576–3589. doi:10.1074/jbc.RA119.011396
29. Kategaya L, Di Lello P, Rougé L, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550(7677):534–538. doi:10.1038/nature24006
30. Zadi S, Javaid S, Atia Tul W, et al. Repurposing of US-FDA-approved drugs as negative modulators of ubiquitin specific protease-7 (USP7). Heliyon. 2024;10(5):e26345. doi:10.1016/j.heliyon.2024.e26345
31. Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550(7677):481–486. doi:10.1038/nature24451
32. Pei Y, Fu J, Shi Y, et al. Discovery of a potent and selective degrader for USP7. Angew Chem Int Ed Engl. 2022;61(33):e202204395. doi:10.1002/anie.202204395
33. Oliveira RI, Guedes RA, Salvador JAR. Highlights in USP7 inhibitors for cancer treatment. Front Chem. 2022;10:1005727. doi:10.3389/fchem.2022.1005727
34. Hu M, Li P, Li M, et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111(7):1041–1054. doi:10.1016/S0092-8674(02)01199-6
35. Kaar JL, Basse N, Joerger AC, Stephens E, Rutherford TJ, Fersht AR. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010;19(12):2267–2278. doi:10.1002/pro.507
36. Gerstenecker S, Haarer L, Schröder M, et al. Discovery of a potent and highly isoform-selective inhibitor of the neglected ribosomal protein S6 kinase beta 2 (S6K2). Cancers. 2021;13(20):5133. doi:10.3390/cancers13205133
37. Schwarz M, Kurkunov M, Wittlinger F, et al. Development of highly potent and selective covalent FGFR4 inhibitors based on S(N)Ar electrophiles. J Med Chem. 2024;67(8):6549–6569. doi:10.1021/acs.jmedchem.3c02483
38. Wydra V, Gerstenecker S, Schollmeyer D, et al. N-(6-Chloro-3-nitropyridin-2-yl)-5-(1-methyl-1H-pyrazol-4-yl)isoquinolin-3-amine. Molbank. 2021;2021(1):M1181. doi:10.3390/M1181
39. Serafim RAM, da Silva Santiago A, Schwalm MP, et al. Development of the first covalent monopolar spindle kinase 1 (MPS1/TTK) inhibitor. J Med Chem. 2022;65(4):3173–3192. doi:10.1021/acs.jmedchem.1c01165
40. Forster M, Liang XJ, Schröder M, et al. Discovery of a novel class of covalent dual inhibitors targeting the protein kinases BMX and BTK. Int J Mol Sci. 2020;21(23):9269. doi:10.3390/ijms21239269
41. Wang G, Seidler NJ, Röhm S, et al. Probing the protein kinases’ cysteinome by covalent fragments. Angew Chem Int Ed Engl. 2024:e202419736. doi:10.1002/anie.202419736.
42. Hillebrand L, Liang XJ, Serafim RAM, Gehringer M. Emerging and re-emerging warheads for targeted covalent inhibitors: an update. J Med Chem. 2024;67(10):7668–7758. doi:10.1021/acs.jmedchem.3c01825
43. Gehringer M, Laufer SA. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62(12):5673–5724. doi:10.1021/acs.jmedchem.8b01153
44. Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2(9):2212–2221. doi:10.1038/nprot.2007.321
45. Gao K, Oerlemans R, Groves MR. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys Rev. 2020;12(1):85–104. doi:10.1007/s12551-020-00619-2
46. Huynh K, Partch CL. Analysis of protein stability and ligand interactions by thermal shift assay. Curr Protoc Protein Sci. 2015;79:28.9.1–28.9.14. doi:10.1002/0471140864.ps2809s79
47. Javaid S, Zadi S, Awais M, et al. Identification of new leads against ubiquitin specific protease-7 (USP7): a step towards the potential treatment of cancers. 10.1039/D4RA06813K. RSC Adv. 2024;14(45):33080–33093. doi:10.1039/D4RA06813K
48. Baud MGJ, Bauer MR, Verduci L, et al. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur J Med Chem. 2018;152:101–114. doi:10.1016/j.ejmech.2018.04.035
49. Finlay MR, Anderton M, Ashton S, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57(20):8249–8267. doi:10.1021/jm500973a
50. Zhao Z, Bourne PE. Progress with covalent small-molecule kinase inhibitors. Drug Discov Today. 2018;23(3):727–735. doi:10.1016/j.drudis.2018.01.035
51. Lanman BA, Allen JR, Allen JG, et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J Med Chem. 2020;63(1):52–65. doi:10.1021/acs.jmedchem.9b01180
52. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi:10.1038/nature12796
53. Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20(9):1061–1073. doi:10.1016/j.drudis.2015.05.005
54. Lonsdale R, Ward RA. Structure-based design of targeted covalent inhibitors. Chem Soc Rev. 2018;47(11):3816–3830. doi:10.1039/c7cs00220c
55. Barf T, Kaptein A. Irreversible protein kinase inhibitors: balancing the benefits and risks. J Med Chem. 2012;55(14):6243–6262. doi:10.1021/jm3003203
56. Serafimova IM, Pufall MA, Krishnan S, et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol. 2012;8(5):471–476. doi:10.1038/nchembio.925
57. Jackson PA, Widen JC, Harki DA, Brummond KM. Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-michael addition reactions. J Med Chem. 2017;60(3):839–885. doi:10.1021/acs.jmedchem.6b00788
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.