")
Back to Journals » International Journal of Nanomedicine » Volume 20
Significantly Increased Aqueous Solubility of Piperine via Nanoparticle Formulation Serves as the Most Critical Factor for Its Brain Uptake Enhancement
Authors Li J, Leung SSY, Chan EHY, Jiang C, Ho ETY, Zuo Z
Received 16 November 2024
Accepted for publication 24 February 2025
Published 30 March 2025 Volume 2025:20 Pages 3945—3959
DOI https://doi.org/10.2147/IJN.S506827
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xing Zhang
Jiahao Li,1 Sharon Shui Yee Leung,1 Edwin Ho Yin Chan,2,3 Cuiping Jiang,4,5 Evelyn Tze Yin Ho,6 Zhong Zuo1
1Guangdong-Hong Kong-Macao Joint Laboratory for New Drug Screening, School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, People’s Republic of China; 2School of Life Sciences, Faculty of Science, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, People’s Republic of China; 3Gerald Choa Neuroscience Institute, The Chinese University of Hong Kong, Shatin, Hong Kong SAR, People’s Republic of China; 4Guangdong Provincial Key Laboratory of Chinese Medicine Pharmaceutics, School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, People’s Republic of China; 5Guangdong Basic Research Center of Excellence for Integrated Traditional and Western Medicine for Qingzhi Diseases, Southern Medical University, Guangzhou, People’s Republic of China; 6Department of Psychological & Brain Sciences, University of California, Santa Barbara, CA, USA
Correspondence: Zhong Zuo, School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong SAR, Tel +852 3943 6832, Fax +852 2603 5295, Email [email protected]
Introduction: Piperine, the major component in Piper retrofractum and Piper nigrum, had potential therapeutic effects on central nervous system diseases such as Alzheimer’s disease, Parkinson’s disease, epilepsy and fragile X-associated tremor/ataxia syndrome. However, its low aqueous solubility (0.04 mg/mL) limits brain uptake and pharmacological investigations at higher doses. In the current study, formulation strategies and routes of administration were assessed to enhance systemic and brain uptake of piperine.
Methods: Formulation of piperine nanoparticles (PIP NPs) was developed to enhance its solubility. PIP NPs were prepared using flash nanoprecipitation via a four-stream Multi-Inlet Vortex Mixer, employing an aqueous solution of poloxamer 188 and an ethanolic solution containing piperine and Eudragit L100-55. The process was optimized using the Design of Experiments to minimize the particle size and maximize the encapsulation efficiency of piperine. Additionally, we investigated the impact of administrating PIP NPs via oral and intranasal routes on its systemic and brain uptake.
Results: The optimized PIP NPs formulation exhibited a particle size of 171.45± 2.38 nm, polydispersity index of 0.27± 0.01, zeta potential of − 43.71± 5.11 mV, encapsulation efficiency of 92.49± 1.92% and drug loading of 15.07± 0.09%. Fourier-transform infrared spectroscopy confirmed the successful encapsulation of piperine into nanoparticles. The PIP NPs could significantly increase the aqueous solubility of piperine from 0.04 mg/mL to 52.31± 0.9 mg/mL and release piperine with a 12.83-fold higher rate than that from piperine suspension. Both oral and intranasal administrations of PIP NPs to C57BL/6 mice at 20 mg/kg demonstrated an increase in AUC0-120min for both plasma (7.9– 10 times) and brain (4.7– 5.0 times) comparing to that from piperine suspension, with no significant difference between these two routes.
Discussion: Our findings suggested that increasing solubility rather than changing the administration route served as the most critical step to enhance the brain uptake of piperine.
Keywords: piperine, nanoparticles, aqueous solubility, brain uptake, oral administration, intranasal administration
Graphical Abstract:

Introduction
Piperine, 1-(5-(1,3-benzodioxol-5-yl)-1-oxo-2,4-pentadienyl), is a natural compound found in Piper retrofractum and Piper nigrum. It had a molecular weight of 285.34 and a water solubility of 0.04 mg/mL.1 Research demonstrated that piperine had potential therapeutic effects on various central nervous system (CNS) diseases including Alzheimer’s disease via improvement of cognitive abilities,2–4 Parkinson’s disease via inhibition of apoptosis and inflammation,5 epilepsy and seizure via activation of the transient receptor potential vanilloid 1 receptor6 and Fragile X-associated tremor/ataxia syndrome (FXTAS) via inhibition of the r(CGG)exp RNA.7
To improve the efficacy of drug treatment for a CNS disease, increasing the drug brain uptake is one of the most adopted strategies. Previous studies have indicated that piperine had a very efficient intestinal permeability with a Papp,a→b of 4.78×10−5 cm/s and efficient distribution with similar AUC0-10h in both brain and plasma.8 In addition, piperine had limited first-pass metabolism with a Clint of 8.15 μL/min/mg for its Phase I hepatic metabolism and limited urine excretion.8,9 The oral bioavailability of piperine was 23.2% in mice10 and 19.7%–24.1% in rat.11,12 Although these ADME properties of piperine suggested that the intestinal/brain permeability and liver first-pass effect may not be the barriers to its brain uptake, the limited aqueous solubility of piperine (0.04 mg/mL) restricted its use at higher dosage levels for further in vivo pharmacological investigations. As a BCS Class II compound, the low solubility and dissolution rate of piperine could limit its in vivo bioavailability.13 Therefore, enhancing the solubility of piperine is hypothesized to be one of the effective approaches to improve its systemic bioavailability and brain uptake so as to enhance its efficacy for CNS disease treatment. Various approaches including SEDDS, liposomes, inclusion complexes and nanoparticles had been tried to increase the solubility of piperine,14 among which formulating it into nanoparticles could achieve the highest piperine concentration of 2 mg/mL with 3.7–4.7 folds increase in AUC0-10h.15
Besides the formulation strategy, changing the route of administration of piperine had also been tried to increase its brain uptake.16,17 In recent years, the intranasal delivery route is well known for its capability of directly delivering drugs to the brain via the olfactory region and trigeminal nerves.18 However, the low aqueous solubility of piperine together with the small handling volume of drug formulation allowed to make the intranasal administration of piperine to achieve therapeutic dosing levels challenging.19 So far, none of the previous approaches to increase the solubility of piperine14 could achieve a target concentration of 40–50 mg/mL for its intranasal use in less than 20 μL. Recently, we had prepared a piperine nanoparticles (PIP NPs) with a concentration of 0.87 mg/mL via a flash nano participation technique to enhance its oral bioavailability for anti-epileptic treatment,15 however, piperine concentration remains insufficient for its intranasal delivery. Considering that solvent evaporation of the formulation could further increase piperine concentration, we aimed to condense the nanocarrier formulations of piperine via such approach. Based on the nature of the formulations, SEDDS and liposome were excluded due to potential instability of the formulation after solvent evaporation, leaving nanoparticles as the only possible nanocarrier for piperine in achieving its targeted concentration. Thus, in the current study, we utilized the four-stream Multi-Inlet Vortex Mixer to refine the preparation method of nanoparticles, aiming to achieve a piperine concentration in the range of 40–50 mg/mL, so as to enable us to conduct further in vivo pharmacokinetic evaluations via both oral and intranasal administration routes. The findings will provide important insights for the selection of a suitable administration route for future pharmacological evaluations of PIP NPs.
Materials and Methods
Materials
Piperine (purity > 97%) was sourced from Sigma-Aldrich Chem. GmbH (Riedstr, Germany). Poloxamer 188 was obtained from Sigma-Aldrich Chem. GmbH (Steinheim, Germany). Sodium phosphate monobasic was purchased from Sigma-Aldrich Chem. GmbH (Riedstr, Germany). Sodium phosphate dibasic was bought from Aladdin (Shanghai, China). Potassium bromide (KBr) was acquired from Sigma-Aldrich Co. (St. Louis, MO). Eudragit L100-55 was purchased from Evonik Röhm GmbH (Darmstadt, Germany). Phosphotungstic acid was purchased from Merck (Merck KGaA, Darmstadt, Germany). Formic acid was obtained from BDH Laboratory Supplied Ltd. (Kampala, Ukraine). Acetonitrile (ACN) was purchased from RCI Labosan Ltd. (Bangkok, Thailand). Ethanol (EtOH) was bought from DAEJUNG (Gyeonggi-do, Korea) and methanol (MeOH) from Merck (Darmstadt, Germany) were both in HPLC grade without further purification. Berberine was bought from Sichuan Weikeqi Biological (Sichuan, China). Milli-Q water from the Millipore water purification system (Millipore, Milford, MA, USA) was used in the preparation of nanoparticles and all other solutions.
Animals
C57BL/6 mice (female, 20–25 g) were provided by the Laboratory Animal Services Centre at The Chinese University of Hong Kong. All procedures were consistently conducted in strict compliance with the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health in USA.20 The animal experiments were conducted under the approval of the Animal Ethics Committee of The Chinese University of Hong Kong (Animal ethical approval number: 20–035-GRF).
Preparation and Optimization of the PIP NPs Formulation via the Flash Nanoprecipitation Technique (FNP)
PIP NPs were prepared through flash nanoprecipitation employing a four-stream Multi-Inlet Vortex Mixer (MIVM) system as shown in Figure 1 with the ethanolic solution containing piperine and Eudragit L100-55 and the aqueous solution containing poloxamer 188. Briefly, a volume of 1 mL ethanolic solution and 2.5 mL aqueous solution was introduced to the mixer chamber of the MIVM via syringe pumps (Model PHD 2000, Harvard Apparatus, MA, USA) to induce particle precipitation.
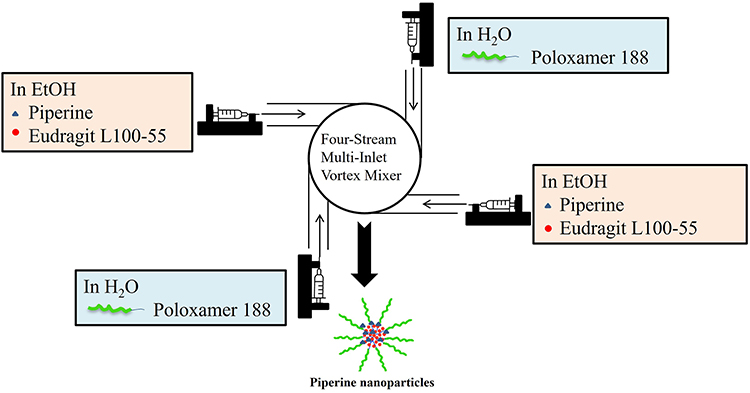 |
Figure 1 Schematic diagram of MIVM set-up for piperine nanoparticle preparation. |
The nanoparticle formulation of piperine was optimized using a response surface methodology-based method through Design-Expert® software, employing a 3-factor, 3-level Box-Behnken design as shown in Table 1. A total of 17 experiments were conducted with three replicates (n=3) for predictive modeling. Preliminary trials were conducted to identify the key factors for formulating nanoparticles. Briefly, the effect of independent variables was studied at three levels, namely high (+1), medium (0) and low (−1) levels, with the concentration of Eudragit L100-55 in EtOH (15–20 mg/mL), the aqueous solution to the ethanolic solution ratio (1.8–3.2, v/v), and Poloxamer 188 to piperine ratio (1–3, w/w) selected as the final independent variables for further optimization. Particle size (Y1) and encapsulation efficiency (Y2) were the dependent variables to serve as the quality control parameters of nanoparticles. The optimization of PIP NPs aimed to minimize the particle size and maximize the encapsulation efficiency. The optimized PIP NPs were concentrated using a vacuum centrifugation concentrator (SpeedVac, Thermo Scientific, USA) at 45 °C for 1 to 2 hours to prepare the oral formulation of piperine and further concentrated another 1 hour to prepare the intranasal formulation of piperine.
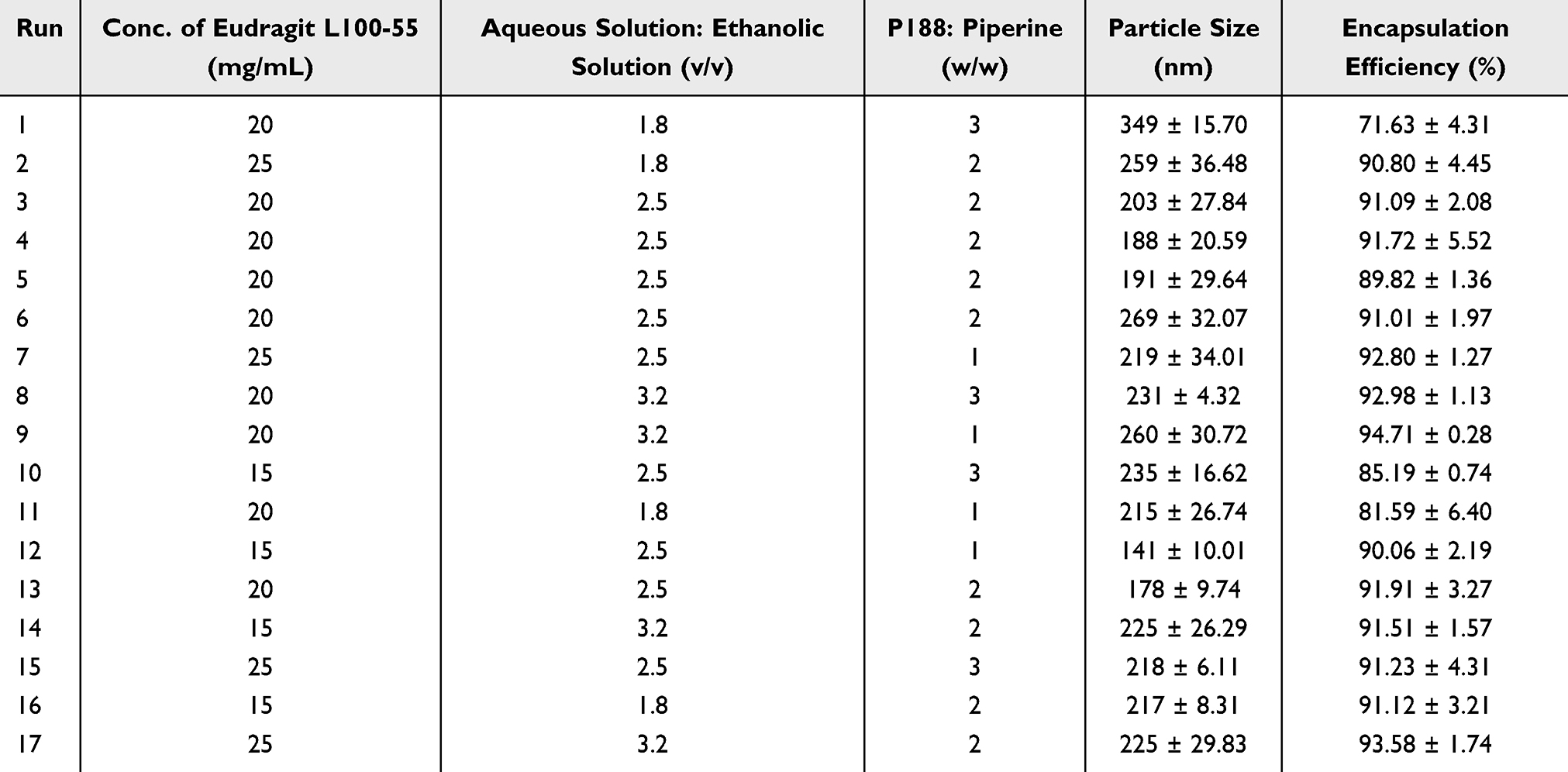 |
Table 1 Matrix of Three-Factor, Three-Level Response Surface Design and Observed Particle Size and Encapsulation Efficiency of PIP NPs |
Characterization of the Optimized PIP NPs Formulation
Quantification of Piperine via High-Performance Liquid Chromatography
To determine the concentrations of piperine in different preparations, HPLC analyses performed on the Agilent HPLC system (Agilent Technologies, Inc., Santa Clara, CA, USA) were adopted. Separations of the analytes were achieved with an Agilent ZORBAX RR Eclipse XDB-C18 column (4.6 × 150 mm, 3.5 µm) via a mobile phase consisting of 0.1% TFA in water (A) and 0.1% TFA in ACN (B). The gradient elution of B started at 50% for 5 minutes and then increased to 59% at 8 minutes, followed by a drop back to 50% at 10 minutes. The flow rate was maintained at 1.0 mL/min with an injection volume of 13 μL. The DAD detector wavelength was set at 343 nm. The auto-sampler and the analytical column temperatures were set at 4 °C and ambient, respectively. Linearity of piperine was observed in the concentration range from 10 to 2560 ng/mL with a correlation coefficient R2 = 0.9997 and 10 ng/mL as the lowest limit of quantitation.
Particle Size, Polydispersity Index and Zeta Potential of PIP NPs
A submicron particle size analyzer (DelsaNano C, Beckman Coulter, Osaka, Japan) was utilized to determine the particle size and polydispersity index (PDI) of the prepared PIP NPs. The optimized PIP NPs or concentrated PIP NPs (diluted 1:40 in water) were diluted 20 times with water, and the dynamic light scattering (DLS) technique was employed for the analysis. Additionally, the zeta potential of PIP NPs was also characterized by using the same instrument under an electrical field.
Encapsulation Efficiency (EE) of PIP NPs
To determine the encapsulation efficiency of the optimized PIP NPs and concentrated PIP NPs, the ultracentrifugation method was employed. The total content of piperine in the nanoparticles was first quantified by HPLC-DAD after dissolving the 5 μL optimized PIP NPs or diluted concentrated PIP NPs (1:40 in water) in 995 μL EtOH. To measure the unencapsulated piperine concentration, 3 mL of the optimized PIP NPs or diluted concentrated PIP NPs (1:40 in water) was centrifuged at 19,700 rpm for 1.5 hours at 4 °C using a Beckman Coulter centrifuge (Fullerton, CA, USA) followed by determining the unencapsulated piperine amount in the supernatants by HPLC/DAD as mentioned above. EE of the piperine into nanoparticles was calculated using the following equation:

Drug Loading of PIP NPs
To evaluate the drug loading, optimized PIP NPs and concentrated PIP NPs were first freeze dried for the testing. About 5 mg of the optimized PIP NPs or concentrated PIP NPs was dissolved in 5 mL EtOH solution containing 0.01 mg/mL berberine (internal standard for HPLC/DAD assay), followed by centrifugation at 13,000 rpm for 10 minutes. The total piperine concentration was then determined using the HPLC/DAD method mentioned above. Based on the total PIP amount in freeze-dried nanoparticles (optimized or concentrated) and its encapsulation efficiency, the mass of encapsulated PIP in the nanoparticles and the drug loading were calculated using the following equation:

Morphology Characterization of PIP NPs by Transmission Electron Microscopy (TEM)
PIP NPs of the optimized formulation before and after evaporative concentration were observed with TEM. The optimized or diluted concentrated PIP NPs (1:40 in water) PIP NPs was first diluted with 50-time volumes of water followed by vortex mixing. A 10 μL aliquot of the diluted sample was placed on a carbon-coated copper grid (230 mesh, 7–20 nm, Beijing Zhongjingkeyi Technology Co., China) and air-dried at room temperature. The resulted sample was subsequently stained with 10 μL of 1% phosphotungstic acid solution (w/v) for 2 minutes with excess stain removed using filter paper. The stained sample was examined using a Hitachi HT7700 TEM system (Hitachi High-Technologies Corporation, Japan).
Fourier Transforms Infrared Spectroscopy Analysis (FT-IR)
FT-IR analyses were performed on piperine (1 mg), Eudragit L100-55 (3.33 mg), Poloxamer 188 (1.66 mg), physical mixture of piperine, Eudragit L100-55, and Poloxamer 188 (1:3.33:1.66, w/w, total 6 mg), optimized PIP NPs (6 mg) and concentrated PIP NPs (6 mg) using a FT-IR spectrometer (Spectrum BX, MA, USA). All samples were freeze-dried before testing. Each freeze-dried sample was mixed with 100 mg KBr to compress into a pellet. Each pellet was scanned against a blank KBr pellet at wavenumbers ranging from 4000 to 400 cm−1 with a resolution of 4 cm−1 for 8 times/scan.
Crystallographic Characterization by Powder X-Ray Diffraction (PXRD)
The crystallographic characterization of piperine, Poloxamer 188, Eudragit L100-55, physical mixture of piperine, Eudragit L100-55, and Poloxamer 188 (1:3.33:1.66, w/w), optimized PIP NPs and concentrated PIP NPs were conducted using an X-ray diffractometer (XRD, Rigaku, mini flex 600, Tokyo, Japan) with a Cu kα radiation source (λ = 0.154 nm), operating at a constant voltage of 40 kV and a tube current of 15 mA, across a 2θ range of at 3–90°. All samples were freeze-dried before testing.
Thermal Properties by Differential Scanning Calorimetry (DSC)
Thermal analysis was performed using DSC (TA Discovery DSC 2500, TA Instruments, New Castle, DE, USA). Optimized PIP NPs and concentrated PIP NPs were first freeze dried for testing. Samples of piperine (1 mg), Eudragit L100-55 (3.33 mg), Poloxamer 188 (1.66 mg), a physical mixture of piperine, Eudragit L100-55, and Poloxamer 188 (1:3.33:1.66, w/w, total 6 mg), optimized PIP NPs (6 mg, equivalent to 1 mg of PIP), and concentrated PIP NPs (6 mg, equivalent to 1 mg of PIP) were placed in aluminium pans with lids, respectively. The samples were first heated to 110 °C, followed by cooling down to 20 °C and heating again to 200 °C at a heating rate of 10 °C/min and nitrogen flow rate of 30 mL/min.
In Vitro Release of Piperine From Various Dosage Forms
In vitro drug releases of piperine from suspension (prepared by dispersing piperine in water), optimized PIP NPs and concentrated PIP NPs were assessed using the dialysis bag-diffusion technique via a dissolution tester (708-DS Dissolution Apparatus, Agilent, Germany). Briefly, about 0.4 mg of piperine from optimized PIP NPs (0.2 mL * 2 mg/mL), 0.4 mg of piperine from concentrated PIP NPs (0.01 mL * 40 mg/mL) or 0.4 mg of piperine was added into a dialysis bag with a molecular weight cut-off of 12–14 kDa (Spectrumlabs, CA, USA), which was then immersed in 500 mL of 0.1 N HCL (pH 1.2) or phosphate buffer (0.1M, pH 6.5, containing 8.868 g sodium phosphate monobasic monohydrate and 5.06 g sodium phosphate dibasic in 1 L Milli-Q water) and maintained at 37 ± 0.5 °C with the paddle speed of 50 rpm or 100 rpm. A volume of approximately 3 mL samples was withdrawn at 0, 5, 10, 20, 30, 60, 120, 240 and 300 minutes, followed by replacement with an equal volume of prewarmed phosphate buffer. In vitro drug release of piperine was also conducted under pH transition conditions. Briefly, the dialysis bag containing 0.4 mg of piperine from optimized PIP NPs (0.2 mL * 2 mg/mL), 0.4 mg of piperine from concentrated PIP NPs (0.01 mL * 40 mg/mL) or 0.4 mg of piperine was first immersed in 500 mL of 0.1 N HCl (pH 1.2). At 0, 5, 10, 20, 30, 60, and 120 minutes, approximately 3 mL samples were withdrawn followed by replacement with an equal volume of prewarmed buffer (pH 1.2). After sampling at 120 minutes, pH of the dissolution media was adjusted to 6.5 by adding 200 mL of 0.3 M dibasic sodium hydrogen phosphate and 10.4 mL of 1 N NaOH solution as described before.21 Subsequently, approximately 3 mL samples were withdrawn at 5, 10, 20, 30, 60, 120 and 180 minutes followed by replacement with an equal volume of prewarmed buffer (pH 6.5). Concentrations of piperine in all collected samples were subjected to HPLC-DAD analyses as described above. As piperine is sensitive to light, the entire process was protected from light as suggested before.22
Stability of PIP NPs
To evaluate their storage stability, the prepared PIP NPs were stored at room temperature (prevent from light) for one month followed by the measurement of their particle size and encapsulation efficiency as described above.
Selection of the Route of Administration for PIP NPs in C57BL/6 Mice
C57BL/6 mouse was selected due to its common use in nasal administration studies23–27 and relevance to the disease model for our future pharmacology evaluation. A total of three groups of C57BL/6 mice (30 mice per group: 6 mice/time point for 5 time points) were utilized in this study, with six mice examined at each time point for a total of five time points at 5, 10, 20, 60, and 120 minutes post-dosing. The first two groups received a bolus dose of the PIP suspension or PIP NPs orally, while the third group received the concentrated PIP NPs intranasally. For the oral administrations, the first and second groups of C57BL/6 mice were administered with PIP NPs and PIP suspension (0.2 mL of 2 mg/mL) via oral gavage at a dose of 20 mg/kg, respectively. For intranasal administration, C57BL/6 mice were administered with PIP NPs of 40 mg/mL piperine at a dose of 20 mg/kg. To facilitate the administration, the mice were briefly anesthetized by inhaling 95% CO2 (recovery within 5 minutes). A total volume of 10 µL of concentrated PIP NPs was then delivered into both nostrils using a micropipette as previously described by us.28
At 5, 10, 20, 60, and 120 minutes post-dosing, mice were sacrificed followed by collecting blood and brain for further analyses. Blood samples were obtained via cardiac puncture, while brain samples were collected after an immediate infusion of 20 mL of ice-cold saline to ensure no blood residue was left in the tissue. The blood samples were then centrifuged at 13,000 rpm for 10 minutes to obtain plasma samples, while the collected whole brain was further rinsed with ice-cold saline followed by removing excess water using tissue paper. All the collected plasma and brain samples were stored at −80 °C till further analyses.
For sample analyses, the collected brain tissue was homogenized with 3 times the volume of water containing 0.2% formic acid to obtain the related brain homogenate. About 50 μL of the collected plasma or brain homogenates was added with 150 μL of berberine (200 ng/mL, as Internal Standard) for piperine quantification by HPLC-MS/MS as described in our previous study.8 The HPLC-MS/MS method was validated following the Bioanalytical Method Validation Guidance for Industry from FDA.29 The pharmacokinetic parameter of AUC0-120 min was calculated based on the obtained piperine plasma and brain concentration versus time profiles using Phoenix WinNonlin 8.1 software.
Data Analyses
All in vitro samples obtained from PIP NPs characterizations were analyzed in triplicate. The mean plus or minus one standard deviation was used to express the experimental data in both in vitro and in vivo studies. Statistically significant difference was evaluated by comparing the means between two groups via the Student’s t-test, whereas ANOVA followed by post hoc Tukey’s test was used to compare the means for multiple groups. All statistical analyses were performed using GraphPad Prism (version 9.5.1, GraphPad Software, La Jolla, CA). A p-value of less than 0.05 was considered statistically significant.
Results
Preparation of PIP NPs and Formulation Optimization
PIP NPs were successfully produced using the flash nanoprecipitation technique via a MIVM system. The PIP NPs were appeared as colloidal dispersions without any precipitation observed. The nanoparticle formulation was optimized based on the formulation and processing parameters yielding the smallest particle size with the high encapsulation efficiency of piperine. During our preliminary experiments, the ratio of piperine to Eudragit L100-55 was fixed at 1:3.33 (w/w) to maximize the drug loading of piperine in nanoparticles. In addition, the concentration of Eudragit L100-55 in EtOH (mg/mL), aqueous solution to ethanolic solution ratio (v/v) and Poloxamer 188 to piperine ratio (w/w) were identified as the most critical factors affecting the stability of the PIP NPs. In addition, the suitability for further concentration by solvent evaporation for the intranasal administration was included for consideration.
Based on the identified critical factors and their possible range, seventeen runs of experiments were conducted to optimize the nanoparticle formation with the concentration of Eudragit L100-55 in EtOH (A, 15–25 mg/mL), aqueous solution to ethanolic solution ratio (B, 1.8–3.2, v/v) and Poloxamer 188 to piperine ratio (C, 1–3, w/w) as independent variables at three levels and particle size (Y1) together with encapsulation efficiency (Y2) as dependent variables. The full design used for optimization was shown in Table 1.
The 3D surface plots and contour plots for particle size and encapsulation efficiency were displayed in Figure 2A–F to visualize interactions between independent and dependent variables. Each plot simultaneously evaluated the combined effect of two variables while keeping the third factor at its central point. The statistical experimental design indicated that the quadratic model was the most suitable model to represent the relationship between independent variables and dependent variables. Figure 2B revealed that decreasing Eudragit L100-55 in EtOH (mg/mL) and/or Poloxamer 188 to piperine ratio resulted in reduced particle size. Figure 2D and E indicated that increasing the aqueous solution to ethanolic solution ratio could result in higher EE. The optimized parameters suggested by the Design Expert to obtain the desired responses were 15 mg/mL of Eudragit L100-55 in EtOH, aqueous solution to ethanolic solution ratio as 2.8:1 (v/v), and Poloxamer 188 to piperine ratio as 1.67 (w/w). The predicted particle size and EE of the optimized formulation were 173.99 nm and 94.10%, respectively.
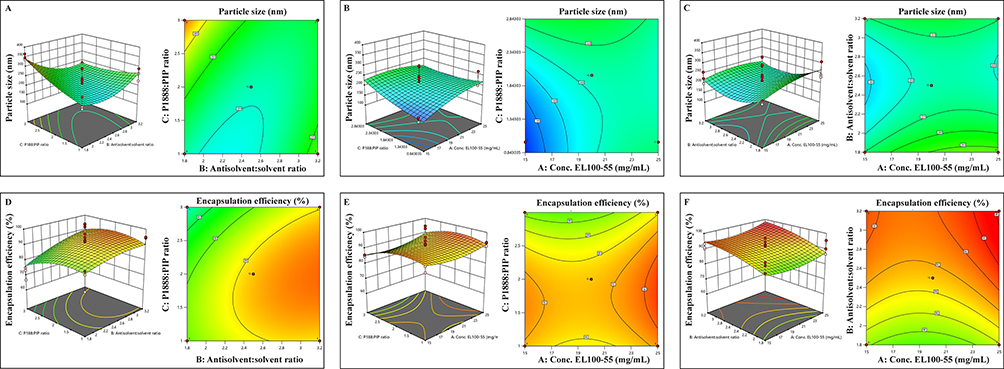 |
Figure 2 Diagrams of the 3D surface and contour plot showing effects of i) Aqueous solution to Ethanolic solution ratio, ii) Poloxamer 188 to piperine ratio and iii) Concentration of Eudragit L100-55 in EtOH on particle size (A): i and ii, (B): ii and iii, (C): i and iii) and encapsulation efficiency (D): i and ii, (E): ii and iii, (F): i and iii). |
Characterization of Optimized PIP NPs
Particle Size, Polydispersity Index, Zeta Potential and Encapsulation Efficiency of PIP NPs
PIP NPs were prepared according to the optimized conditions identified in Preparation of PIP NPs and Formulation Optimization. The optimized PIP NPs had an average particle size of 171.45 ± 2.38 nm with a PDI value of 0.27 ± 0.01, an encapsulation efficiency of 92.49 ± 1.92% and a drug loading of 15.07 ± 0.09%, which were comparable to the predicted values of particle size (173.99 nm) and EE 94.10%), confirming the reliability of our optimization processes. Since the zeta potential (−43.71 ± 5.11 mV) of the nanoparticles was greater than |30| mV, the optimized PIP NPs were considered stable colloidal suspensions that could prevent nanoparticle aggregation as suggested before.30 After solvent evaporation, no significant difference was observed in particle size, PDI, encapsulation efficiency and drug loading, indicating that the process of solvent evaporation had not changed the parameter of PIP NPs. The decrease of zeta potential in concentrated PIP NPs (−22.51 ± 4.29 mV, p<0.05) compared with it in the optimized PIP NPs suggested that the stability of PIP NPs could be decreased after solvent evaporation. The maximum concentration of piperine in concentrated PIP NPs was 52.31 ± 0.9 mg/mL.
Morphology Characterization of Optimized PIP NPs by Transmission Electron Microscopy (TEM)
The surface morphology was shown in Figure 3A for the optimized PIP NPs (i) and concentrated PIP NPs (ii, 1:40 in water). The TEM indicated that the particles were discrete, uniform, and exhibited a spherical shape with a smooth surface. It is noted that their particle sizes in the TEM image were smaller than 200 nm, which were similar to those obtained from DLS detection (171.45 ± 2.38 nm and 185.24 ± 11.91 nm respectively).
 |
Figure 3 Characterization of the PIP NPs via (A) morphology image of (i) optimized PIP NPs and (ii) concentrated PIP NPs, (B) FT-IR spectra; (C) PXRD patterns(i) Poloxamer 188, (ii) Eudragit L100-55, (iii) Piperine, (iv) Physical mixture of Piperine, Eudragit L100-55 and Poloxamer 188 (1:3.33:1.66, w/w/w), (v) Optimized PIP NPs and (vi) Concentrated PIP NPs; (D) DSC thermograms of (i) Poloxamer 188, (ii) Eudragit L100-55, (iii) Piperine, (iv) Physical mixture of Piperine, Eudragit L100-55 and Poloxamer 188 (1:3.33:1.66, w/w/w), (v) Optimized PIP NPs and (vi) Concentrated PIP NPs, and (E) in vitro release profiles at different paddles speed (50 or 100 rpm) in buffer of different pH (pH 1.2 or 6.5) under 37 °C. |
Fourier Transforms Infrared Spectroscopy Analysis (FT-IR) of Optimized PIP NPs
The FT-IR spectra of piperine, Eudragit L100-55, Poloxamer 188, physical mixture, optimized PIP NPs and concentrated PIP NPs were presented in Figure 3B. The FT-IR spectrum of piperine showed characteristic peaks at various wavenumbers, which correspond to different functional groups.31,32 These peaks were observed at different wavenumbers: 3009 cm−1 (aromatic –C–H stretching), 2940 cm−1 (C–H symmetric and asymmetric stretching), 2862 cm−1 (C–H stretching), 1634 cm−1 (stretching of –CO–N), 1612 cm−1 (symmetric and asymmetric stretching of C=C diene), 1584 cm−1 (aromatic stretching of C=C), 1492 cm−1 (aromatic stretching of C=C), 1449 cm−1 (methylenedioxy CH2 bending), 1253 cm−1 (asymmetric stretching of =C–O–C), 1194 cm−1 (asymmetric stretching of =C–O–C), 1114 cm−1 (stretching of C-O), 1032 cm−1 (symmetric stretching of =C–O–C), 997 cm−1 (C-H bending of trans –CH=CH-), 929 cm−1 (C–O stretching), and 847 ~ 805 cm−1 (out of plane C–H bending). In the spectrum of the physical mixture of piperine, Eudragit L100-55 and Poloxamer 188 (1:3.33:1.66, w/w), peaks observed from piperine at 3009 cm−1 and 2862 cm−1 were absent. In the spectrum of PIP NPs, the following characteristic peaks of piperine as: 3009 cm−1, 2862 cm−1, 1612 cm−1, 1584 cm−1, 1492 cm−1, 1194 cm−1, 1032 cm−1, 997 cm−1, 929 cm−1, and 847 ~ 805 cm−1 were absent, indicating the encapsulation of piperine into nanoparticles carriers.
Crystallographic Characterization of PIP NPs by PXRD
The PXRD method is commonly utilized to identify the crystallinity of solids and to characterize amorphous substances by observing amorphous halo patterns. Crystalline PIP appeared characteristic diffraction peaks at 14.14, 14.72, 19.22, 19.63, 22.28, 22.61 and 25.75 ° in both piperine and physical mixture as shown in Figure 3C. In contrast, the characteristic amorphous halo, indicating a lack of crystallinity, was observed in the PXRD patterns of both optimized and concentrated PIP nanoparticles, demonstrating the amorphization of piperine in these nanoparticles as shown in Figure 3C.
Thermal Properties of PIP NPs by DSC
As shown in Figure 3D, the melting temperature of piperine was observed in the pure piperine and the physical mixture of piperine, Eudragit L100-55 and Poloxamer 188 (1:3.33:1.66, w/w), whereas it was absent in the optimized PIP NPs and concentrated PIP NPs. Additionally, the glass transition temperature was observed in the optimized PIP NPs and the concentrated PIP NPs as shown in Figure 3D, indicating the amorphization of piperine in these nanoparticles.
In Vitro Release of PIP NPs
The release profiles of piperine from its suspension and nanoparticles were shown in Figure 3E, and the accumulated percentage of drug release from PIP NPs under different conditions was listed in Table S1. It was noted that, regardless of the pH or paddle speed, in vitro releases of piperine at 300 minutes from optimized PIP NPs (ranged from 48.55% to 80.79%) and concentrated PIP NPs (ranged from 52.48% to 85.79%) were all significantly higher (p<0.001) than that from piperine suspension (ranged from 5.36% to 8.40%). For the dissolution study in pH 1.2 with a paddle speed of 100 rpm, the drug release after 300 minutes from piperine suspension, optimized PIP NPs, and concentrated PIP NPs was 5.36%, 48.55% and 52.49%, respectively. For the dissolution study in pH 6.5 with the paddle speed of 100 rpm, the drug release after 300 minutes from piperine suspension, optimized PIP NPs and concentrated PIP NPs were 6.69%, 80.79% and 85.79%, respectively. Although there was no significant difference observed in in vitro piperine release optimized PIP NPs and concentrated PIP NPs at the same pH, significantly higher drug release was observed in optimized PIP NPs or concentrated PIP NPs in pH 6.5 than that in pH 1.2 at all time points till 240 minutes (p<0.001), suggesting that the in vitro release of PIP NPs and concentrated PIP NPs could be significantly influenced by the pH. Such phenomenon was double confirmed by the in vitro release of piperine from both optimized PIP NPs and concentrated PIP NPs under the pH transition method shown in Figure 3E. The piperine release under pH 1.2 after 120 minutes from optimized PIP NPs and concentrated PIP NPs was 27.54% and 29.20%, respectively, which was significantly increased to 76.51% and 71.09%, respectively, under pH 6.5 at 300 minutes. Besides the impact of pH, a significantly increased in vitro release of piperine was also observed in PIP NPs or concentrated PIP NPs under the paddle speed of 100 rpm than that under 50 rpm at all time points till 240 minutes (p<0.05), suggesting that the in vitro release of piperine from optimized PIP NPs and concentrated PIP NPs could be significantly influenced by the paddle speed.
Stability of PIP NPs
After one month of storage, no significant difference was observed in particle size and encapsulation efficiency in both the optimized PIP NPs and concentrated PIP NPs, with no aggregation and sedimentation observed, suggesting the favorable stabilities of both PIP NPs.
Significantly Enhanced Systemic and Brain Exposures of Piperine by PIP NPs with No Difference Between Oral and Intranasal Administrations
The maximum concentration of 52.31 ± 0.9 mg/mL piperine in concentrated PIP NPs could be achieved through the solvent evaporation method. Such significantly increased concentrations of piperine could lead to an intranasal formulation of concentrated PIP NPs at 40 mg/mL. The plasma Figure 4A and brain Figure 4B concentration profiles of piperine over time following oral administration of the piperine suspension and the optimized PIP NPs, as well as intranasal administration of the concentrated PIP NPs were obtained and compared. Following oral administration of the piperine suspension at 20 mg/kg, the AUC0-120min was 7519.25 ± 1263.33 ng⋅min/mL in plasma and 5992.88 ± 1945.48 ng⋅min/g in the brain, which was much lower than that after oral administration of PIP NPs (AUC0-120min of 75,256.67 ± 16,111.65 ng⋅min/mL for the plasma and 29,694.63 ± 6936.30 ng⋅min/g for the brain) (p<0.0001 for both plasma and brain). Regarding the intranasal administration of the concentrated PIP NPs, the AUC0-120min was determined to be 59308.84 ± 13,772.13 ng⋅min/mL in plasma and 27,899.90 ± 6763.10 ng⋅min/g in the brain, which were significantly higher than those observed in the oral administration of piperine suspension group (p<0.0001 for both plasma and brain). Regardless, no significant difference in AUC0-120min (for both plasma and brain) was observed between the intranasally administered and the orally administered concentrated PIP NPs.
 |
Figure 4 Comparisons of plasma (A) and brain (B) concentration versus time profiles of piperine in mice after oral administrations of the piperine suspension and the optimized PIP NPs, and intranasal administration of the optimized PIP NPs at 20 mg/kg. Each point represents mean ± S.D. (n = 6). |
Discussion
Since brain uptake of a drug is highly dependent on its physical chemical properties as well as its route of administration, strategies to improve drug brain uptake commonly included increasing the drug aqueous solubility, enhancing drug intestinal/brain permeability or utilizing alternative route of administration such as intranasal administration, etc. Based on the physical/chemical and ADME properties of piperine, we for the first time adopted the approach of increasing its aqueous solubility together with administration via intranasal route to enhance its brain uptake. Our findings indicated that increasing the solubility of piperine rather than changing the route of administration served as the most critical factor for its brain uptake enhancement, which provided essential guidance for its further pharmacological effect evaluations.
We started with converting piperine into nanoparticles to enhance its aqueous solubility. In our previous study, we prepared a PIP NPs via the FNP technique by adding the solvent (piperine and Eudragit S100 in EtOH) drop-wisely via a syringe pump into the stirring anti-solvent (Poloxamer in water) and obtained a piperine solution of 0.86 mg/mL for oral administration.15 However, such solvent delivery procedure could lead to batch-to-batch inconsistency. Moreover, the use of Eudragit S100 could result in delayed drug release at the pH of 7 in the distal ileum and colon of human gastrointestinal tract.33,34 In our current study, PIP NPs were further optimized by utilizing Eudragit L100-55 as a stabilizer instead of Eudragit S100. Eudragit L100-55, being readily soluble at pH above 5.5, leads to rapid drug release in the proximal small bowel.34,35 During the formulation optimization, we have also tried to use other types of stabilizers or surfactants, including ionic polymer (Eudragit RS 100), linear polymer (PEG 400), and non-ionic surfactant (TPGS). However, none of them could form nanoparticles or remain as stable nanoparticles after solvent evaporation. Our currently observed significant increase in piperine released from nanoparticles under pH 6.5 (than that under pH 1.2) could possibly be due to the pH-sensitive of Eudragit L100-55 as previously described.36 The use of Eudragit L100-55 can also prevent the gastric release of piperine, which may induce hemorrhagic ulceration in the stomach.37 In the meantime, considering the intranasal administration and pH in human nasal mucosa (ranged from 5.5 to 6.5),38 Eudragit L100-55 was more suitable for the current development of NPs instead of Eudragit S100 which was soluble at pH levels above 7.15,33 Once we selected the stabilizer and surfactants, PIP NPs were then prepared via a four-stream MIVM-driven FNP technique. MIVM was selected due to its high mixing efficiency39 and scalability,40 making it highly suitable for commercial applications.40
For the PIP NPs preparation optimization, 17 experiments were designed by Design of Experiments (DOE) in our current investigations. The DOE was a widely recognized and efficient method that offers several benefits, including the reduction of required experiments for formulation optimization. It enables the construction of mathematical models to assess the relevance and statistical significance of the factor effects under study and evaluate the interaction effects between these factors.41 Response surface designs were experimental designs used to model the curvature in the relationship between the factors and the response. A comparative analysis of the Box-Behnken Design (BBD) with other response surface designs (central composite, Doehlert matrix and three-level full factorial design) demonstrated that the BBD and Doehlert matrix were marginally more efficient than the central composite design and much more efficient than the three-level full factorial designs.42 Another advantage associated with the BBD was that it avoids combinations for which all factors were at their highest or lowest levels simultaneously, thereby preventing experiments under extreme conditions that might produce unsatisfactory results.42 Considering the need for further concentration of PIP NPs by solvent evaporation for the intranasal administration, the factors for formulating PIP NPs were limited to a specific range to maintain their stability and suitability for such process. Therefore, BBD was chosen to minimize the particle size and maximize the encapsulation efficiency of PIP NPs. During the optimization, it was noted that the particle size of run #12 was smaller and had lower encapsulation efficiency compared to that from the optimized PIP NPs. However, such differences between the two formulations were considered to have no impact on the penetration across olfactory region due to their small particle sizes (<200 nm). Previous cellular uptake studies indicated that nanoparticles smaller than 200 nm could effectively penetrate across both the gastrointestinal and blood–brain barrier.43 The optimized PIP NPs were eventually selected for the subsequent pharmacokinetic evaluation due to its higher encapsulation efficiency. After solvent evaporation concentration, the final piperine concentration in optimized PIP NPs could reach to over 50 mg/mL, achieving 1250-fold enhancement, which could enable us to further increase the oral dose of PIP NPs in rats from 3.5 mg/kg in our previous study15 to up to 1000 mg/kg. Such a significant increase in the dose of piperine would definitely help to further explore its efficacy. In addition, compared to our previous research, the optimized and concentrated PIP NPs demonstrated a significantly faster release, reaching a plateau at 4 hours, 20 hours quicker than our earlier PIP NPs.15 Such significantly increased in vitro release of piperine from PIP NPs could be possibly due to the use of surfactant of Poloxamer 188 in PIP NPs and amorphization of piperine shown in Figure 3C and D.
In addition to further increasing the solubility, our current studies also evaluated whether intranasal administration can further increase the brain uptake of piperine. After administrations of PIP NPs or piperine suspension to mice, our current findings indicated that formulating piperine into nanoparticles can significantly increase the AUC0-120min in both plasma (p<0.0001) and brain (p<0.0001), whereas changing the route of administration to intranasal administration does not lead to additional improvement in brain uptake. It was noted in our current study that the peak concentration of piperine was reached in about 5 minutes after oral administration. Such fast absorption of PIP NPs was due to not only the fast intestinal permeability of piperine but also the quick arrival of our prepared PIP NPs at duodenum via oral gavage of such liquid formulations.44 In the meantime, the pH of 6.8 at the duodenum in mice45 would also allow Eudragit L100-55 in PIP NPs to dissolve so as to release piperine as shown in Figure 3E. Additionally, almost identical piperine concentration versus time profiles were found in both plasma and brain tissue, which could be explained by the efficient tissue distribution of piperine to brain as we described previously.8 Moreover, it was found that the concentration of piperine dropped quickly in both plasma and brain tissue from 10 to 20 minutes, which could be also contributed by the fast distribution of piperine to other tissues as we observed in our previous study.15
Although there was no significant difference in AUC0-120min, it was noticed that both plasma and brain concentrations of piperine at the initial 5 minutes via intranasally administrated PIP NPs (1197.06 ±159.98 ng/mL in plasma and 375.79 ± 78.85 ng/g in brain) were lower (p<0.001) than that via orally administrated PIP NPs (3062 ± 778.60 ng/mL in plasma and 941.90 ± 185.34 ng/g in brain). To facilitate the intranasal administration, we had tried to use short-term anaesthesia to minimize the impact of anaesthesia in pharmacokinetic study in nasal drug delivery, as we suggested before.46 Thus, the absorption of piperine via intranasal administration would not be impacted by the short-term anaesthesia. On the other hand, it was noted that the in vitro releases of our prepared PIP NPs were significantly increased under the paddle speed of 100 rpm compared with that under 50 rpm, which was consistent with previous findings on the higher dissolution rate resulted from the enhanced fluid velocity via increasing paddle speed.47,48 The observed lower piperine concentration at 5 minutes via nasal administration could possibly be related to the weaker motility of nasal cavity (with pressures ranged from 4.6 to 6.6 mm Hg49) compared to that of peristaltic movement in the gastrointestinal tract (with pressure of small intestinal contractions exceed 20 mm Hg).[50] Nevertheless, the similar AUC0-120min between orally administrated PIP NPs and intranasally administrated PIP NPs can be attributed to the high brain penetration potency of piperine as demonstrated in our previous study.8 Therefore, in our future pharmacological evaluations for piperine, orally administered PIP NPs would be used as it is more convenient for the long-term drug administrations in mouse models.
Conclusion
We had successfully formulated piperine into nanoparticles to further enhance its solubility to 52.31 ± 0.9 mg/mL after vacuuming solvent evaporation. Both oral and intranasal administrations of the optimized PIP NPs at 20 mg/kg could significantly increase systemic (7.9–10 times) and brain (4.7–5.0 times) exposures of piperine than that from its suspension dosage form, with no significant difference between the two routes of administrations. Our findings suggested that increasing the solubility of piperine rather than changing the route of administration as the most critical step to enhance the brain uptake of piperine in vivo.
Data Sharing Statement
Data will be made available on request.
Acknowledgment
This work was supported by the General Research Fund from the University Grant Council of Hong Kong SAR [Reference number: 1145732].
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. O’Neil, C MJ. Royal Society of, the Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals.
2. Nazifi M, Oryan S, Esfahani DE, Ashrafpoor M. The functional effects of piperine and piperine plus donepezil on hippocampal synaptic plasticity impairment in rat model of Alzheimer’s disease. Life Sci. 2021;265:118802. doi:10.1016/j.lfs.2020.118802
3. Khalili-Fomeshi M, Azizi MG, Esmaeili MR, et al. Piperine restores streptozotocin-induced cognitive impairments: insights into oxidative balance in cerebrospinal fluid and hippocampus. Behav Brain Res. 2018;337:131–138. doi:10.1016/j.bbr.2017.09.031
4. Wang C, Cai Z, Wang W, et al. Piperine attenuates cognitive impairment in an experimental mouse model of sporadic Alzheimer’s disease. J Nutr Biochem. 2019;70:147–155. doi:10.1016/j.jnutbio.2019.05.009
5. Shrivastava P, Vaibhav K, Tabassum R, et al. Anti-apoptotic and anti-inflammatory effect of Piperine on 6-OHDA induced Parkinson’s rat model. J Nutr Biochem. 2013;24:680–687. doi:10.1016/j.jnutbio.2012.03.018
6. Chen C-Y, Li W, Qu K-P, Chen C-R. Piperine exerts anti-seizure effects via the TRPV1 receptor in mice. Eur J Pharmacol. 2013;714:288–294. doi:10.1016/j.ejphar.2013.07.041
7. Verma AK, Khan E, Mishra SK, Jain N, Kumar A. Piperine modulates protein mediated toxicity in fragile X-associated tremor/ataxia syndrome through interacting expanded CGG repeat (r (CGG) exp) RNA. ACS Chem Neurosci. 2019;10:3778–3788. doi:10.1021/acschemneuro.9b00282
8. Ren T, Wang Q, Li C, Yang M, Zuo Z. Efficient brain uptake of piperine and its pharmacokinetics characterization after oral administration. Xenobiotica. 2018;48:1249–1257. doi:10.1080/00498254.2017.1405293
9. Bhat BG, Chandrasekhara N. Studies on the metabolism of piperine: absorption, tissue distribution and excretion of urinary conjugates in rats. Toxicology. 1986;40:83–92. doi:10.1016/0300-483X(86)90048-X
10. Roy S, Gupta A, Chopra H, et al. Pharmacokinetic study of piperine in mice plasma after orally and intravenous administration. Int J Drug Delivery. 2012;4:107.
11. Sahu PK, Sharma A, Rayees S, et al. Pharmacokinetic study of piperine in Wistar rats after oral and intravenous administration. Int J Drug Delivery. 2014;6:82.
12. Li C, Wang Q, Ren T, et al. Non-linear pharmacokinetics of piperine and its herb-drug interactions with docetaxel in Sprague-Dawley rats. J Pharm Biomed Anal. 2016;128:286–293. doi:10.1016/j.jpba.2016.05.041
13. Reddy BBK, Karunakar A. Biopharmaceutics classification system: a regulatory approach. Dissolution Technol. 2011;18:31–37. doi:10.14227/DT180111P31
14. Salsabila H, Fitriani L, Zaini E. Recent strategies for improving solubility and oral bioavailability of piperine. Int J Appl Pharm. 2021;13:31–39. doi:10.22159/ijap.2021v13i4.41596
15. Ren T, Hu M, Cheng Y, et al. Piperine-loaded nanoparticles with enhanced dissolution and oral bioavailability for epilepsy control. Eur J Pharm Sci. 2019;137:104988. doi:10.1016/j.ejps.2019.104988
16. Priprem A, Chonpathompikunlert P, Sutthiparinyanont S, Wattanathorn J. Antidepressant and cognitive activities of intranasal piperine-encapsulated liposomes. Adv Biosci Biotechnol. 2011;2:108–116. doi:10.4236/abb.2011.22017
17. Gupta I, Adin SN, Rashid MA, Alhamhoom Y, Aqil M, Mujeeb M. Spanlastics as a potential approach for enhancing the nose-to-brain delivery of piperine: in vitro prospect and in vivo therapeutic efficacy for the management of epilepsy. Pharmaceutics. 2023;15:641. doi:10.3390/pharmaceutics15020641
18. Crowe TP, Greenlee MHW, Kanthasamy AG, Hsu WH. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018;195:44–52. doi:10.1016/j.lfs.2017.12.025
19. Maigler F, Ladel S, Flamm J, et al. Selective CNS targeting and distribution with a refined region-specific intranasal delivery technique via the olfactory mucosa. Pharmaceutics. 2021;13:1904. doi:10.3390/pharmaceutics13111904
20. Council NR. Guide for the Care and Use of Laboratory Animals.
21. Hedaya MA, El-Masry SM, Helmy SA. Physiologically relevant model to establish the in vivo-in vitro correlation for etamsylate controlled release matrix tablets. J Drug Delivery Sci Technol. 2021;66:102864. doi:10.1016/j.jddst.2021.102864
22. Hashimoto K, YAOI T, KOSHIBA H, et al. Photochemical isomerization of piperine, a pungent constituent in pepper, Food Sci. Technol Int Tokyo. 1996;2:24–29. doi:10.3136/fsti9596t9798.2.24
23. Wang Q, Peng S, Hu Y, et al. Efficient brain uptake and distribution of an expanded CAG RNA inhibitor DB213 via intranasal administration. Eur J Pharm Sci. 2019;127:240–251. doi:10.1016/j.ejps.2018.10.025
24. Hanson LR, Fine JM, Renner DB, et al. Intranasal delivery of deferoxamine reduces spatial memory loss in APP/PS1 mice, Drug Delivery Transl. Res. 2012;2:160–168. doi:10.1007/s13346-011-0050-2
25. Jiang Y, Zou Y, Chen S, et al. The anti-inflammatory effect of donepezil on experimental autoimmune encephalomyelitis in C57 BL/6 mice. Neuropharmacology. 2013;73:415–424. doi:10.1016/j.neuropharm.2013.06.023
26. Jin Z, Han Y, Zhang D, et al. Application of intranasal administration in the delivery of antidepressant active ingredients. Pharmaceutics. 2022;14:2070. doi:10.3390/pharmaceutics14102070
27. Gartziandia O, Herran E, Pedraz JL, Carro E, Igartua M, Hernandez RM. Chitosan coated nanostructured lipid carriers for brain delivery of proteins by intranasal administration. Colloids Surf B. 2015;134:304–313. doi:10.1016/j.colsurfb.2015.06.054
28. Wang Q, Wong C-H, Chan HE, Lee W-Y, Zuo Z. Statistical design of experiment (DoE) based development and optimization of DB213 in situ thermosensitive gel for intranasal delivery. Int J Pharm. 2018;539:50–57. doi:10.1016/j.ijpharm.2018.01.032
29. U.S. FDA, Bioanalytical Method Validation Guidance for Industry, US Department of Health and Human Services Food and Drug Administration. Center for Drug Evaluation and Research and Center for Veterinary Medicine; 2018.
30. Raval N, Maheshwari R, Kalyane D, Youngren-Ortiz SR, Chougule MB, Tekade RK. Importance of physicochemical characterization of nanoparticles in pharmaceutical product development. Basic Fundamentals Drug Delivery. 2019;2019:369–400. Elsevier.
31. Moraru AC, Roşca I, Crăciun B, Nicolescu A, Chiriac AE, Voicu V. Insights of the antimicrobial activity of piperine extracted from Piper nigrum L. Farmacia. 2019;67:1099–1105. doi:10.31925/farmacia.2019.6.24
32. Khatri S, Awasthi R. Piperine containing floating microspheres: an approach for drug targeting to the upper gastrointestinal tract, Drug Delivery Transl. Res. 2016;6:299–307. doi:10.1007/s13346-016-0285-z
33. Sheskey RCRPJ, Quinn ME. Handbook of Pharmaceutical Excipients. 2009.
34. Hadji H, Bouchemal K. Advances in the treatment of inflammatory bowel disease: focus on polysaccharide nanoparticulate drug delivery systems. Adv Drug Deliv Rev. 2022;181:114101. doi:10.1016/j.addr.2021.114101
35. Nikam A, Sahoo PR, Musale S, Pagar RR, Paiva-Santos AC, Giram PS. A systematic overview of Eudragit® based copolymer for smart healthcare. Pharmaceutics. 2023;15:587. doi:10.3390/pharmaceutics15020587
36. Pérez‐Ibarbia L, Majdanski TC, Schubert S, Windhab N, Schubert US. Synthesis and characterization of colored EUDRAGIT® as enteric coating material. J Polym Sci Part A. 2016;54:2386–2393. doi:10.1002/pola.28113
37. Tripathi AK, Ray AK, Mishra SK. Molecular and pharmacological aspects of piperine as a potential molecule for disease prevention and management: evidence from clinical trials. Beni Suef Univ J Basic Appl Sci. 2022;11:16. doi:10.1186/s43088-022-00196-1
38. England R, Homer J, Knight L, Ell S. Nasal pH measurement: a reliable and repeatable parameter. Clin Otolaryngol Allied Sci. 1999;24:67–68. doi:10.1046/j.1365-2273.1999.00223.x
39. Tao J, Chow SF, Zheng Y. Application of flash nanoprecipitation to fabricate poorly water-soluble drug nanoparticles. Acta Pharm Sin B. 2019;9:4–18. doi:10.1016/j.apsb.2018.11.001
40. Chan HW, Chow S, Zhang X, Kwok PCL, Chow SF. Role of particle size in translational research of nanomedicines for successful drug delivery: discrepancies and inadequacies. J Pharm Sci. 2023;112:2371–2384. doi:10.1016/j.xphs.2023.07.002
41. Box GE, Hunter WH, Hunter S. Statistics for Experimenters. John Wiley and Sons New York;1978.
42. Ferreira SC, Bruns R, Ferreira HS, et al. Box-Behnken design: an alternative for the optimization of analytical methods, Anal. Chim Acta. 2007;597:179–186. doi:10.1016/j.aca.2007.07.011
43. Kulkarni SA, Feng -S-S. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharma Res. 2013;30:2512–2522. doi:10.1007/s11095-012-0958-3
44. Bennink RJ, De Jonge WJ, Symonds EL, et al. Validation of gastric-emptying scintigraphy of solids and liquids in mice using dedicated animal pinhole scintigraphy. J Nucl Med. 2003;44:1099–1104.
45. Sun Y, Koyama Y, Shimada S. Measurement of intraluminal pH changes in the gastrointestinal tract of mice with gastrointestinal diseases. Biochem Biophys Res Commun. 2022;620:129–134. doi:10.1016/j.bbrc.2022.06.061
46. Wong YC, Qian S, Zuo Z. Pharmacokinetic comparison between the long-term anesthetized, short-term anesthetized and conscious rat models in nasal drug delivery. Pharma Res. 2014;31:2107–2123. doi:10.1007/s11095-014-1312-8
47. McCarthy LG, Bradley G, Sexton JC, Corrigan OI, Healy AM. Computational fluid dynamics modeling of the paddle dissolution apparatus: agitation rate, mixing patterns, and fluid velocities. AAPS Pharm Sci Tech. 2004;5:50–59. doi:10.1208/pt050231
48. Todaro V, Persoons T, Grove G, Healy AM, D’Arcy DM. Characterization and simulation of hydrodynamics in the paddle, basket and flow-through dissolution testing apparatuses-a review. Dissolution Technol. 2017;24:24–36. doi:10.14227/DT240317P24
49. J.m. G Jr, Hendley JO, Phillips CD, Bass CR, Mygind N, Winther B. Nose blowing propels nasal fluid into the paranasal sinuses, Clin. Infect Dis. 2000;30:387–391. doi:10.1086/313661
50. Camilleri M, Hasler WL, Parkman HP, Quigley EM, Soffer E. Measurement of gastrointestinal motility in the GI laboratory. Gastroenterology. 1998;115:747–762. doi:10.1016/S0016-5085(98)70155-6
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.