")
Back to Journals » Drug Design, Development and Therapy » Volume 19
SHR-1916: A Novel PEGylated Interleukin-2 Analogue with Altered Cellular Selectivity and Improved Pharmacokinetic Profiles for Cancer Immunotherapy
Authors Kong X , Lin Y, Ouyang C, Chen H, Gao X
Received 21 October 2024
Accepted for publication 6 February 2025
Published 24 February 2025 Volume 2025:19 Pages 1251—1270
DOI https://doi.org/10.2147/DDDT.S493011
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Xianglin Kong,1,2 Yuan Lin,2 Chao Ouyang,2 Hao Chen,2 Xiangdong Gao1
1Jiangsu Key Laboratory of Druggability of Biopharmaceuticals and State Key Laboratory of Natural Medicines, School of Life Science and Technology, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China; 2Jiangsu Hengrui Pharmaceuticals Co., Ltd, Lianyungang, 222047, People’s Republic of China
Correspondence: Xiangdong Gao, Jiangsu Key Laboratory of Druggability of Biopharmaceuticals and State Key Laboratory of Natural Medicines, School of Life Science and Technology, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China, Email [email protected]
Purpose: Human interleukin-2 (IL-2) stimulates the differentiation and expansion of diverse immune cells dose-dependently. As an immunotherapy agent to treat metastatic cancers, IL-2 has been used in clinical practice and has demonstrated clear antitumor effects; however, its short half-life, the risk of capillary leak syndrome, and the unintended activation of immunosuppressive Treg cells hinder its clinical application. To address these challenges, a novel PEGylated interleukin-2 analogue, SHR-1916, was designed. Its cellular selectivity, efficacy, and improved pharmacokinetic profiles were investigated.
Methods: The binding affinities were characterized by surface plasmon resonance (SPR) in vitro. Subsequently, the stimulatory properties were investigated in a murine cell line (CTLL-2), a human cell line (M07e), and human peripheral blood mononuclear cells (PBMCs). To assess the anti-tumor efficacy, a CT-26 colon carcinoma syngeneic model in BALB/c mice and a A375 human melanoma xenograft model using PBMC humanized NCG mice were used in vivo. Moreover, the pharmacokinetic behavior following a single intravenous or subcutaneous dose was evaluated in Sprague-Dawley rats.
Results: SHR-1916 abolished binding to its receptor IL-2Rα, as evidenced by SPR assays, and exerted its activity mainly through binding to IL-2Rβγ, as confirmed by CTLL-2 and M07e cell proliferation assays. In contrast to IL-2, SHR-1916 exhibited a more biased activation of CD8+ T and NK cells compared to Treg cells and stimulated an increase in IFNγ secretion in PBMCs dose-dependently without triggering the release of other potential side effect-associated cytokines. In CT26 colon carcinoma and A375 melanoma models, SHR-1916 significantly reduced the tumor burden. Pharmacokinetic results showed that SHR-1916 had a significantly prolonged half-life in rats.
Conclusion: SHR-1916 exhibited excellent cellular selectivity, anti-tumor efficacies, and improved pharmacokinetics. It has the potential to serve as a novel immunotherapeutic agent designed to enhance IL-2’s immune-stimulating activities and promote its tolerability while reducing the immunoregulatory function of Treg cells.
Keywords: interleukin-2 mutein, long-acting, receptor-binding bias, antitumor effect
Introduction
Human interleukin-2 (IL-2) was initially characterized as a cytokine for its capacity to promote T cell proliferation.1 It was identified as a T cell growth factor that enabled continuous propagation of human T lymphocytes from bone marrow in suspension culture.2 It is a critical growth factor to promote proliferation of CD8+, CD4+, and NK cells.3
IL-2 stimulates the differentiation and proliferation of various immune cell types. It is able to stimulate memory and effector T cell differentiation and expansion at high doses, while boosting the function and survival of regulatory T cells (Treg) at low doses.4 The dose-dependent effect can be linked to the structural and compositional features of its receptor (IL-2R). IL-2R comprises three subunits: IL-2Rα (CD25), IL-2Rβ (CD122), and IL-2Rγ (CD132).5 Different cell types and their activation status may exhibit different combinations of the subunits. IL-2Rαβγ exhibits high affinities to IL-2, while IL-2Rα and IL-2Rβγ exhibit low and intermediate affinities.6 Treg cells, constitutively expressing the high-affinity IL-2Rαβγ, are more sensitive to IL-2 (Kd ≈ 10−11 M) than other effector cells, such as NK and memory CD8+ T cells, which express the intermediate-affinity receptor IL-2Rβγ (Kd ≈ 10−9 M).3
Aldesleukin (Proleukin®), a high-dose interleukin-2, was approved in 1992 for metastatic renal cell carcinoma by the FDA, followed by its approval for metastatic melanoma in 1998. However, aldesleukin can only be applied in patients exhibiting intact cardiac and pulmonary function, owing to the risk of capillary leak syndrome (CLS), which is defined as a pathological condition characterized by increased permeability of blood vessels, leading to the escape of fluid and proteins from vascular compartment into surrounding tissues.7 Recent research indicates that IL-2’s direct interaction with CD25+ endothelial cells predominantly mediates the CLS.8 This observation suggests that preserving IL‑2’s interaction with CD122 while selectively inhibiting its interaction with CD25, may reduce the onset of CLS.9
IL-2’s short half-life is another limitation, resulting from its relatively small molecular weight and rapid excretion through the kidneys. When administered intravenously, IL-2 undergoes rapid elimination, exhibiting a 6–12 minute initial elimination half-life and a 40–80 minute terminal elimination half-life.10 Therefore, it must be administered frequently, significantly affecting overall treatment adherence among patients and limiting its clinical use.11,12
Improved understanding of the structure and function of IL-2 and its receptor complex has led to the development of several strategies to enhance IL-2 therapeutic efficacy and reduce toxicity. These strategies include: IL-2/monoclonal antibody complexes (eg, IL-2/S4B6, which alters receptor selectivity by masking the IL-2 binding site on IL-2Rα (CD25));13 IL-2 mutants (eg, no-α mutein,14 MK-6,15 FSD13,16 and H917 mutants, which alter receptor selectivity through key amino acid mutations); IL-2 fusion proteins (eg, EMD 521873,18 GA504/GA501,19,20 and Hu14.18-IL-2,21,22 which enhance targeting and half-life through fusion with specific antibodies; or, as in ALKS 4230,23 which alters receptor selectivity through fusion with the extracellular domain of IL-2Rα); and PEGylated IL-2 proteins (eg, NKTR-214,24 THOR-707,25 and TransCon IL-2 β/γ,26 which extend half-life and block IL-2 binding to IL-2Rα through PEGylation of key amino acids or regions).
In this study, we integrated IL-2 mutant and PEGylation strategies and developed a PEGylated IL-2 mutein (SHR-1916). To reduce IL-2’s binding ability to Treg and CD25+ cells, we introduced key residue mutations in SHR-1916, and to prolong its half-life, we selected a 20 kDa polyethylene glycol (PEG) to covalently conjugate to it. The design and construction of SHR-1916 are based on principles disclosed in PCT Patent WO 2020/125743.This study aimed to evaluate SHR-1916 as a novel candidate for a long-acting interleukin-2 analogue. In vitro, we characterized the binding affinities and stimulatory properties in murine and human cell lines, and in PBMCs. In vivo, we evaluated the anti-tumor efficacies in mouse tumor models and assessed pharmacokinetic profiles in rats.
Material and Methods
Binding Affinity Measurements
A Biacore® 8K instrument (GE Healthcare) was utilized to measure the receptor binding affinities. A sensor chip (CM5, GE Healthcare, 29149603) was utilized and coated with 15 μg/mL anti-His antibody until it achieved 12,000 RUs following a standard amine coupling procedure (Biacore® protocol). The human CD25/IL-2Rα with a His-tag (Sino Biological, 10165-H08H) or CD122/IL-2Rβ with a His-tag (Sino Biological, 10696-H08B) was then immobilized. Analytes were prepared in eight 2-fold serial dilutions, starting at concentrations of 2000 nM or 125 nM. Each analyte’s exposure time to the receptor-modified chip was 30 seconds, and the dissociation was tracked for 30 seconds. After each cycle, 10 mmol glycine-HCl (pH 1.5) was used to remove residual samples on the IL-2R surfaces to return to the baseline level. To determine the KD values of IL-2Rα, a 1:1 Langmuir binding model was applied using Biacore 8K Evaluation Software, and a Steady State Affinity model was utilized to determine the KD values of IL-2Rβ.
In vitro Cell Proliferation Assay
The cytokine activities of IL-2 and SHR-1916 were assessed using CTLL-2 (ATCC, TIB-214) and M07e (Chinese Academy of Medical Sciences, Beijing, China, 3111C0001CCC000112) cell lines, which were cytokine growth-dependent. A total of 3×104 cells (50 μL) per well were seeded in 96-well plates, and 50 μL of the analytes with ten 3-fold serial dilutions were added to each well. For CTLL-2 cells, the initial concentration of SHR-1916 was 8000 ng/mL, and the initial concentration of IL-2 was 15 ng/mL. The cells were incubated at 37°C and 5% CO2 for 18 to 24 hours. For M07e cells, the initial concentration of SHR-1916 was 108000 ng/mL, and the initial concentration of IL-2 was 5400 ng/mL. The cells were incubated at 37°C and 5% CO2 for 44 to 48 hours. After incubation, 20 μL of MTT solution (0.5% w/v MTT dissolved in PBS) was added to each well, and the plates were incubated at 37°C and 5% CO2 for 4 to 6 hours. Subsequently, 150 μL of lysis buffer (15% w/v SDS dissolved in purified water) was added to each well, and the plates were kept at 37°C and 5% CO2 for 18 to 24 hours. The mixture was then mixed by shaking for 10 minutes, followed by measurement of absorbance at 570 nm using a microplate reader (Synergy HT, BioTek), with 630 nm as the reference wavelength. GraphPad Prism software was used to plot the proliferation curves and calculate the half-maximal effective concentration (EC50). The curve-fitting method was log(concentration of analytes) vs corresponding absorbance value (OD570-OD630).
In vitro pSTAT5 Assay in PBMCs
PBMCs were isolated from three normal, healthy volunteers by Ficoll-Paque (GE Healthcare). 1×106 cells (80 μL) per well were seeded in 96-well plates, and 20 μL of the analytes with nine 10-fold serial dilutions were added to each well, resulting in final concentrations ranging from 0 to 1000 nM. Incubate the cells at 37°C and 5% CO2 for 15 minutes. Subsequently, 100 μL of preheated Cytofix Fixation Buffer (BD Biosciences, 554655) at 37°C was added per well, mixed, and fixed at 37°C and 5% CO2 for 15 minutes, followed by an additional 15 minutes at 4°C. After centrifugation at 500 g to pellet the cells, they were washed once using the FACS buffer. To resuspend the cells, add 150 μL of Phosflow Perm Buffer III (BD Biosciences, 558050) per well, followed by overnight incubation at −20°C. Then, centrifugation at 500 g to pellet the cells, wash the cells with FACS buffer twice and resuspend the cells with 100 μL of Fc block (BD Biosciences, 564220, 200-fold dilution) per well, followed by 20 minutes of incubation at 4°C. The cells were divided equally into two parts: one for T cell staining and the other for NK cell staining. The cells were collected by centrifugation at 500 g and resuspended with the prepared staining antibody (50 µL per well), followed by incubation at 4°C for 60 minutes. For flow cytometry analysis, wash the cells twice and resuspend them with FACS buffer (150 µL per well).
The staining antibodies included the following: anti-human CD3 APC-Cy7 SK7 (1:100, BD Biosciences, 557832), CD16 FITC (1:40, BioLegend, 302006), CD8 PerCP-Cy5.5 SK1 (1:100, BD Biosciences, 565310), CD4 FITC RPA-T4 (1:40, BD Biosciences, 555346), CD25 PE M-A251 (1:20, BD Biosciences, 555432), CD56 PE (1:40, BioLegend, 318306), Alexa Fluor®647 anti-human FOXP3 (1:40, BioLegend, 320214), and phosphoStat5 (pY694) BV421 47 (1:20, BD Biosciences, 562984). The FACScelesta system (BD Biosciences) and FlowJo software were utilized for collecting and analyzing the data. NK cells are identified as CD3−CD16+CD56+, CD8+ T cells are identified as CD3+CD8+, and Treg cells are identified as CD3+CD4+CD25+Foxp3+. Analyze the median fluorescence intensities (MFI) of pSTAT5 in all subsets and calculate the EC50 values with GraphPad Prism software for each analyte.
PBMC Cytokine Secretion Assay
A total of 1×105 cells (90 μL) per well were seeded in each well of the 96-well plates, and SHR-1916 was subsequently added to each well (10 μL), resulting in final concentrations of 0.15, 0.5, and 1.5 μg/mL. Lipopolysaccharides (Sigma, L2880) and CD3 monoclonal antibody (eBioscience, 16-0037-85) were used as positive controls with final concentrations of 50 ng/mL and 1 μg/mL, respectively. RPMI-1640 medium (GE Healthcare, SH30809.01) and 1.69 μg/mL of PEG solution (JenKem Technology, 20 kDa) served as negative controls. The mixture of cells and analytes was cultured for 24 hours at 37°C and 5% CO2 and centrifuged for 10 minutes at 1000 g to remove the cells. The supernatants were collected to analyze the concentrations of IFNγ, IL-1β, IL-4, IL-8, IL-10, IL-12p70, IL-13, and TNFα using the V-PLEX Proinflammatory Panel 1 (Human) Kit (Meso Scale Diagnostics, K15049D-2). IL-5 and IL-6 concentrations were determined with a separate ELISA kit (Neobioscience, EHC148, EHC007).
In vivo Mouse Tumor Model of CT-26 Colon Carcinoma
Female BALB/c mice (6–8 weeks old, 18–24 g) were obtained from Jihui Laboratory Animal Care Company (Shanghai, China). The CT-26 cells were obtained from Phenotek Biotechnology Company (Shanghai, China). Following a one-week acclimatization period, the mice were weighed and injected subcutaneously with 5×105 CT-26 cells. Daily monitoring was performed, with body weight and tumor volume recorded weekly. According to the body weight and tumor volume, the mice were randomized to four groups (10 per group) once the average tumor volume reached 70–80 mm3: the vehicle group (water for injection), the low-dose group (1 mg/kg SHR-1916), the medium-dose group (3 mg/kg SHR-1916), and the high-dose group (9 mg/kg SHR-1916). The mice received three injections on days 0, 4, and 8. Record the body weights and tumor volumes three times a week until day 13. Measure the tumor volumes by a vernier caliper and calculate them as follows: π/6 × a × b2, where a and b represent the longest and shortest diameters of the tumor, respectively. Tumor growth inhibition (TGI%) was calculated as follows: TGI% = [1 - (T-T0) / (C-C0)] × 100%, where T0 and T represent the average tumor volume before and after treatment in the test group, and C0 and C represent the average tumor volume before and after treatment in the control group, respectively.
In vivo Mouse Tumor Model of A375 Melanoma Cells
Female NCG mice (6–8 weeks old, 18–22 g) were obtained from GemPharmatech Company (Nanjing, China) and used for tumor studies in the A375 melanoma model. A375 human melanoma cells were obtained from Lide Biotech Company (Shanghai, China) and cultured in DMEM (Gibco, 11965–092) supplemented with 10% FBS (Gibco, 10099–141). The cells were harvested in the exponential phase, treated with mitomycin C for 2 hours, washed with PBS three times, and co-cultured with PBMCs from a healthy donor in RPMI 1640 medium (Gibco, 11875–093) containing 10% FBS and IL-2 for 6 days. The fresh A375 cells and co-cultured PBMCs were collected and suspended in a 1:1 volume mixture of HBSS (BasalMedia, B420KJ) and Matrigel® (Corning, 354234). Thirty-six female NCG mice were randomized to four groups (9 per group) according to body weight, receiving a subcutaneous injection with 200 μL of cell suspension containing 4×106 A375 cells and 8×105 PBMCs in the right flank. The treatment groups included a vehicle (water for injection), a low-dose group (0.04 mg/kg), a medium-dose group (0.2 mg/kg), and a high-dose group (1 mg/kg). SHR-1916 was injected subcutaneously six times on days 0, 3, 6, 9, 12, and 15 in the neck. Measure the body weights two times a week. All mice were euthanized 21 days after administration, and the tumors were collected, weighed, and photographed. Tumor growth inhibition (TGI%) was calculated as follows: TGI% = (1 - T/C) × 100%, where T and C represent the average tumor weight of the test and control groups, respectively.
Pharmacokinetic Study in Rats
The pharmacokinetic behavior of SHR-1916 following a single intravenous (i.v.) or a single subcutaneous (s.c.) dose was evaluated in Sprague-Dawley rats. Six to eight-week-old rats (165–215 g) were obtained from Vital River Laboratory Animal Technology Company (Beijing, China) and acclimatized for 5 days. Twenty-four rats were randomized to four groups according to body weight and gender, with three males and three females per group. Rats from each group received just one injection of 1.5 mg/kg SHR-1916 intravenously (via tail vein) or 1.5 mg/kg, 3 mg/kg, and 6 mg/kg of SHR-1916 subcutaneously (in the dorsal neck region). Blood samples were collected at the following times: for the intravenous dose: 0, 1, 30 minutes, and 2, 4, 8, 24, 48, 72, 120, and 168 hours after injection; for the subcutaneous doses: 0, 0.5, 2, 4, 8, 24, 48, 72, 120, and 168 hours after injection. Plasma samples were analyzed with an IL-2 sandwich-type ELISA kit (BD Biosciences, 555190). The standard curve was generated using SHR-1916 as the standard. The method was validated for precision, accuracy, selectivity, dilution linearity, and the absence of hook effect, all meeting acceptable criteria.The lower limit of quantification was validated to be 3.13 ng/mL, and the standard curve range was 3.13–200 ng/mL. Phoenix WinNonlin 6.4 software (Pharsight) was used to calculate the pharmacokinetic parameters with the non-compartmental model. Calculate the bioavailability as follows: F = (average AUClast of 1.5 mg/kg i.v.) / (average AUClast of 1.5 mg/kg s.c.).
Statistical Analysis
Data were analyzed using SPSS software. One-way analysis of variance (ANOVA) was employed to assess the differences among groups. Prior to ANOVA, Levene’s test was conducted to evaluate the homogeneity of variances. If the variances were homogeneous (p > 0.05), post-hoc comparisons were performed using the Least Significant Difference (LSD) test. If the variances were heterogeneous (p ≤ 0.05), Dunnett’s T3 test was used for post-hoc comparisons. Differences were considered statistically significant at p < 0.05.
Results
Design of PEGylated IL-2
Based on the crystal structure of the human IL-2/IL-2R complex,27,28 we selected Phe42 and Leu72 as the key residues involved in IL-2’s interaction with IL-2Rα. Phe42 was mutated to Ala (F42A), and Leu72 was mutated to Gly (L72G) in order to reduce the affinity to high-affinity IL-2Rαβγ. A free Cys at position 125 was substituted with Ala (C125A), as Cys125 does not participate in a disulfide bond and is not required for receptor binding.29 The C125A mutation is also present in the NMPA-approved product “Recombinant Human Interleukin-2 (125Ala) Injection”, which is manufactured by SL Pharm. Additionally, during the stability study, we observed that residues Asn26, Asn29, and Asn71 in IL-2 are particularly susceptible to deamidation and degradation (Figure 1). Based on the crystal structure analysis, these three residues were found not to interact with IL-2Rβγ.28 Therefore, we introduced the N26Q/N29S/N71Q mutations to enhance the chemical stability of IL-2. Furthermore, to prolong the elimination half-life in vivo, we employed a site-specific modification to the N-terminal amine group of IL-2 with a 20 kDa PEG. In conclusion, SHR-1916 is designed as an IL-2Rβ-binding biased compound containing six mutations (N26Q/N29S/F42A/N71Q/L72G/C125A) and a 20 kDa PEG at the N-terminus (Figure 2).
 |
Figure 1 Deamidation stability study of Asn26, Asn29, and Asn71 residues in IL-2. IL-2 refers to a wild-type sequence and contains the C125A mutation. IL-2 (F42A/L72G) refers to a mutant variant of IL-2, which contains the F42A, L72G, and C125A mutations. IL-2 (SHR-1916) refers to a mutant variant of IL-2 used in SHR-1916, which includes the F42A, L72G, C125A, N26Q, N29S, and N71Q mutations. After 10 and 30 days of storage at 40°C, the samples were subjected to trypsin digestion, and the deamidation peptide segments were monitored by LC-MS. (A) The increased levels of deamidation observed in the Thr10-Lys32 peptide segment during the stability study. This segment comprises six potential deamidation sites: Q11, Q13, Q22, N26, N29, and N30. The presence of N26Q and N29S mutations significantly reduces deamidation levels in this Thr10-Lys32 peptide segment. (B) The increased levels of deamidation observed in the His55-Lys76 peptide segment during the stability study. This segment contains three potential deamidation sites: Q57, N71, and Q74. The presence of the N71Q mutation significantly reduces deamidation levels in this His55-Lys76 peptide segment. |
 |
Figure 2 The design and receptor binding mechanism of SHR-1916. |
SHR-1916 Exhibits Significant IL-2Rβ-Binding Bias
In order to confirm that the modifications on IL-2 meet our design goals, we first measured the binding affinities of SHR-1916 to IL-2Rα and IL-2Rβ by SPR. As shown in Figure 3 and Table 1, IL-2 binds to IL-2Rα and IL-2Rβ with affinities of 1.29×10−8 M and 2.80×10−7 M, respectively, consistent with previous reports.24,30,31 SHR-1916 maintained its binding ability to IL-2Rβ (KD = 1.71×10−6 M), while exhibiting no or very weak binding to IL-2Rα. These findings suggest that SHR-1916 exhibits a significant IL-2Rβ-binding bias compared to IL-2 and has reduced binding affinity to IL-2Rα.
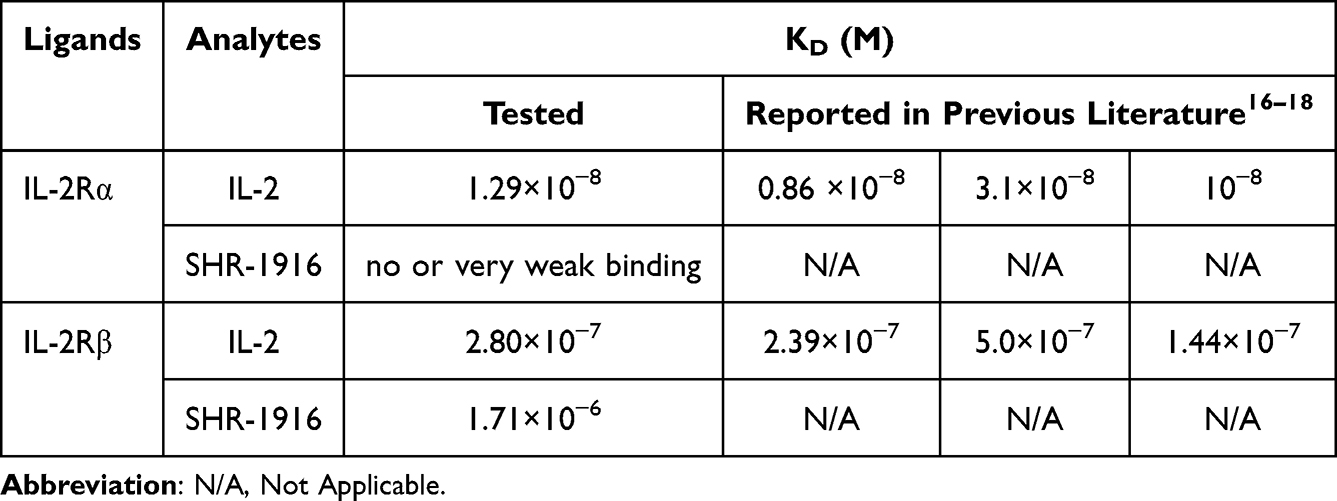 |
Table 1 Affinity Analysis of IL-2 and SHR-1916 to IL-2Rα and IL-2Rβ |
 |
Figure 3 Surface plasmon resonance analysis of IL-2 and SHR-1916 binding to IL-2Rα and IL-2Rβ. (A) IL-2 binds to IL-2Rα. (B) SHR-1916 binds to IL-2Rα. (C) IL-2 binds to IL-2Rβ. (D) SHR-1916 binds to IL-2Rβ. |
SHR-1916 Exhibits a Significantly Weaker Proliferation Efficiency in CTLL-2 Cells but is Less Affected in M07e Cells
To further demonstrate that the changes in affinities to IL-2 receptors can be translated into cellular activities, we evaluated the proliferation activities of IL-2 and SHR-1916 in the cell lines CTLL-2 and M07e. CTLL-2 is a cell line of mouse cytotoxic T lymphocytes that expresses the α, β, and γ chains of IL-2R on its surface, while M07e is a cell line of human megakaryocytic leukemia, which only expresses the β and γ chains.32–34 The results indicated that both IL-2 and SHR-1916 could dose-dependently promote the proliferation of CTLL-2 and M07e cells (Figure 4). The EC50 values of IL-2 and SHR-1916 for CTLL-2 cells were 0.0111 ± 0.00309 nM and 6.099 ± 1.597 nM, respectively, whereas for M07e cells, the EC50 values were 2.989 ± 0.484 nM and 55.080 ± 13.590 nM, respectively (Table 2). Further analysis of EC50 indicated that SHR-1916 had a significantly weaker proliferation efficiency than IL-2 in CTLL-2 cells (549.5-fold), whereas it was less affected in M07e cells (18.4-fold). The results indicate that SHR-1916 demonstrates significantly lower proliferation efficiency in cell lines which express the high-affinity IL-2Rαβγ, while its impact on proliferation efficiency is less pronounced in cell lines which express the intermediate-affinity IL-2Rβγ.
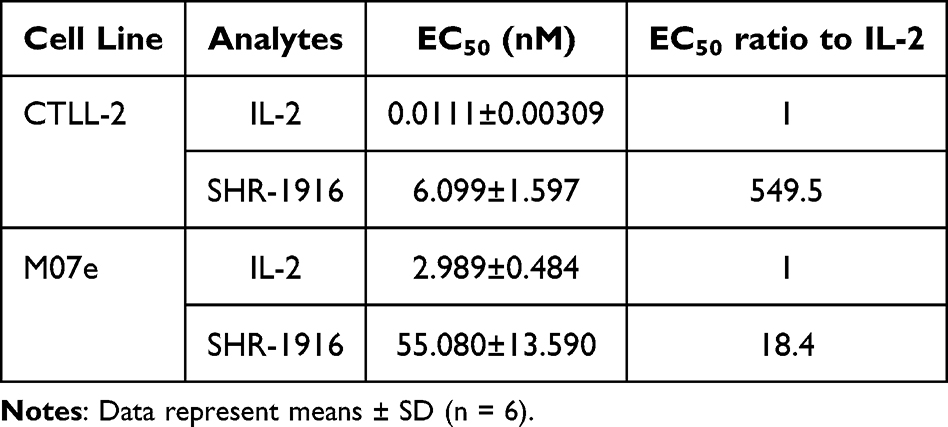 |
Table 2 EC50 Values of Cell Proliferation Induced by IL-2 and SHR-1916 |
 |
Figure 4 Dose-dependent curves of IL-2 and SHR-1916 in the proliferation activity on CTLL-2 (A) and M07e cells (B). |
SHR-1916 Exhibits Selective Activation of CD8+ T and NK Cells Compared to Treg Cells in PBMCs
To evaluate the selective stimulatory effect of SHR-1916 on Treg, NK, and CD8+ T cells, flow cytometry was employed to assess the phosphorylation of STAT5 in these immune cells from PBMC samples of three healthy donors after treatment with SHR-1916 or IL-2. The results showed that both SHR-1916 and IL-2 can dose-dependently stimulate the phosphorylation of STAT5 in Treg, CD8+ T, and NK cells (Figure 5). The EC50 values were summarized in Table 3. The results revealed that IL-2 exhibited a strong stimulating ability on Treg cells in PBMCs (EC50: 0.000696–0.001202 nM), while SHR-1916 significantly reduced this ability (EC50: 3.514–4.967 nM), resulting in a 3029.1–7136.5-fold difference. Additionally, IL-2 also stimulated CD8+ T cells (EC50: 0.7836–1.048 nM) and NK cells (EC50: 0.3206–0.3613 nM) in PBMCs. In comparison to IL-2, SHR-1916 showed a 15.5–19.7-fold difference in stimulating CD8+ T cells (EC50: 12.17–20.6 nM) and a 14.6–18.0-fold difference in stimulating NK cells (EC50: 4.667–6.517 nM). Compared to IL-2, SHR-1916 significantly reduced the stimulating ability in Treg cells, while the reduction in CD8+ T and NK cells was relatively limited. In conclusion, the results suggest that SHR-1916 exhibits better selectivity for the stimulation of CD8+ T and NK cells versus Treg cells.
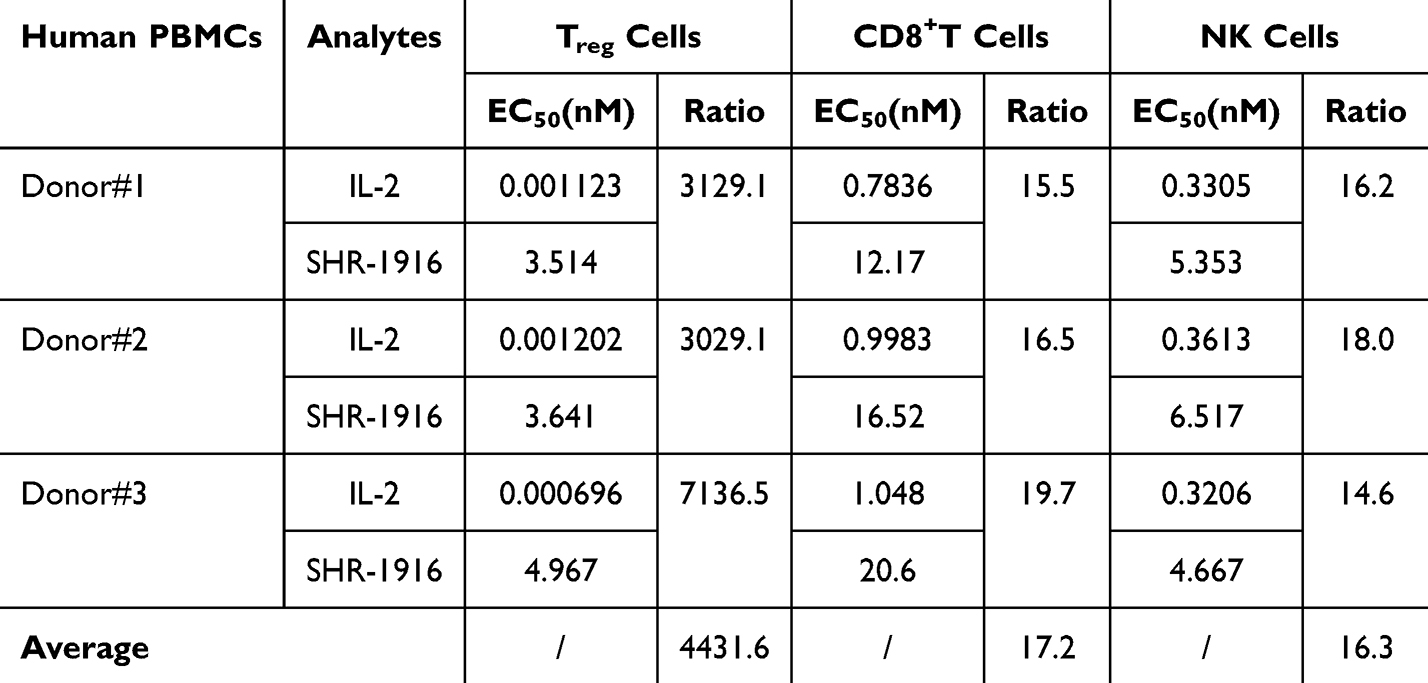 |
Table 3 EC50 Values of the pSTAT5 Assay in PBMCs Induced by IL-2 and SHR-1916 |
 |
Figure 5 Dose-dependent curves of IL-2 and SHR-1916 in the pSTAT5 assay in PBMCs of three healthy donors. |
SHR-1916 Induces Dose-Dependent Secretion of IFNγ by PBMCs
To further test the effect of SHR-1916 on primary immune cells, the response of cytokine secretion after SHR-1916 treatment was investigated using PBMCs from three healthy volunteers. Positive controls (anti-CD3 antibody and lipopolysaccharides) significantly increased the secretion of cytokines including IFNγ, IL-1β, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, IL-13, and TNFα (Figure 6). In contrast to negative controls (RPMI-1640 medium and PEG solution), SHR-1916 showed a dose-dependent increase in IFNγ secretion, while having no significant effect on the secretion of other cytokines, including IL-1β, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, IL-13, and TNFα. Prior research has reported that IL-2 could stimulate PBMCs to secrete IFNγ.35,36 IFNγ is primarily generated by NK and T cells and is crucial for tumor immunity.37 Our findings suggest that SHR-1916 retains the ability to promote IFNγ secretion while avoiding potential side effects associated with increasing levels of other cytokines.
 |
Figure 6 The response of cytokine secretion to SHR-1916 treatment by PBMCs (n = 3). |
SHR-1916 Exhibits Antitumor Effects in CT-26 Colon Carcinoma Model
The in vivo efficacy of SHR-1916 was first evaluated in mice with subcutaneously implanted CT-26 colon carcinomas. Three different doses of SHR-1916 were compared with control animals on day 13. Compared to the vehicle group, the medium and high-dose groups showed a significant reduction in tumor growth (Figure 7A). As shown in Table 4, the tumor volumes of the vehicle, low-dose (1 mg/kg), medium-dose (3 mg/kg), and high-dose (9 mg/kg) groups were 1728.1 ± 233.1 mm3, 1062.2 ± 136.0 mm3, 854.1 ± 150.8 mm3, and 443.6 ± 85.7 mm3, respectively. Clear antitumor efficacy was presented in the medium and high-dose groups, and the antitumor activity showed a positive correlation with the doses. Between the low-dose and vehicle groups, no significant differences were observed in tumor volume, but a significant tumor volume reduction in the medium and high-dose groups was observed, with tumor growth inhibition (TGI%) of 53.0% (p < 0.05) and 77.8% (p < 0.01), respectively. The body weight changes among the mice in each group showed no significant difference (Figure 7B), suggesting that these tumor-bearing mice tolerated the tested doses of SHR-1916 well.
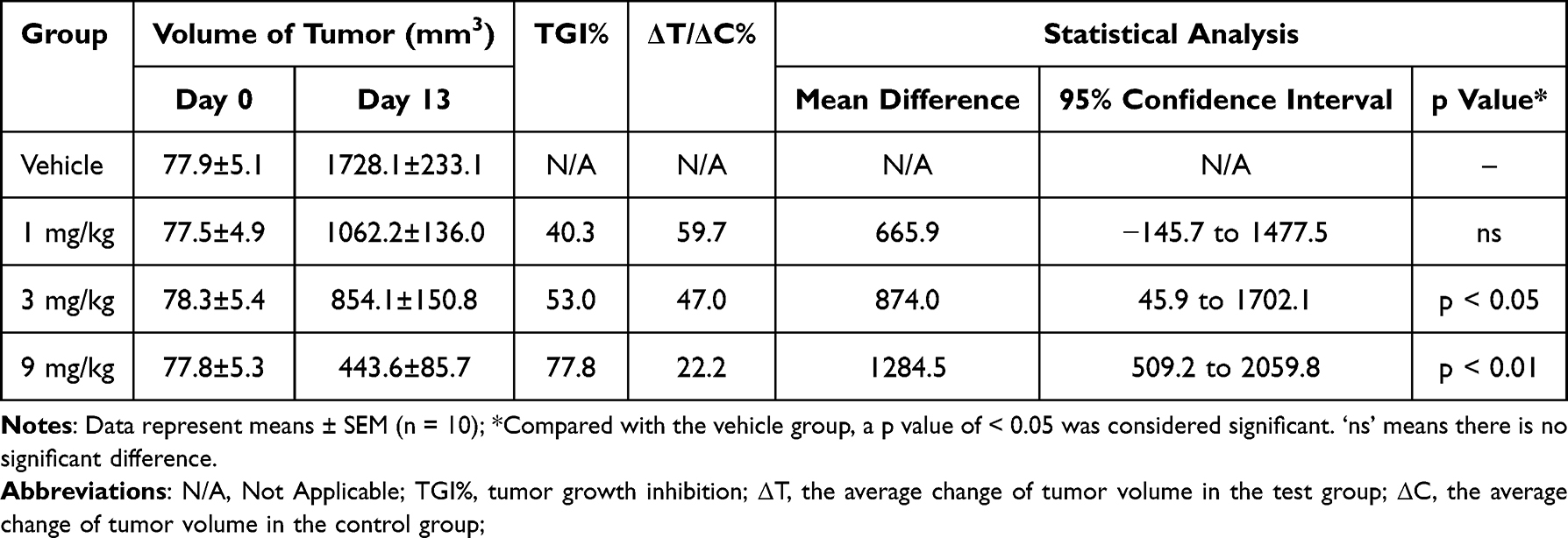 |
Table 4 Tumor Volume and TGI% in the CT-26 Colon Carcinoma Model |
 |
Figure 7 Antitumor activity of SHR-1916 in the CT-26 colon carcinoma model. (A) The tumor volumes in different groups (n = 10). “ns” represents no significant difference between the two groups. ‘*’ and “**” represent significant differences between the two groups (p < 0.05 and p < 0.01, respectively). (B) The body weights of mice in different groups (n = 10). |
SHR-1916 Exhibits Antitumor Effects in the A375 Melanoma Model
PBMC humanized NCG mice were used in a subcutaneous A375 human melanoma xenograft model to evaluate the anticancer effects of SHR-1916. The animals were treated with three different doses of SHR-1916 as well as a placebo. On day 21, a significant tumor growth reduction was observed (Figure 8A and C). As shown in Table 5, the tumor weights of the vehicle, low-dose (0.04 mg/kg), medium-dose (0.2 mg/kg), and high-dose (1 mg/kg) groups at the end of the experiment were 1.347 ± 0.189 g, 0.059 ± 0.008 g, 0.110 ± 0.008 g, and 0.147 ± 0.012 g, respectively. The tumor weights was significantly reduced in the three treatment groups, with tumor growth inhibition (TGI%) of 95.6% (p < 0.001), 91.8% (p < 0.001), and 89.1% (p < 0.01), respectively. The body weights of mice were measured twice a week throughout the study duration (Figure 8B and Table 6). Compared to the vehicle group, no significant differences were observed in the low and medium-dose groups, while we observed that the mice in the high-dose group exhibited a decreasing trend in body weight after day 10. These findings suggest that mice exhibited good tolerance to the middle and low doses of SHR-1916, while the high dose of SHR-1916 may exert a potential impact to some extent.
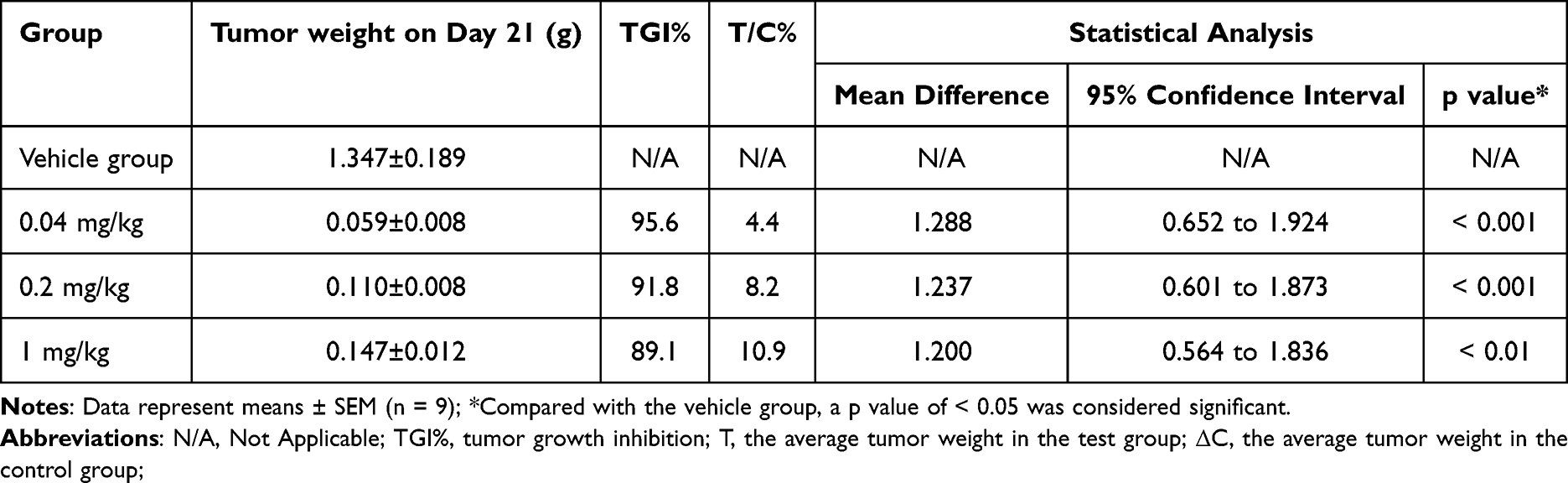 |
Table 5 The Tumor Growth Inhibition (TGI%) of SHR-1916 in the A375 Melanoma Model |
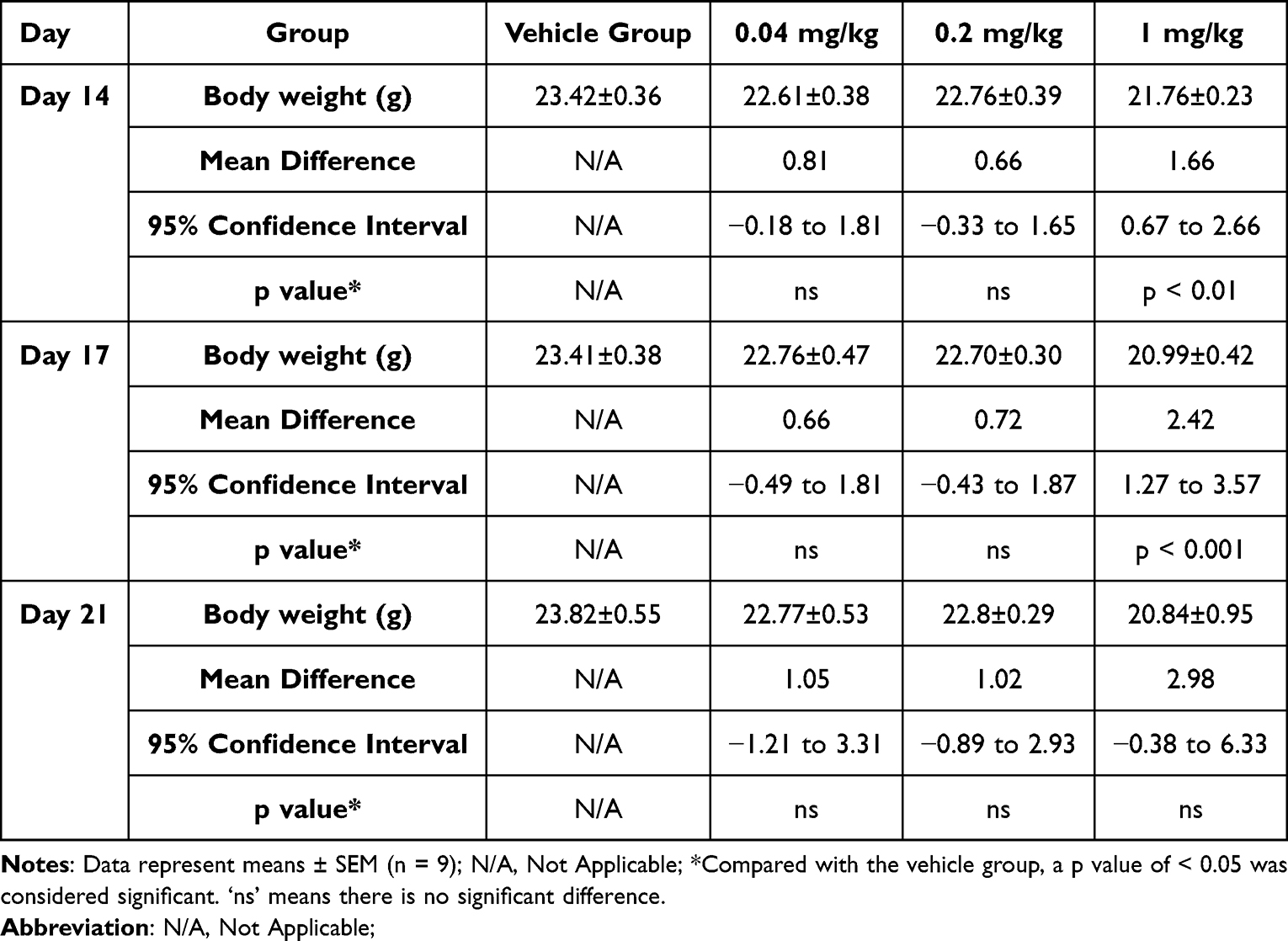 |
Table 6 The Body Weights of Mice in the A375 Melanoma Model |
 |
Figure 8 Antitumor activity of SHR-1916 in the A375 melanoma model. (A) The tumor weight on day 21 after administration (n = 9). “***” represents a significant difference between the two groups (p < 0.001). (B) The body weight of mice in different groups (n = 9). “**” represents a significant difference between the two groups (p < 0.01). (C) The tumor images at day 21 after administration. |
SHR-1916 Exhibits Prolonged Half-Life in Rats
A pharmacokinetic (PK) study was performed in rats to evaluate the PK profile and bioavailability of SHR-1916. Twenty-four SD rats were randomized to four groups according to body weight and gender, receiving a single s.c. dose (1.5 mg/kg, 3 mg/kg, or 6 mg/kg) or an i.v. dose (1.5 mg/kg). The plasma concentrations of all rats before injection were tested, and the results were below 3.13 ng/mL (LLOQ). The pharmacokinetic parameters detailed in Tables 7 and 8 and plasma concentration-time profiles are shown in Figure 9A. After s.c. injection, the Cmax, AUClast, and AUC0-∞ increased with dose, and a concentration peak was observed at 4 to 8 hours (Tmax). There was no significant gender difference in pharmacokinetic parameters among the different administration groups (p > 0.05). The dosage ratios in different groups were 1:2:4. The average Cmax was 2.96, 6.38, and 9.55 μg/mL, respectively. The average AUClast was 78.0, 148, and 260 h μg/mL, respectively. The average AUC0-∞ was 78.2, 148, and 261 h μg/mL, respectively. The logarithms of the main pharmacokinetic parameters AUClast, AUC0-∞, and Cmax were linearly regressed against the logarithms of the administered doses (Figure 9B, C, and D). The obtained regression equations were y = 0.8707x + 4.011, y = 0.8695x + 4.014, and y = 0.8542x + 0.7776, with correlation coefficients of 0.9689, 0.9685, and 0.8488, respectively. The results suggested that the average increases in AUClast, AUC0-∞, and Cmax were consistent with the increase in dose in the range of 1.5 to 6 mg/kg. After just one intravenous injection of 1.5 mg/kg SHR-1916 in SD rats, the average C1min was 57.3 μg/mL, the average AUClast was 274 h μg/mL, and the average AUC0-∞ was 274 h μg/mL. The bioavailability of the drug in the serum after subcutaneous and intravenous injections of SHR-1916 (1.5 mg/kg) was 28.5%. Comparison of key pharmacokinetic (PK) parameters of SHR-1916 and previously reported IL-2 data in rats are shown in Table 9. The elimination half-lives (t1/2) for 1.5 mg/kg (i.v.), 1.5 mg/kg (s.c.), 3 mg/kg (s.c.), and 6 mg/kg (s.c.) dose levels were 19±8.8 h, 11±2.0 h, 10±1.5 h, and 11±5.5 h, respectively. Compared to IL-2 (19 to 40 minutes by i.v. and 23 to 52 minutes by s.c.) reported previously,38 SHR-1916 showed a significantly prolonged half-life. The clearance (Cl) of intravenously administered IL-2 in rats ranged from 558.89 to 716.77 mL/h/kg. In comparison, SHR-1916 exhibited a significantly lower clearance of 5.70 ± 1.30 mL/h/kg, suggesting a slower elimination rate. Furthermore, the mean residence time (MRT) for intravenously and subcutaneously administered IL-2 was 0.18–0.23 h and 0.65–1.34 h, respectively. In contrast, SHR-1916 demonstrated significantly longer MRT values of 6.4 h (intravenous) and 15–17 h (subcutaneous), indicating a substantially longer residence time in vivo.
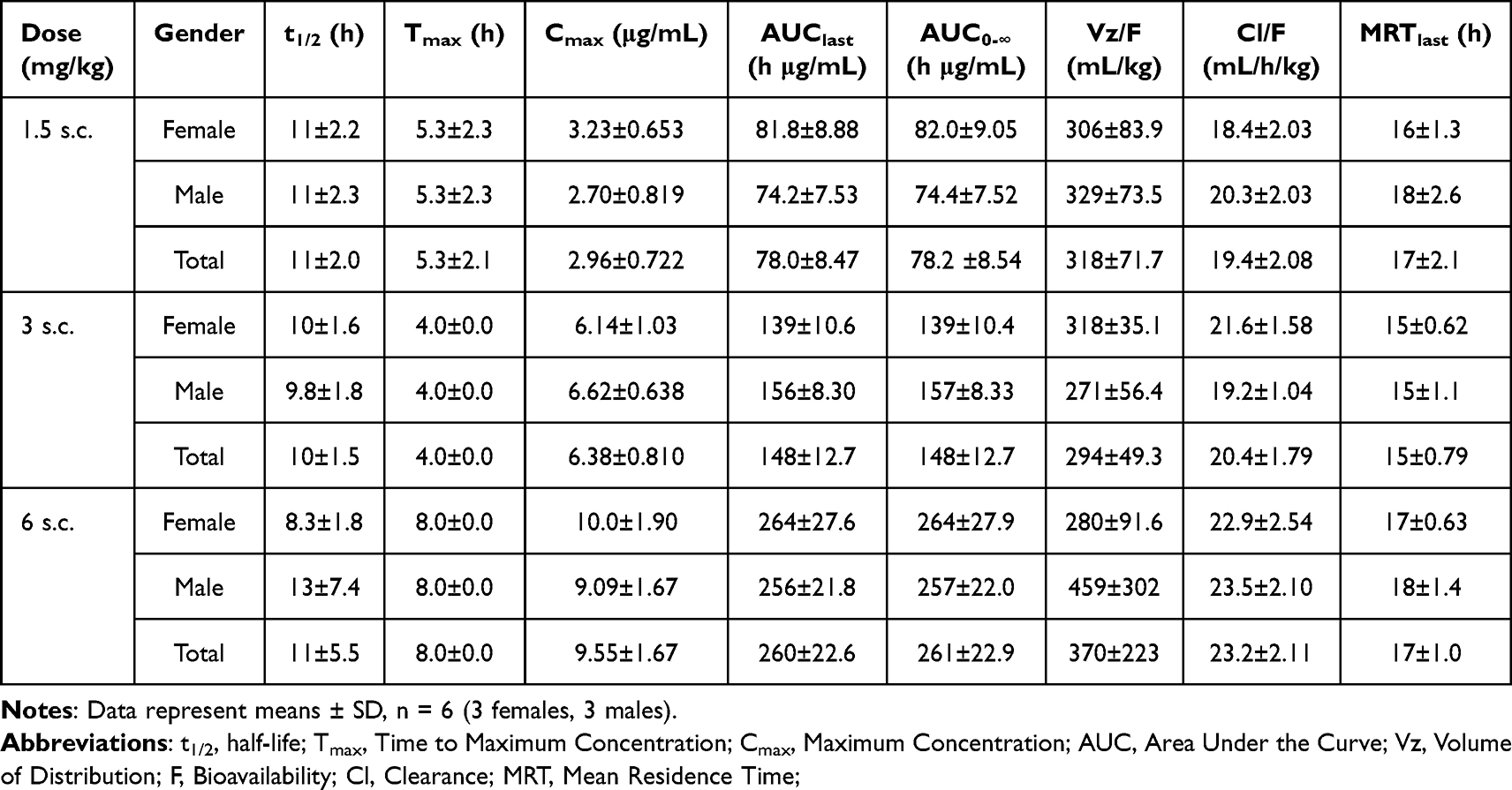 |
Table 7 Pharmacokinetic Parameters Following s.c. Dosing with SHR-1916 in Rats |
 |
Table 8 Pharmacokinetic Parameters Following i.v. Dosing with SHR-1916 in Rats |
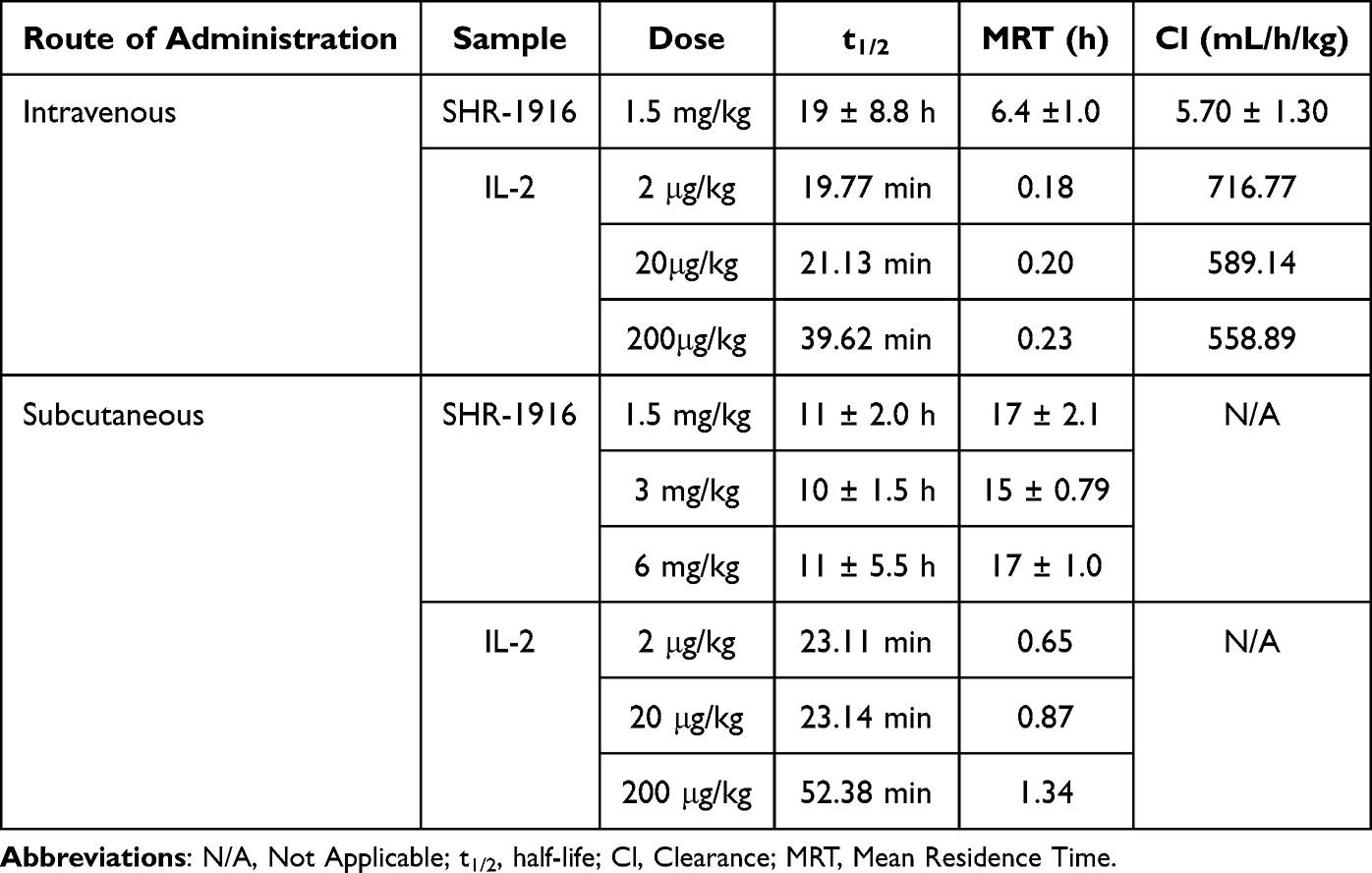 |
Table 9 Comparison of Key Pharmacokinetic (PK) Parameters of SHR-1916 and Previously Reported IL-2 Data in Rats |
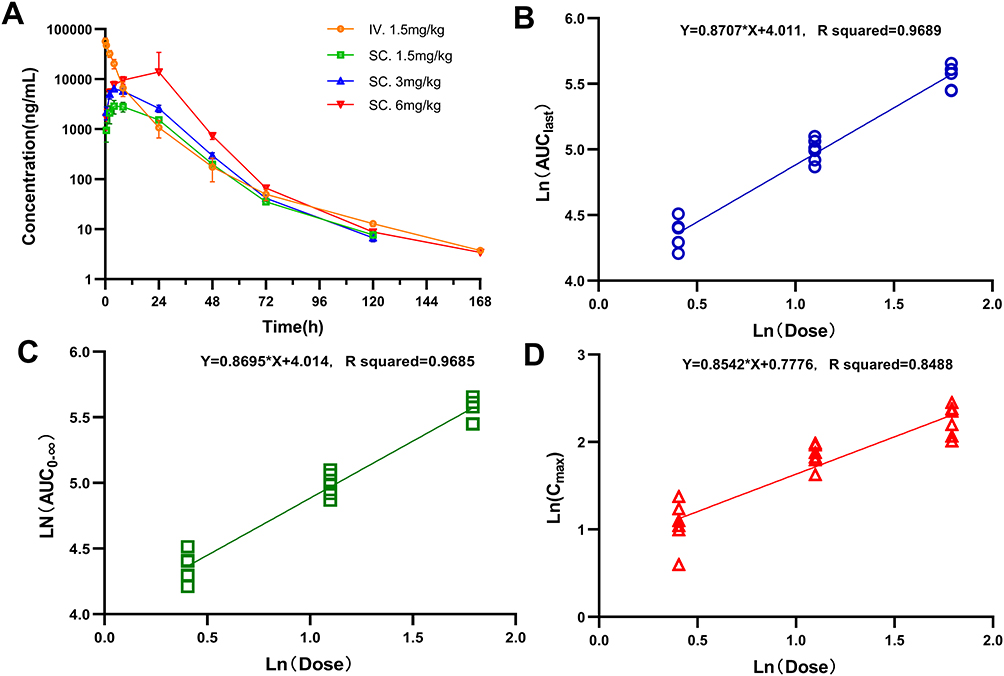 |
Figure 9 Pharmacokinetic profile of SHR-1916 following i.v. and s.c. administration in rats. (A) Plasma concentration of SHR-1916 (n = 6). (B) Linear relationship between AUClast and dose (natural logarithm taken) (n = 6). (C) Linear relationship between AUC0-∞ and dose (natural logarithm taken) (n = 6). (D) Linear relationship between Cmax and dose (natural logarithm taken) (n = 6). |
Discussion
Aldesleukin has been licensed for the treatment of metastatic melanoma and metastatic renal cell carcinoma (RCC). A clinical study comprising 283 patients diagnosed with renal cancer or metastatic melanoma has shown a significant antitumor effect, with a 7% complete response (CR) and a 10% partial response (PR) in the metastatic melanoma group, and a 7% CR and a 13% PR in the metastatic RCC group.39 In another study on IL-2 therapy that included a consecutive cohort of 509 patients diagnosed with RCC or progressive metastatic melanoma, the objective response rates were found to be 22.6% and 16.3%, respectively.40
Despite its effectiveness in some patients, the adverse effects, notably capillary leak syndrome, constrained IL-2’s clinical application. Consequently, its application is confined to a carefully selected group of patients who require inpatient care at specialized facilities. High-dose IL-2 treatment is often connected to a variety of side effects, including fever, chills, diarrhea, dyspnea, arrhythmia, and renal dysfunction.41 These complications primarily arise from vascular leaks and potentially life-threatening edema, affecting many organs, including the kidney, lung, heart, and central nervous system. Recent findings indicate that CLS is predominantly mediated by the interaction of IL‑2 to CD25+ endothelial cells.8 Another limitation of this therapy is the proliferation of Treg cells, which could potentially result in suppressing the responses of anti-tumor immunity. Due to IL-2’s short half-life,42 it is usually infused continuously every eight hours at a dose of 600,000 IU/kg (0.037 mg/kg), delivered over a 15-minute intravenous infusion for up to 14 cycles, ensuring sufficient concentrations of IL-2 to promote T cell proliferation in vivo.
Considering the aforementioned limitations of high-dose IL-2 treatment, we developed a novel long-acting interleukin-2 analogue, SHR-1916. It is a potential immunotherapeutic agent aiming to selectively target the intermediate-affinity IL-2R in order to reduce the inevitable toxicity related to traditional high-dose IL-2 treatment and attenuate Treg expansion. Pegylation can improve the PK properties and extend the elimination half-life, enabling decreased injection frequency and improved patient compliance.
In vitro, we characterized the binding affinities and stimulatory properties of SHR-1916 in murine and human cell lines, as well as PBMCs.
Using the SPR assay, SHR-1916 exhibited no or very weak binding to IL-2Rα while reducing the binding affinity to IL-2Rβ by only 6.1-fold. We evaluated the cytokine activity of IL-2 and SHR-1916 using a cell line of murine cytotoxic T lymphocytic, CTLL-2, and a cell line of human megakaryocytic leukemia, M07e. SHR-1916 showed significantly weaker proliferation efficiency than IL-2 in CTLL-2 cells (549.5-fold), which express IL-2Rαβγ, while exhibiting less affected proliferation efficiency in M07e cells (18.4-fold), which only express IL-2Rβγ. This indicates that SHR-1916 exerts its proliferation activity mainly through binding to intermediate-affinity IL-2Rβγ. SHR-1916 showed a weak decrease in affinity to IL-2Rβ (6.1-fold) and M07e cell proliferation efficiency (18.4-fold), likely due to non-specific intramolecular blockage of PEG moieties.43 Previous studies have reported similar findings. Deborah Charych et al in their study on NKTR-214, found that 1-PEG-IL-2 binds to IL-2Rβ with an affinity of 1769.5 nM, whereas IL-2 binds to IL-2Rβ with an affinity of 238.9 nM, a difference of 7.4-fold.24 Additionally, PEG modification reduced the cellular biological activity by 5 to 50-fold.44 David B. Rosen et al in their study on TransCon IL-2 β/γ, showed that a 20 kDa PEG modification decreased the affinity of IL-2 to IL-2Rβ by 16-fold (280 nM and 4500 nM for IL-2 and 20 kDa pegylated IL-2, respectively).26 Although PEGylation decreased the in vitro potency of IL-2, its activity in vivo was compensated by the increased drug exposure.
A selective stimulatory effect on immune cell subsets was observed in PBMCs. Compared to IL-2, the phosphorylation of STAT5 in Treg cells was activated by SHR-1916, showing a significant increase in EC50 (3029.1–7136.5-fold). However, the EC50 of the two analytes was less affected in NK and CD8+ T cells (14.6–18.0-fold and 15.5–19.7-fold, respectively). SHR-1916 showed a selective activation of CD8+ T and NK cells compared to Treg cells in PBMCs. Elevated ratios of CD8+ T to Treg cells were associated with better therapeutic responses in patients who were diagnosed with urothelial carcinoma.45 Conversely, lower baseline ratios were linked to unfavorable results in patients with severe ovarian cancer.46 Consequently, promoting a shift toward favoring CD8+ T over Treg cells in the immune cell balance could improve therapeutic efficacy.47–49 SHR-1916, which can activate and expand effector cells without significant expansion of Treg cells, may correlate with improved efficacy relative to IL-2.
IL-2Rα is a low-affinity receptor (Kd ~10⁻8 M), IL-2Rβγ is an intermediate-affinity receptor (Kd ~10⁻9 M), and IL-2Rαβγ is a high-affinity receptor (Kd ~10⁻¹¹ M).50 Furthermore, previous studies have shown that the affinity of the IL-2/IL-2Rα complex for IL-2Rβ (Kd ~3 × 10⁻¹¹ M) is significantly higher than that of IL-2 alone (Kd ~10⁻7 M).31,51 While IL-2Rα alone does not trigger downstream signaling upon IL-2 binding,50 its presence is crucial for the formation of the signaling-competent high-affinity IL-2Rαβγ receptor. IL-2Rαβγ is highly expressed on Treg cells, activated effector T cells, and innate lymphoid cells,52 making Treg cells highly sensitive to IL-2, thus allowing low doses of IL-2 to preferentially activate Treg cells and mediate immunosuppression. The intermediate-affinity IL-2Rβγ is predominantly expressed on resting NK cells and CD8+ T cells.50 The amino acid mutation in SHR-1916, by eliminating its binding to IL-2Rα, results in a significantly weaker stimulatory effect on cells expressing the high-affinity receptor IL-2Rαβγ compared to IL-2. Therefore, compared to IL-2, SHR-1916 exhibits a selectivity for the stimulation of CD8+ T and NK cells over Treg cells.
A cytokine secretion assay was conducted using PBMCs. SHR-1916 displayed a dose-dependent increase in IFNγ secretion, while having no significant effect on the secretion of other cytokines, including IL-1β, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, and TNFα. This suggests that SHR-1916 retains the ability to promote IFNγ secretion similarly to the original IL-2, while avoiding potential side effects associated with elevated levels of other cytokines. Treatment with IL-2 elicited significantly elevated levels of cytokines commonly linked to cytokine release syndrome, such as IFNγ, IL-6, and TNFα.53–55 SHR-1916 has not stimulated the secretion of proinflammatory cytokines such as IL-5, which is associated with eosinophilia resulting from high-dose IL-2.56 These variations in systemic proinflammatory cytokine profiles suggest that SHR-1916 may possess a superior capacity for improving tolerability.
In vivo, we evaluated the efficacy of SHR-1916 in mouse tumor models and assessed its pharmacokinetic profiles in rats. For the CT-26 colon carcinoma syngeneic model, the doses of the drug were primarily determined based on previously reported doses of IL-2 and its analogs. For example, Ptacin et al reported doses of 1, 3, and 6 mg/kg for THOR-707 in a syngeneic mouse model.25 Charych et al reported doses of 2 mg/kg for NKTR-214 and 3 mg/kg for IL-2.44 Rosen et al reported a dosage of 3 mg/kg for TransCon IL-2 β/γ.26 Based on these reports, we selected low, medium, and high dose groups of 1 mg/kg, 3 mg/kg, and 9 mg/kg, respectively, in the syngeneic model, maintaining a 3-fold dose increment between groups. For the A375 tumor model in PBMC-humanized NCG mice, considering that IL-2 exhibits higher binding affinity to human IL-2Rβγ than to murine IL-2Rβγ,57 and given the current lack of reports on the antitumor activity of IL-2 and its analogs in this model, we employed a lower dosage regimen with a 5-fold increment between groups: 0.04 mg/kg, 0.2 mg/kg, and 1.0 mg/kg.
SHR-1916 dramatically decreased tumor burden in mice with CT-26 colon carcinoma and A375 melanoma at the selected doses. A significant reduction of tumor volume in mice injected with medium and high doses of SHR-1916 was observed in the CT-26 colon carcinoma model, with tumor growth inhibition (TGI%) of 53.0% (p < 0.05) and 77.8% (p < 0.01), respectively. In the CT-26 model, SHR-1916 demonstrated a dose-dependent antitumor effect. No significant weight loss was observed in any treatment group compared to vehicle control, indicating good tolerability. In the A375 melanoma model using PBMC humanized NCG mice, SHR-1916 showed significant tumor growth inhibition (TGI%) of 95.6% (p < 0.001), 91.8% (p < 0.001), and 89.1% (p < 0.01) in the low-dose (0.04 mg/kg), medium-dose (0.2 mg/kg), and high-dose (1 mg/kg) groups, respectively. In contrast to the CT-26 model, a plateau effect was observed with no further increase in TGI% at doses above 0.04 mg/kg. This humanized mouse model better mimics the human immune response to tumor cells, and PBMC immune cells are sensitive to IL-2. The lack of further improvement at 1 mg/kg (a 25-fold increase) may be due to potential reduced tolerance at this higher dose, as suggested by a trend towards weight loss. However, at the effective doses of 0.04 mg/kg and 0.2 mg/kg, no significant weight difference was observed compared to the control group, indicating good tolerability of SHR-1916 in NCG mice at effective doses.
IL-2’s short half-life is another important factor limiting its clinical application. Previous studies have indicated that PEGylated IL-2 or its analogues can lead to a marked extension of the half-life.24,58 In the pharmacokinetic study conducted in rats, the elimination half-lives of SHR-1916 for the doses of 1.5 mg/kg (i.v.), 1.5 mg/kg (s.c.), 3 mg/kg (s.c.), and 6 mg/kg (s.c.) were 19 ± 8.8 h, 11 ± 2.0 h, 10 ± 1.5 h, and 11 ± 5.5 h, showing a significantly extended half-life compared to IL-2 (19 to 40 minutes by i.v. and 23 to 52 minutes by s.c.) reported previously.38 Significantly lower clearance (Cl) and longer mean residence time (MRT) were also observed. The bioavailability in the serum after subcutaneous and intravenous injections of SHR-1916 (1.5 mg/kg) was 28.5%. SHR-1916 showed decreased clearance and exhibited an extended half-life relative to IL-2, facilitating less frequent administration.
This study has several limitations. First, preclinical efficacy data were generated using in vitro cell lines and in vivo murine tumor models. Due to limitations of these models, these results may not fully predict the clinical efficacy of SHR-1916. Second, because of the inherent differences between the CT-26 syngeneic mouse model and human patients, we only assessed the macroscopic tumor growth inhibition rate. Cytokine release or immune cell stimulation in mice was not analyzed. Third, in the PBMC-humanized A375 mouse model, further analysis of cytokine release or immune cell stimulation was not performed as in vitro studies on these parameters using PBMCs had already been conducted.
PEGylation is commonly used to extend the in vivo half-life of proteins and reduce glomerular filtration clearance while also potentially reducing immunogenicity and improving stability.59 Several marketed PEGylated protein therapeutics, such as pegaspargase (Oncaspar®)60 and pegademase bovine (Adagen®),61 demonstrate significantly reduced immunogenicity compared to their non-PEGylated counterparts. Numerous other PEGylated drugs approved by the FDA, including pegfilgrastim (Neulasta®), PEG-interferon-α-2b (PegIntron®), PEG-IFNα-2a (Pegasys®), and pegaptanib (Macugen®), have been used extensively for years. A previous study suggested that PEGylation can reduce the immunogenicity of IL-2,62 but these findings are limited to preclinical animal models and lack clinical validation. We will investigate the immunogenicity of SHR-1916 in future studies and clinical trials.
Conclusion
We have designed a novel PEGylated IL-2 analogue, SHR-1916. The comprehensive study design and key findings of this research were summarized in Figure 10.The results in vivo and in vitro have confirmed that SHR-1916 exhibits excellent cellular selectivity, anti-tumor efficacies and improved pharmacokinetics. It has the potential to become a novel candidate immunotherapeutic agent that is designed to improve IL-2’s tolerability and enhance its immune-stimulating effects, while limiting its immune-regulatory role of promoting Treg cell function. We anticipate that treatment with SHR-1916 in cancer patients may achieve a remission rate comparable to IL-2, yet with a considerably lower incidence of severe adverse effects. Based on this study, SHR-1916 has entered into Phase I clinical trials for patients with solid tumors. In future studies, we will investigate the pharmacokinetic (PK) properties and immune cell activation selectivity of SHR-1916 in both human and animal models. We will also assess its immunogenicity and safety profile and conduct long-term toxicology studies in animals. Our current results may provide a foundation for the development of effective cancer immunotherapies.
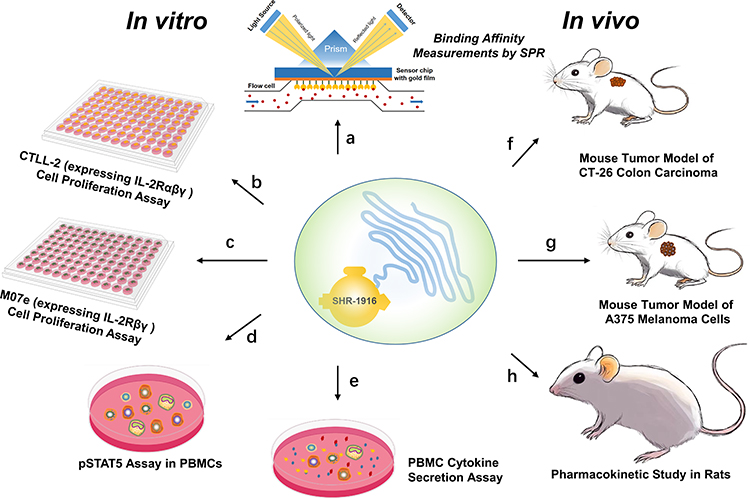 |
Figure 10 Comprehensive study design and key findings of this research. (a) Binding affinity measurements by SPR show that SHR-1916 exhibits a significant IL-2Rβ-binding bias compared to IL-2 and reduced binding affinity to IL-2Rα. (b) and (c) In vitro cell proliferation assays using CTLL-2 and M07e cells demonstrate that SHR-1916 shows significantly lower proliferation efficiency in cell lines that express the high-affinity IL-2Rαβγ, while its impact on proliferation efficiency is less pronounced in cell lines that express the intermediate-affinity IL-2Rβγ. (d) In vitro pSTAT5 assay in PBMCs shows that SHR-1916 exhibits better selectivity for the stimulation of CD8+ T and NK cells versus Treg cells compared to IL-2. (e) PBMC cytokine secretion assay shows that SHR-1916 retains the ability to promote IFNγ secretion while avoiding potential side effects associated with increasing levels of other cytokines. (f) In vivo mouse tumor model of CT-26 colon carcinoma demonstrates significant antitumor effects in the medium and high-dose groups, with tumor growth inhibition (TGI%) of 53.0% (p < 0.05) and 77.8% (p < 0.01), respectively. (g) In vivo mouse tumor model of A375 melanoma cells demonstrates significant antitumor effects in the three treatment groups, with tumor growth inhibition (TGI%) of 95.6% (p < 0.001), 91.8% (p < 0.001), and 89.1% (p < 0.01), respectively. (h) Pharmacokinetic study in rats SHR-1916 showed a significantly prolonged half-life, a lower clearance (Cl), and a longer mean residence time (MRT) compared to previously reported IL-2 data in rats. |
Ethical Statement
Human peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors, following the approval by the Ethics Committee of Shanghai Zhaxin Traditional Chinese and Western Medicine Hospital and the acquisition of written informed consent from all donors (in accordance with the Declaration of Helsinki). All animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The studies were approved by the following Institutional Animal Care and Use Committees (IACUCs): Phenotek Biotechnology (Shanghai) Co., Ltd. (for the CT-26 colon carcinoma mouse tumor model); LIDE Shanghai Biotech, Ltd. (for the A375 melanoma mouse tumor model); and West China-Frontier Pharma Tech Co., Ltd. (for the rat pharmacokinetic (PK) study).
Acknowledgments
We would like to thank LIDE Shanghai Biotech, Ltd., Phenotek Biotechnology (Shanghai) Co., Ltd., and West China-Frontier Pharma Tech Co., Ltd. for their assistance on animal experiments.
Disclosure
Financial support for this study was provided by Jiangsu Hengrui Pharmaceuticals Co., Ltd. The authors, Xianglin Kong, Yuan Lin, Chao Ouyang, and Hao Chen, are employees and shareholders of Jiangsu Hengrui Pharmaceuticals Co., Ltd. The authors report no other conflicts of interest in this work.
References
1. Dhupkar P, Gordon N. Interleukin-2: old and New Approaches to Enhance Immune-Therapeutic Efficacy. Adv Exp Med Biol. 2017;995:33–51. doi:10.1007/978-3-319-53156-4_2
2. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. 1976;193(4257):1007–1008. doi:10.1126/science.181845
3. Orozco Valencia A, Camargo Knirsch M, Suavinho Ferro E, Antonio Stephano M. Interleukin-2 as immunotherapeutic in the autoimmune diseases. Int Immunopharmacol. 2020;81:106296. doi:10.1016/j.intimp.2020.106296
4. Yuan Y, Kolios AGA, Liu Y, et al. Therapeutic potential of interleukin-2 in autoimmune diseases. Trends Mol Med. 2022;28(7):596–612. doi:10.1016/j.molmed.2022.04.010
5. Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010;33(2):153–165. doi:10.1016/j.immuni.2010.08.004
6. Taniguchi T, Minami Y. The IL-2/IL-2 receptor system: a current overview. Cell. 1993;73(1):5–8. doi:10.1016/0092-8674(93)90152-g
7. McDermott DF, Atkins MB. Application of IL-2 and other cytokines in renal cancer. Expert Opin Biol Ther. 2004;4(4):455–468. doi:10.1517/14712598.4.4.455
8. Krieg C, Letourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A. 2010;107(26):11906–11911. doi:10.1073/pnas.1002569107
9. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. doi:10.1038/nri3156
10. Konrad MW, Hemstreet G, Hersh EM, et al. Pharmacokinetics of recombinant interleukin 2 in humans. Cancer Res. 1990;50(7):2009–2017.
11. Rosalia RA, Arenas-Ramirez N, Bouchaud G, Raeber ME, Boyman O. Use of enhanced interleukin-2 formulations for improved immunotherapy against cancer. Curr Opin Chem Biol. 2014;23:39–46. doi:10.1016/j.cbpa.2014.09.006
12. Qian M, Zhang Q, Lu J, et al. Long-Acting Human Interleukin 2 Bioconjugate Modified with Fatty Acids by Sortase A. Bioconjug Chem. 2021;32(3):615–625. doi:10.1021/acs.bioconjchem.1c00062
13. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311(5769):1924–1927. doi:10.1126/science.1122927
14. Carmenate T, Pacios A, Enamorado M, et al. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. J Immunol. 2013;190(12):6230–6238. doi:10.4049/jimmunol.1201895
15. Kobayashi M, Kojima K, Murayama K, et al. MK-6, a novel not-alpha IL-2, elicits a potent antitumor activity by improving the effector to regulatory T cell balance. Cancer Sci. 2021;112(11):4478–4489. doi:10.1111/cas.15127
16. Chen X, Ai X, Wu C, et al. A novel human IL-2 mutein with minimal systemic toxicity exerts greater antitumor efficacy than wild-type IL-2. Cell Death Dis. 2018;9(10):989. doi:10.1038/s41419-018-1047-2
17. Levin AM, Bates DL, Ring AM, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484(7395):529–533. doi:10.1038/nature10975
18. Gillessen S, Gnad-Vogt US, Gallerani E, et al. A phase I dose-escalation study of the immunocytokine EMD 521873 (Selectikine) in patients with advanced solid tumours. Eur J Cancer. 2013;49(1):35–44. doi:10.1016/j.ejca.2012.07.015
19. Klein C, Waldhauer I, Nicolini VG, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):e1277306. doi:10.1080/2162402X.2016.1277306
20. Klein C. S41. Novel CEA-targeted IL2 variant immunocytokine for immunotherapy of cancer. J Immunother Cancer. 2014;2(2):I8. doi:10.1186/2051-1426-2-S2-I8
21. Hank JA, Gan J, Ryu H, et al. Immunogenicity of the hu14.18-IL2 immunocytokine molecule in adults with melanoma and children with neuroblastoma. Clin Cancer Res. 2009;15(18):5923–5930. doi:10.1158/1078-0432.CCR-08-2963
22. Shusterman S, London WB, Gillies SD, et al. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children’s Oncology Group (COG) Phase II study. J Clin Oncol. 2010;28(33):4969–4975. doi:10.1200/JCO.2009.27.8861
23. Lopes JE, Fisher JL, Flick HL, et al. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J Immunother Cancer. 2020;8(1):e000673. doi:10.1136/jitc-2020-000673
24. Charych D, Khalili S, Dixit V, et al. Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PLoS One. 2017;12(7):e0179431. doi:10.1371/journal.pone.0179431
25. Ptacin JL, Caffaro CE, Ma L, et al. An engineered IL-2 reprogrammed for anti-tumor therapy using a semi-synthetic organism. Nat Commun. 2021;12(1):4785. doi:10.1038/s41467-021-24987-9
26. Rosen DB, Kvarnhammar AM, Laufer B, et al. TransCon IL-2 beta/gamma: a novel long-acting prodrug with sustained release of an IL-2Rbeta/gamma-selective IL-2 variant with improved pharmacokinetics and potent activation of cytotoxic immune cells for the treatment of cancer. J Immunother Cancer. 2022;10(7):e004991. doi:10.1136/jitc-2022-004991
27. Stauber DJ, Debler EW, Horton PA, Smith KA, Wilson IA. Crystal structure of the IL-2 signaling complex: paradigm for a heterotrimeric cytokine receptor. Proc Natl Acad Sci U S A. 2006;103(8):2788–2793. doi:10.1073/pnas.0511161103
28. Wang X, Rickert M, Garcia KC. Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors. Science. 2005;310(5751):1159–1163. doi:10.1126/science.1117893
29. Wang A, Lu SD, Mark DF. Site-specific mutagenesis of the human interleukin-2 gene: structure-function analysis of the cysteine residues. Science. 1984;224(4656):1431–1433. doi:10.1126/science.6427925
30. Myszka DG, Arulanantham PR, Sana T, Wu Z, Morton TA, Ciardelli TL. Kinetic analysis of ligand binding to interleukin-2 receptor complexes created on an optical biosensor surface. Protein Sci. 1996;5(12):2468–2478. doi:10.1002/pro.5560051209
31. Rickert M, Boulanger MJ, Goriatcheva N, Garcia KC. Compensatory energetic mechanisms mediating the assembly of signaling complexes between interleukin-2 and its alpha, beta, and gamma(c) receptors. J mol Biol. 2004;339(5):1115–1128. doi:10.1016/j.jmb.2004.04.038
32. Arai T, Matsui K. A purified protein from Salmonella typhimurium inhibits high-affinity interleukin-2 receptor expression on CTLL-2 cells. FEMS Immunol Med Microbiol. 1997;17(3):155–160. doi:10.1111/j.1574-695X.1997.tb01008.x
33. Meazza R, Basso S, Gaggero A, et al. Interleukin (IL)-15 induces survival and proliferation of the growth factor-dependent acute myeloid leukemia M-07e through the IL-2 receptor beta/gamma. Int J Cancer. 1998;78(2):189–195. doi:10.1002/(sici)1097-0215(19981005)78:2<189::aid-ijc12>3.0.co;2-6
34. Kermer V, Baum V, Hornig N, Kontermann RE, Muller D. An antibody fusion protein for cancer immunotherapy mimicking IL-15 trans-presentation at the tumor site. mol Cancer Ther. 2012;11(6):1279–1288. doi:10.1158/1535-7163.MCT-12-0019
35. Fuji A, Kakumu S, Ohtani Y, Murase K, Hirofuji H, Tahara H. Interferon-gamma production by peripheral blood mononuclear cells of patients with chronic liver disease. Hepatology. 1987;7(3):577–581. doi:10.1002/hep.1840070327
36. Matsubayashi S, Akasu F, Kasuga Y, Snow K, Keystone E, Volpe R. In vitro production of interferon-gamma by peripheral blood from patients with Graves’ disease, Hashimoto’s thyroiditis and rheumatoid arthritis. Clin Exp Immunol. 1990;82(1):63–68. doi:10.1111/j.1365-2249.1990.tb05404.x
37. Ni L, Lu J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018;7(9):4509–4516. doi:10.1002/cam4.1700
38. LeBel CP, Langlois L, Bell DP, et al. Pharmacokinetic, pharmacodynamic, and pharmacotoxic profiles of recombinant-methionyl human interleukin-2[alanine-125] (r-metHuIL-2[ala-125]) following intravenous and subcutaneous administration in rats. Hum Exp Toxicol. 1995;14(11):909–915. doi:10.1177/096032719501401109
39. Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271(12):907–913. doi:10.1001/jama.1994.03510360033032
40. Royal RE, Steinberg SM, Krouse RS, et al. Correlates of response to IL-2 therapy in patients treated for metastatic renal cancer and melanoma. Cancer J Sci Am. 1996;2(2):91–98.
41. Kammula US, White DE, Rosenberg SA. Trends in the safety of high dose bolus interleukin-2 administration in patients with metastatic cancer. Cancer. 1998;83(4):797–805. doi:10.1002/(SICI)1097-0142(19980815)83:4<797::AID-CNCR25>3.0.CO;2-M
42. Lotze MT, Frana LW, Sharrow SO, Robb RJ, Rosenberg SA. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J Immunol. 1985;134(1):157–166. doi:10.4049/jimmunol.134.1.157
43. Kubetzko S, Sarkar CA, Pluckthun A. Protein PEGylation decreases observed target association rates via a dual blocking mechanism. Mol Pharmacol. 2005;68(5):1439–1454. doi:10.1124/mol.105.014910
44. Charych DH, Hoch U, Langowski JL, et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin Cancer Res. 2016;22(3):680–690. doi:10.1158/1078-0432.CCR-15-1631
45. Starkebaum G, Loughran Jr TP, Waters CA, Ruscetti FW. Establishment of an IL-2 independent, human T-cell line possessing only the p70 IL-2 receptor. Int J Cancer. 1991;49(2):246–253. doi:10.1002/ijc.2910490218
46. Preston CC, Maurer MJ, Oberg AL, et al. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS One. 2013;8(11):e80063. doi:10.1371/journal.pone.0080063
47. Gajewski TF, Woo SR, Zha Y, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25(2):268–276. doi:10.1016/j.coi.2013.02.009
48. Zhou P, Zheng X, Zhang H, Liu Y, Zheng P. B7 blockade alters the balance between regulatory T cells and tumor-reactive T cells for immunotherapy of cancer. Clin Cancer Res. 2009;15(3):960–970. doi:10.1158/1078-0432.CCR-08-1611
49. Hadrup S, Donia M, Thor Straten P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. 2013;6(2):123–133. doi:10.1007/s12307-012-0127-6
50. Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 2018;18(10):648–659. doi:10.1038/s41577-018-0046-y
51. Arima N, Kamio M, Imada K, et al. Pseudo-high affinity interleukin 2 (IL-2) receptor lacks the third component that is essential for functional IL-2 binding and signaling. J Exp Med. 1992;176(5):1265–1272. doi:10.1084/jem.176.5.1265
52. Abbas AK, Trotta E, Rs D, Marson A, Bluestone JA. Revisiting IL-2: biology and therapeutic prospects. Sci Immunol. 2018;3(25):1482. doi:10.1126/sciimmunol.aat1482
53. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
54. Panelli MC, Martin B, Nagorsen D, et al. A genomic- and proteomic-based hypothesis on the eclectic effects of systemic interleukin-2 administration in the context of melanoma-specific immunization. Cells Tissues Organs. 2004;177(3):124–131. doi:10.1159/000079986
55. Panelli MC, White R, Foster M, et al. Forecasting the cytokine storm following systemic interleukin (IL)-2 administration. J Transl Med. 2004;2(1):17. doi:10.1186/1479-5876-2-17
56. van Haelst Pisani C, Kovach JS, Kita H, et al. Administration of interleukin-2 (IL-2) results in increased plasma concentrations of IL-5 and eosinophilia in patients with cancer. Blood. 1991;78(6):1538–1544. doi:10.1182/blood.V78.6.1538.1538
57. Silva DA, Yu S, Ulge UY, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. 2019;565(7738):186–191. doi:10.1038/s41586-018-0830-7
58. Yang JC, Topalian SL, Schwartzentruber DJ, et al. The use of polyethylene glycol-modified interleukin-2 (PEG-IL-2) in the treatment of patients with metastatic renal cell carcinoma and melanoma. A phase I study and a randomized prospective study comparing IL-2 alone versus IL-2 combined with PEG-IL-2. Cancer. 1995;76(4):687–694. doi:10.1002/1097-0142(19950815)76:4<687::aid-cncr2820760424>3.0.co;2-m
59. Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs. 2008;22(5):315–329. doi:10.2165/00063030-200822050-00004
60. Heo YA, Syed YY, Keam SJ. Pegaspargase: a Review in Acute Lymphoblastic Leukaemia. Drugs. 2019;79(7):767–777. doi:10.1007/s40265-019-01120-1
61. Levy Y, Hershfield MS, Fernandez-Mejia C, et al. Adenosine deaminase deficiency with late onset of recurrent infections: response to treatment with polyethylene glycol-modified adenosine deaminase. J Pediatr. 1988;113(2):312–317. doi:10.1016/s0022-3476(88)80271-3
62. Katre NV. Immunogenicity of recombinant IL-2 modified by covalent attachment of polyethylene glycol. J Immunol. 1990;144(1):209–213. doi:10.4049/jimmunol.144.1.209
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.