")
Back to Journals » International Journal of Nanomedicine » Volume 20
Small Extracellular Vesicles from Young Healthy Human Plasma Inhibit Cardiac Fibrosis After Myocardial Infarction via miR-664a-3p Targeting SMAD4
Authors Wang W, Li Y, Zhang C, Zhou H, Li C, Cheng R, Chen X, Pu Y, Chen Y
Received 23 July 2024
Accepted for publication 5 January 2025
Published 13 January 2025 Volume 2025:20 Pages 557—579
DOI https://doi.org/10.2147/IJN.S488368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Krishna Nune
Weiwei Wang,1,* Ying Li,1,* Cheng Zhang,2,* Haoyang Zhou,1 Chunyu Li,3 Rong Cheng,1 Xufeng Chen,1 Yanan Pu,4 Yan Chen1,5,6
1Department of Emergency and Critical Care Medicine, The First Affiliated Hospital of Nanjing Medical University, Nanjing, 210029, People’s Republic of China; 2Long Jiang Central Laboratory, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210029, People’s Republic of China; 3Long Jiang Intensive Care Unit, The First Affiliated Hospital of Nanjing Medical University, Nanjing, 210029, People’s Republic of China; 4Department of Clinical Laboratory, Nanjing Chest Hospital, Affiliated Nanjing Brain Hospital, Nanjing Medical University, Nanjing, 210029, People’s Republic of China; 5Department of Emergency and Critical Care Medicine, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital, Gusu School, Nanjing Medical University, Suzhou, 215000, People’s Republic of China; 6Department of Emergency Management, School of Health Policy & Management, Nanjing Medical University, Nanjing, 211166, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Chen; Yanan Pu, Email [email protected]; [email protected]
Purpose: Cardiac fibrosis, a key contributor to ventricular pathologic remodeling and heart failure, currently lacks effective therapeutic approaches.
Patients and Methods: Small extracellular vesicles from young healthy human plasma (Young-sEVs) were characterized via protein marker, transmission electron microscopy, and nanoparticle tracking analysis, then applied in cellular models and mouse models of cardiac fibrosis. Western blotting and qRT-PCR were used to identify protective signaling pathways in cardiac fibroblasts (CFs).
Results: Young-sEVs significantly inhibited cardiac fibrosis and subsequent cardiac dysfunction post-myocardial infarction (MI) in mice. The main findings included that echocardiographic assessments four weeks post-MI indicated that Young-sEVs improved left ventricular ejection fraction (LVEF) and fractional shortening (LVFS), and reduced left ventricular internal diameter in diastole (LVIDd) and systole (LVIDs). Treatment with Young-sEVs also decreased Masson-positive fibroblast areas and collagen synthesis in cardiac tissue. However, sEVs from the old control group did not achieve the above effect. Consistent with in vivo results, Young-sEVs could also inhibit the proliferation, migration, and collagen synthesis of CFs in the TGF-β 1-induced cellular fibrosis model. High-throughput microRNA (miRNA) sequencing and qRT-PCR analysis revealed that miR-664a-3p was abundant in Young-sEVs. The high expression of miR-664a-3p significantly inhibited the proliferation, migration, and collagen synthesis of TGF-β 1-induced CFs. However, suppressing the expression of miR-664a-3p in Young-sEVs eliminated their therapeutic effect on cardiac fibrosis in mice. Further studies confirmed SMAD4 as a direct downstream target of miR-664a-3p, whose overexpression could reverse the anti-fibrotic effects of miR-664a-3p.
Conclusion: In summary, these findings firstly revealed that Young-sEVs could directly bind to the 3′-untranslated region of SMAD4 mRNA through miR-664a-3p, thereby inhibiting the TGF-β/SMAD4 signaling pathway to protect heart from fibrosis and improve cardiac function. Considering the ease of obtaining plasma-derived sEVs, our study offers a promising therapeutic strategy for heart failure, with the potential for rapid clinical translation in the near future.
Keywords: extracellular vesicles, young human plasma, cardiac fibrosis, MiR-664a-3p, SMAD4
Graphical Abstract:

Introduction
In 2022, heart failure affected approximately 64 million people worldwide.1 Heart failure was the leading cause of hospitalization among individuals aged 65 and above.2 Cardiac fibrosis is the fundamental pathological process of heart failure, characterized by the activation and proliferation of cardiac fibroblasts and excessive deposition of extracellular matrix.3 In the early stages, myocardial fibrosis serves as a compensatory mechanism to maintain the structure and mechanical function of the damaged myocardium.4 However, persistent myocardial fibrosis can lead to the accumulation of scar tissue, decreased myocardial elasticity, pathological remodeling of the ventricles, and ultimately result in heart failure.5 Currently, there are no specific treatment methods available to satisfactorily reduce the incidence of cardiac fibrosis. Therefore, the search for effective new strategies and medications to inhibit cardiac fibrosis is of crucial significance in the treatment of heart failure.
In recent years, the utilization of extracellular vesicles (EVs)-based therapy has emerged as a promising approach to tackle organ fibrosis. Exosomes or small EVs (sEVs), cell-derived nanoscale bilayer extracellular vesicles, range in size from 50–200 nm and are present in all tissue spaces, body fluids, and cell culture media.3 The sEVs play an vital role in intercellular communication during various physiological processes and pathological conditions by delivering biological regulatory information (proteins, lipids, DNAs, and RNAs, particularly microRNA) to recipient cells in surrounding and distant sites and organs.6,7 In numerous preclinical studies, sEVs-based nanomedicines had been demonstrated to prevent multiple types of organ fibrosis through intricate mechanisms, including promoting tissue regeneration, resolving inflammation, inactivating myofibroblasts, and degrading extracellular matrix (ECM).8 Furthermore, studies had indicated that sEVs play a role in the occurrence and progression of cardiac fibrosis through intercellular communication.9 For example, exosomes derived from cardiosphere-derived cells (CDCs) had been shown to reduce the formation of fibrotic scars and alleviate adverse ventricular remodeling in a porcine model of myocardial infarction.10 Exosomes derived from mesenchymal stem cells (MSCs) had been shown to possess anti-fibrotic properties in cardiac injury models through various pathways.6,11
The sEVs are present in circulation, and their levels in plasma (or serum) are significantly higher than those secreted by individual cell types.12 Recent studies have provided evidence that blood-derived sEVs exhibit various therapeutic effects,13,14 particularly those derived from young healthy plasma. It has been reported that such plasma holds significant value in suppressing inflammatory responses and providing neuroprotection following cerebral hemorrhage.12 Furthermore, studies had shown that plasma-derived exosomes from neonatal mice or healthy people had beneficial effects in mediating angiogenesis and protecting myocardial tissue from ischemia-reperfusion injury following acute myocardial infarction (AMI).13,15 However, the impact of sEVs from young healthy human plasma on cardiac fibrosis after MI and their underlying mechanisms remain unclear.
As one of the important cargoes of sEVs, microRNAs (miRNAs) are a class of endogenous, short non-coding RNA molecules present in all eukaryotic cells.16 MiRNAs interact with the 3′-untranslated region (3′-UTR) of their target mRNA to regulate the expression of the target gene.9 In the field of cardiovascular research, miRNAs derived from sEVs often play a role in intercellular communication and information transfer among cardiovascular cells.6 According to reports, miRNAs could regulate cardiac fibrosis. For example, miR-125b17 and miR-43318 could promote fibroblast proliferation and differentiation into myofibroblasts in cardiac fibrosis models, while exosomal miR-294 derived from embryonic stem cells had been found to suppress cardiac fibrosis and prevent heart failure after myocardial infarction.6 However, it is currently unclear whether specific miRNAs derived from sEVs of young healthy individuals’ plasma can serve as a novel therapeutic strategy for treating heart failure and improving cardiac fibrosis. Further research is needed to explore this potential avenue of treatment.
Therefore, in this research, we administered Young-sEVs via myocardial injection into a mouse model of cardiac fibrosis and introduced them into a cellular model of transforming growth factor beta 1 (TGF-β1)-induced CFs fibrosis to investigate their therapeutic potential. Echocardiography, Masson staining, Western blotting and quantitative real-time polymerase chain reaction (qRT-PCR) analysis indicated that Young-sEVs improved cardiac dysfunction post-MI and reduced fibrosis in vivo. In vitro, these sEVs inhibited proliferation, migration, and fibrotic proteins expression in TGF-β1-induced CFs. High-throughput miRNA sequencing and qRT-PCR identified miR-664a-3p was abundant in Young-sEVs. The high expression of miR-664a-3p significantly inhibited proliferation, migration, and collagen synthesis in CFs. Suppressing the expression of miR-664a-3p in Young-sEVs eliminated their therapeutic effect on cardiac fibrosis in mice. Further, miR-664a-3p was found to bind directly to the 3′-UTR of SMAD family member 4 (SMAD4) mRNA, thereby inhibiting the TGF-β/SMAD4 signaling transduction. This research highlights the potential of Young-sEVs as a therapy for cardiac fibrosis and lays groundwork for future clinical translation.
Materials and Methods
Young Healthy Human Plasma Collection
According to previous research,12 peripheral blood samples were collected from thirteen healthy young male individuals aged 18–25 years and thirteen healthy old male individuals aged 65–70 years as controls. The basic information of the 26 subjects was shown in Table S1. Referring to previous studies,19–22 healthy human plasma was defined as plasma collected from individuals with no symptoms of disease, no medication use, no metabolic disorders, and normal physical examinations. Blood collection was conducted with the approval of the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University (ethics number: 2023-SRFA-401), and informed consent was obtained from each participant. The blood samples were placed in ethylenediaminetetraacetic acid (EDTA) anticoagulant tubes, gently mixed, centrifuged at 3000 rpm for 10 minutes at 4°C, and the liquid above the sediment was taken. The supernatant was further centrifuged at 3000 rpm for 15 minutes, and the supernatant plasma was collected and stored at −80°C for subsequent use.
The sEVs Isolation and Identification
As previously described,12 sEVs were isolated from freshly thawed plasma using ultracentrifugation (Beckman Coulter Optima L-100 XP ultracentrifuge, Miami, FL). In brief, the plasma was centrifuged at 2000 g for 10 minutes and 12,000 g for 30 minutes at 4°C to remove cell debris and larger vesicles. Subsequently, the plasma was collected and filtered through a 0.45 μm filter. The filter was washed with 3 mL of phosphate buffer saline (PBS) and diluted to reduce viscosity. The diluted plasma was ultracentrifuged at 120,000 g for 2 hours at 4 °C. The precipitate was resuspended in 100 μL PBS to obtain sEVs solution and stored at −80°C. The sEVs characterization was conducted by analyzing the expression of specific markers CD63, CD81, and tumor susceptibility gene 101 (TSG101) using Western blotting. The diameter and concentration were measured using Nanoparticle tracking analysis (NTA, Malvern Instruments Ltd., Malvern, UK), and the morphology of sEVs was characterized using transmission electron microscopy (TEM, JEM-1200EX, EOL Ltd., Tokyo, Japan).
The sEVs Uptake Examination
The sEVs were labeled with Dil (red, 1 μM, Beyotime, China) at a concentration of 10μg/mL, followed by ultracentrifugation to remove excess dye, and washed twice. CFs were incubated for 20 minutes at 37°C in a medium infused with DiO (green, Beyotime, China) solution, followed by washed twice with PBS. CFs labeled with DiO underwent a 2-hour incubation with sEVs marked with Dil. This was followed by stabilization using 4% paraformaldehyde (PFA) at room temperature for a duration of 10 minutes. Subsequently, the cells were washed three times with PBS. Thereafter, CFs were treated with DAPI (blue, Beyotime, China) at room temperature for 5 minutes. All reagents used were supplied by Invitrogen Corporation (Carlsbad, CA, USA). The samples were examined using the Nikon Eclipse Ti confocal laser scanning microscope.
Animals and Experimental Design
Male C57BL/6 mice aged 6–8 weeks were purchased from Vital River Biological Co., Ltd (Beijing, China) and acclimated for one week. All animals were kept in controlled environments with a temperature range of 12–22°C and a 24-hour light/dark cycle. They were provided with unrestricted access to food and water. After one week of acclimation, all mice (approximately weighing 25 g) were randomly divided into four groups: (a) Sham, (b) MI, (c) MI-Young-sEVs, and (d) MI-sEVs from old healthy human plasma (Old-sEVs). The surgical procedure was carried out as described previously.23,24 In brief, anesthesia was induced in mice by intraperitoneal injection of pentobarbital sodium (45mg/kg), followed by endotracheal intubation connected to a rodent ventilator for ventilation. Next, the chest was opened between the second and third intercostal spaces to expose the heart. Subsequently, the left anterior descending coronary artery (LAD) was exposed. It was then ligated using 7–0 silk sutures. The successful establishment of the MI model was confirmed by observing the whitening of the surrounding myocardium in the ligation area. In the sham surgery group, mice underwent a similar surgical procedure, but without ligation of the LAD. The sEVs from young healthy human plasma (200 μg suspended in 10 μL of PBS, dose reference to previous studies23) and sEVs from old healthy human plasma (200 μg suspended in 10 μL of PBS) were respectively injected into four different sites surrounding the infarcted myocardium after LAD, forming the MI-Young-sEVs group and MI-Old-sEVs group. An equal volume of PBS (10 μL) was injected into the same sites in the MI group. At 28 days post-surgery, mice were subjected to echocardiographic assessment. After 28 days, the mice were euthanized to obtain cardiac tissue for further research purposes.
To verify the role of miR-664a-3p in anti-cardiac fibrosis mediated by Young-sEVs, mice were randomly divided into four groups: (a) Sham, (b) MI, (c) MI-negative control inhibitor (NC inhibitor)-Young-sEVs, and (d) MI-miR-664a-3p inhibitor-Young-sEVs. Young-sEVs were transfected with a miR-664a-3p inhibitor or NC inhibitor via electroporation, and the engineered sEVs were injected into the mice with MI. The methods of myocardial injection and the treatment of the Sham and MI groups were consistent with the descriptions above. Electroporation was conducted according to the instructions provided by the Gene Pulse Xcell Electroporation System (Bio-Rad, Hercules, CA, USA). After electroporation, the ExoQuick-TC Kit (System Biosciences, Palo Alto, CA, USA) was used to isolate sEVs that had been transfected with the miR-664a-3p inhibitor or the NC inhibitor. All animal experimental protocols followed the guidelines provided by the National Institutes of Health (NIH). The experimental procedures adhered to the guidelines of the Animal Care and Use Committee of Nanjing Medical University and were approved by the same committee (ethics number: IACUC-2111018).
Echocardiography
Echocardiographic measurements were performed at 4 weeks after surgery using the Vevo3100 Echocardiography System (VisualSonics, Toronto, Canada) to assess cardiac function. The following parameters were measured: left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS), left ventricular internal dimension in diastole (LVIDd), and left ventricular internal dimension in systole (LVIDs). Briefly, mice were anesthetized using inhaled isoflurane and secured on the ultrasound machine’s platform. Cardiac functional parameters were measured using M-mode tracking, a technique that provided high-resolution images of the heart in motion by capturing a single plane of ultrasound waves. All measurements were obtained as the average of 3 to 5 consecutive cardiac cycles.
Masson’s Staining
The cardiac tissue obtained was preserved in 4% PFA for a period of 48 hours. It was then subjected to a dehydration process, encased within a paraffin block, and cut into sections measuring 5 micrometers in thickness. These sections were prepared for detailed histopathological analysis. As previously described,23 Masson’s trichrome staining was utilized to quantify the extent of fibrosis in the left ventricle (LV). Images were captured using a scanning electron microscope (SU8010, Japan), and ImageJ software (NIH, USA) was employed to quantify the collagen protein area of the infarcted zone (CAIZ), by calculating the proportion of the blue-stained area within the infarcted region relative to the area stained in the region not affected by infarction.
Immunofluorescence Staining
The heart tissues were collected as described above, fixed in 4% PFA, and embedded in paraffin for sectioning. For immunofluorescence staining, the heart tissues were stained with a primary antibody against cardiac Troponin T (cTnT, 1:1000, P50752, Servicebio) and incubated overnight. This was followed by the addition of a fluorescein-conjugated secondary antibody (1:500, GB25303, Servicebio) and incubation for 1 hour at room temperature in the dark. After counterstaining the nuclei with DAPI, images were captured under a fluorescence microscope.
Primary Mouse CFs Isolation and Culture
As described in the previous study,4 euthanasia was performed on 50 neonatal C57BL/6 mice aged 1–3 days after disinfection with 75% ethanol, followed by isolation of primary CFs. In brief, collect and mince the ventricular myocardial tissue from 50 neonatal mice. Then, add a preprepared myocardial digestion solution containing 0.6% trypsin (Sigma, USA) and 0.4% collagenase (Worthington, USA) for digestion. Every 6–8 minutes, remove the digested liquid and add 6 mL of horse serum (Invitrogen, USA) to terminate digestion. The ratio of myocardial digestion solution to horse serum is 6:1. After complete digestion of the myocardial tissue, centrifuge the mixture at 1000 rpm for 5 minutes and discard the supernatant. Incubate the remaining material for 40 minutes in complete culture medium composed of Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Shanghai, China) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin-neomycin (Gibco, Shanghai, China). Next, remove the supernatant by aspiration, harvest the CFs utilizing the differential adhesion technique, and culture them in complete culture medium. All studies were conducted using passage two to three CFs. CFs at passage 2–3 were cultured in serum-free medium for 24 hours, followed by treating CFs with 10 ng/mL recombinant human TGF-β1 (PeproTech, USA) for 24 hours to establish the cellular model of cardiac fibrosis.
Cell Proliferation Assay
Proliferation of CFs was assessed by 5-ethynyl-20-deoxyuridine (EdU) and Cell Counting Kit-8 (CCK-8) assays. For the EdU assay, according to the operating instructions of the EdU kit (Beyotime, Shanghai, China), inoculate 9 × 104 CFs per well into a 6-well plate. After the cells were cultured overnight and returned to normal condition, they were co-incubated with 10 μM EdU for 2 hours. After the EdU labeling of the cells was complete and fix, remove the culture medium, add 1 mL of 4% PFA, and fix at room temperature for 15 minutes. Remove the fixative, wash the cells three times with 1 mL of washing solution (PBS containing 3% bovine serum albumin, BSA, Beyotime, Shanghai, China) per well, each time for 3–5 minutes. Remove the washing solution, add 1 mL of permeabilization solution (PBS containing 0.3% Triton X-100 Solution, Beyotime, Shanghai, China) per well, and incubate at room temperature for 10–15 minutes. Remove the permeabilization solution, wash the cells 1–2 times with 1 mL of washing solution per well, each time for 3–5 minutes. Afterwards, prepare the staining solution according to the EdU instructions, add 0.5 mL of the staining solution to each well, and incubate for 30 minutes in the dark at room temperature. Remove the staining solution, wash the cells three times with PBS, and finally add the nuclear stain Hoechst 33342 (Beyotime, Shanghai, China) to stain the nucleus. Images were captured by the Leica DMi8 fluorescent microscope with the Leica LAS-X software. The incorporation rate of EdU was calculated as the ratio of [EdU stained cells (red fluorescence) / Hoechst 33342 stained cells (blue fluorescence)] ×100%.25 For the CCK-8 assay, CFs (5 × 10³ cells/well) were seeded into a 96-well culture plates and incubated overnight. Subsequently, they were incubated for 24, 48, and 72 hours in a complete medium in the absence or presence of sEVs and/or various concentrations of TGF-β1. After incubation, 10 μL of CCK-8 (Biosharp, Hefei, China) was added, and the absorbance was measured at 24, 48, and 72-hour time points using a spectrophotometer set (ELx800, BioTek, Winooski, VT, USA).
Cell Migration Assay
Migration of CFs was measured by transwell and wound healing assays. For the transwell assay, CFs (1 × 104 cells/well) were seeded into the upper chamber of a 24-well cell culture chamber (8μm pore size, Corning, New York, USA) and incubated in serum-free DMEM with the absence or presence of sEVs and/or TGF-β1, while the lower chamber was filled with DMEM containing 10% FBS. After 24 hours of incubation, cells were fixed with 4% PFA for 30 minutes. Following PFA removal, cells were stained with a 0.1% crystal violet staining solution (Beyotime, Shanghai, China) for 20 minutes. After the removal of the crystal violet staining solution, cells were washed twice with PBS, and finally images were captured using an inverted microscope. For the wound healing assay, CFs were seeded in 6-well plates with complete culture medium and incubated overnight to achieve full monolayer confluence. A vertical scratch was made using a 10 μL sterile pipette tip, followed by washing the 6-well plates twice with PBS to remove the scratched cell fragments. Images of the scratched area were captured immediately after the scratching (time 0) and again after 24 hours using an inverted microscope. Wound width was measured using ImageJ software (NIH, USA), and the wound closure rate was calculated as the percentage of final wound width relative to the initial wound width.
Protein Extraction and Western Blotting
Proteins were extracted from human plasma sEVs, mouse cardiac tissue and cultured primary CFs utilizing a protein extraction kit (Thermo Massachusetts, USA). The concentration of each protein was detected using a bicinchoninic acid protein assay kit (Beyotime, Shanghai, China). Western blotting was performed according to the process described in previous studies.7,16 An equivalent amount of total protein extract was fractionated via sodium dodecyl sulphate polyacrylamide gel electrophoresis and subsequently transferred onto polyvinylidene fluoride membranes. The membranes were treated with a 5% skim milk solution to block nonspecific binding, followed by overnight incubation at 4°C with different primary antibodies. They were then incubated with corresponding secondary antibodies matched to the primary antibody species at room temperature for two hours. After washing three times with PBS, an enhanced chemiluminescence Western blotting kit (Beyotime, Shanghai, China) were used to detect immunoreactive bands, and ImageJ software (NIH, USA) was employed to quantify the bands. The antibodies used were as follows: Collagen Type I (1:1000, WL0088, Wanleibio), Collagen Type III (1:1000, T510299S, Abmart), Alpha smooth muscle actin (α-SMA, 1:1000, T55295F, Abmart), Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH, 1:1000, P60037F, Abmart), SMAD4 (1:1000, A19116, ABclonal), CD63 (1:1000, ab134045, Abcam), CD81 (1:1000, T55742, Abmart), TSG101 (1:1000, ab133586, Abcam), and Anti-rabbit IgG, HRP-linked Antibody (1:5000, WLA023a, Wanleibio).
Quantitative Real-Time PCR Analysis
Total RNA was extracted from human plasma sEVs, mouse cardiac tissue and cultured primary CFs using the Trizol reagent (Invitrogen), followed by reverse transcription into complementary DNA (cDNA) with the HiScript III RT SuperMix for quantitative PCR (qPCR, R302-01, Vazyme, Nanjing, China), following the guidelines provided by the supplier. MiRNAs were isolated from the human plasma sEVs, mouse cardiac tissue and CFs utilizing the mirVana RNA Isolation Kit (Ambion, Austin, TX) as directed by the manufacturer’s instructions. The synthesis of cDNA, along with qRT-PCR, was conducted in accordance with the All-in-One miRNA qRT-PCR Detection Kit (GeneCopoeia, Rockville, MD, USA). Subsequently, qPCR for mRNA was conducted with the ChamQ SYBR qPCR Master Mix (Low ROX Premixed) (Q331-02, Vazyme, Nanjing, China), and for miRNA, qPCR was performed with the 2 × All-in-One qPCR Mix (GeneCopoeia), utilizing the LightCycler96 PCR system (Roche, Inc., Switzerland). Primers for real-time quantitative PCR, including miR-1304-3p, miR-6885-5p, miR-183-3p, miR-183-5p, miR-3188, miR-6750-5p, miR-1914-5p, miR-664a-3p, miR-1323, miR-150-3p, miR-2116-3p, miR-483-3p, miR-877-3p, miR-182-5p and miR-6803-3p and U6, were all acquired from GeneCopoeia. The relative expression levels of mRNA and miRNA were quantified using the 2^−ΔΔCt calculation method. For qPCR, the quantification of mRNA was standardized against GAPDH levels, and quantification of miRNA was benchmarked to U6 expression.
RNA Sequencing and Analysis
The sEVs from human plasma were extracted using ultracentrifugation, and RNA was isolated according to the manufacturer’s instructions using an Exosomal RNA Isolation Kit (Invitrogen, Carlsbad, CA, USA). After extracting total RNA, the purity of RNA was measured using NanoDrop 2000 (ThermoScientific, USA), and the integrity of RNA was assessed using Agilent 2100 (Agilent Technologies, USA). Following the protocol of the TruSeq Small RNA Sample Prep Kit (Illumina, San Diego, California, USA), a small RNA library was prepared using approximately 10 ng of total RNA. Subsequently, paired-end sequencing (150 bp) was performed on an Illumina Nova-seq 6000 system (Illumina, California, USA) at Gene Denovo Biotechnology Co., Ltd (Guangzhou, China), following the supplier’s recommended protocol. We used three bioinformatics databases (miRWalk, TargetScanHuman 8.0 and miRBase) to predict the common target genes of differentially expressed miRNAs. Subsequently, perform functional enrichment analysis of Gene Ontology (GO) terms (http://www.geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) to annotate the targets of differentially expressed miRNAs.
Cell Transfection with miRNA Mimic/Inhibitor and Plasmids
The Synthesis and purification of hsa-miR-664a-3p mimic, negative control mimic (NC mimic), its inhibitor, and NC inhibitor were conducted through GeneCopoeia. Plasmids containing the SMAD4 expression gene and control vector plasmid were manufactured by GeneChem (Shanghai, China). The transfection procedure was as follows: Seed the CFs into a 6-well plate until they reached 70–80% confluence. Then, transfect the CFs with 100 nM of miR-664a-3p mimic or NC mimic, as well as its respective inhibitor or NC inhibitor (GenePharma, Shanghai, China), utilizing Lipofectamine 3000 (Invitrogen) in combination with Opti-MEM media (Invitrogen). For plasmid DNA, transfect into CFs using X-treme GENE HP DNA Transfection Reagent (6366236001, Roche, Shanghai, China). Replace the cell culture medium 6 hours post-transfection. Guidelines provided by the manufacturers were followed for all transfections.
Luciferase Activity Assay
The CFs were plated at a density of 5×104 cells per well in a 24-well plate overnight. Subsequently, they were transfected with wild-type (WT) or mutant-type (MUT) 3′-UTR luciferase reporter vectors of SMAD4, along with mimic-NC, miR-664a-3p mimic, inhibitor-NC, and miR-664a-3p inhibitor using Lipofectamine 3000 (Invitrogen). After 36 hours post-transfection, cells were lysed, and luciferase activity was measured by the Luc-Pair™ Dual-Luciferase Assay Kit 2.0 (GenePharma). Luciferase activity was standardized based on the Renilla/Firefly luciferase signal in CFs.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software (Version 9.5; La Jolla, California). Continuous variables were expressed as mean ± standard deviation (SD). The significance of differences between groups was determined using unpaired Student’s t-test, while differences among multiple groups were assessed by one-way analysis of variance (ANOVA), followed by Tukey’s test. P values < 0.05 were considered statistically significant.
Results
Characterization of sEVs and Internalized by CFs and cardiac tissue
To begin with, we isolated and extensively characterized sEVs from young healthy human plasma. The transmission electron microscopy and NanoSight analysis revealed the typical cup- or round-shaped morphology of the sEVs, with a size range of 50 to 200 nm (Figure 1A and B, TEM and NTA of the old control group were shown in Figure S1A and B). Further analysis of the particle size of Young-sEVs and Old-sEVs was conducted. The results showed that the average size of the Young-sEVs was 107 nm ± 23 nm, while the average size of the Old-sEVs was 95 nm ± 21 nm, with no significant differences observed between the two groups (Figure S1C). Western blotting confirmed the presence of sEVs-associated protein markers CD63, CD81, and TSG101 (Figure 1C). To assess the internalization of sEVs by cells, Dil-labeled sEVs were co-cultured with DiO-labeled CFs for 2 hours, followed by fixation with 4% polyformaldehyde and observation under confocal laser scanning microscopy. After co-incubating with CFs, we observed a significant internalization of sEVs by the CFs (Figure 1D). Dil-labeled sEVs were administered to mice via intramyocardial injection to evaluate their uptake by cardiac tissues. As shown in Figure 1E, Dil-labeled sEVs were detectable in cardiac tissues six hours post-injection, indicating efficient in vivo internalization of sEVs by the heart tissues. These findings indicated that the vesicles extracted by ultracentrifugation were indeed sEVs, and they were capable of being taken up and internalized both in vivo and in vitro.
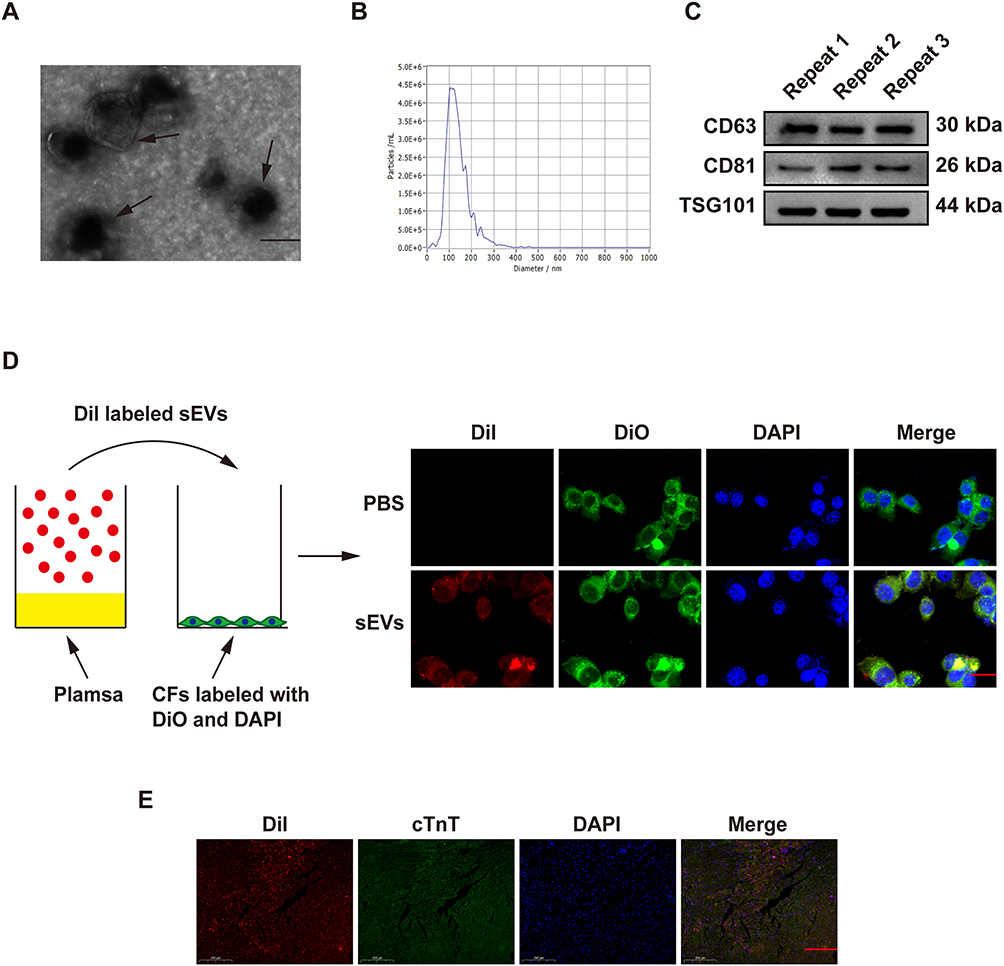 |
Figure 1 Characterization and internalization of sEVs. (A) Transmission electron microscopy imaging of young healthy human plasma-derived sEVs (scale bar = 100 nm). (B) Particle size distribution analysis using NanoSight tracking analysis. (C) Western blotting analysis of the sEVs-associated markers CD63, CD81 and TSG101. (D) Dil-labeled sEVs derived from human plasma were taken up by CFs (scale bar = 25 µm). (E) Six hours after injecting Dil-labeled sEVs (red) into mouse cardiac tissue, immunofluorescence staining and identification were performed on the heart tissues (n = 5, magnification: 20×; scale bar = 200 μm). Data are presented from three independent experiments. Abbreviations: CFs, cardiac fibroblasts; cTnT, cardiac Troponin T; PBS, phosphate buffer saline; sEVs, small extracellular vesicles; TSG 101, tumor susceptibility gene 101. |
Intramyocardial Delivery of Young-sEVs Inhibit Cardiac Fibrosis After MI
To investigate the potential role of Young-sEVs in cardiac fibrosis following myocardial infarction, we employed a mouse model of myocardial infarction in vivo. Echocardiography was performed four weeks post-MI to assess cardiac structure and function. The echocardiographic examination revealed that MI impaired cardiac structure and function of the mice, characterized by reduced LVEF and LVFS, as well as increased LVIDd and LVIDs values. Importantly, these effects were ameliorated by treatment with Young-sEVs (Figure 2A–C). Furthermore, cardiac fibrosis was also significantly alleviated after treatment with Young-sEVs, accompanied by a reduction in the Masson-positive fibroblast area (Figure 2D). In addition, there was a reduction in the mRNA and protein expression of collagen type I (Col-I), collagen type III (Col-III), and α-smooth muscle actin (α-SMA) (Figure 2E and F). However, Old-sEVs did not achieve the aforementioned effects. These data suggested that Young-sEVs could ameliorate cardiac fibrosis and functional impairment following myocardial infarction.
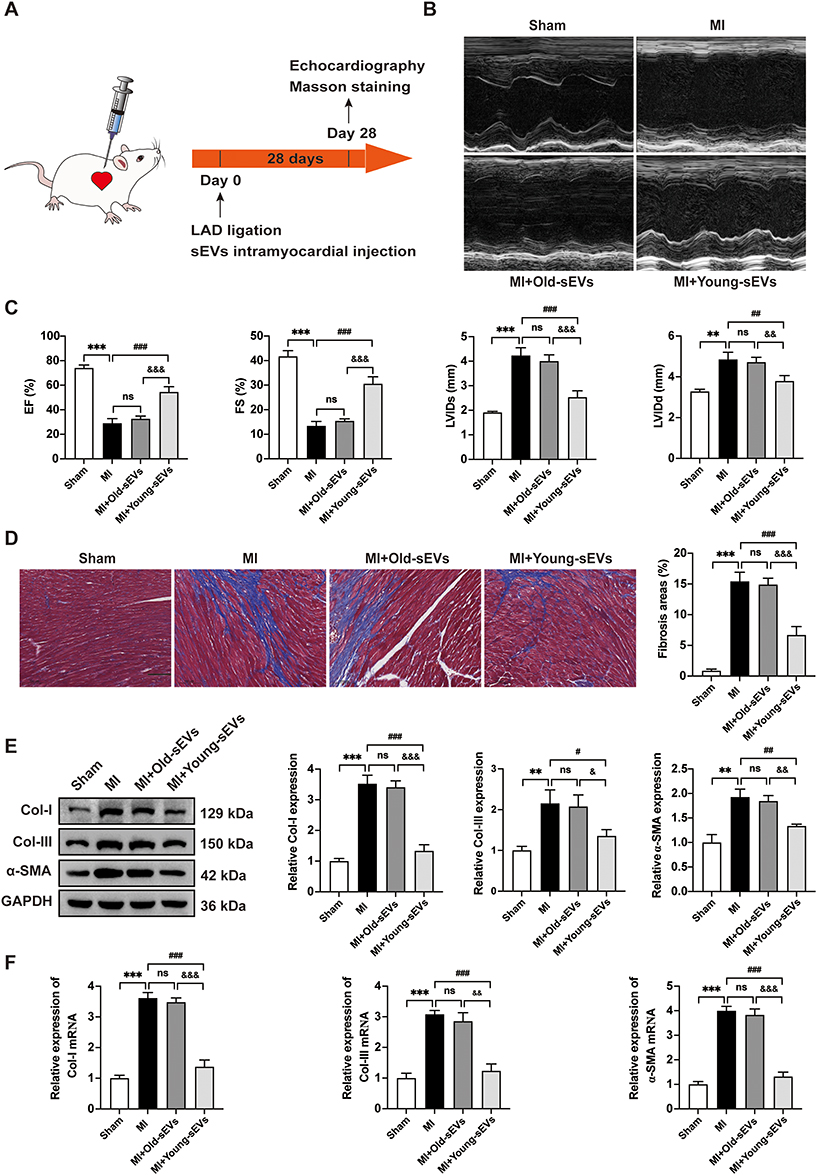 |
Figure 2 Young-sEVs inhibit cardiac fibrosis after MI. (A) The flowchart of experimental design in vivo. (B) Representative echocardiograms of mouse hearts in each group at 28 days post-MI. (C) Echocardiographic measurements of EF, FS, LVIDs and LVIDd (n = 6). (D) Masson’s trichrome staining of representative cardiac cross-sections and the collagen volume fraction (n = 6, magnification: 20 ×; scale bar = 100 μm). (E) Western blotting analysis and quantification of Col-I, Col-III and α-SMA protein levels in cardiac tissues (n = 3). (F) qRT-PCR analysis of the mRNA expressions of Col-I, Col-III and α-SMA in cardiac tissues (n = 3). Data were presented as mean ± SD. **P < 0.01, ***P < 0.001, compared with the sham group. #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the MI group. &P < 0.05, &&P < 0.01, &&&P < 0.001, compared with the MI + Old-sEVs group. Abbreviations: α-SMA, alpha smooth muscle actin; Col-I, collagen I; Col-III, collagen III; Old-sEVs, small extracellular vesicles from old healthy human plasma; Young-sEVs, small extracellular vesicles from young healthy human plasma; EF, ejection fraction; FS, fractional shortening; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; LAD, left anterior descending coronary artery; LVIDd, left ventricular internal dimension in diastole; LVIDs, left ventricular internal dimension in systole; MI, myocardial infarction; ns, no statistical difference; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation. |
Young-sEVs Inhibit Fibrotic Responses of CFs in vitro
Young-sEVs have been shown to inhibit cardiac fibrosis in mice after MI. Our subsequent investigation aims to explore the role of young plasma sEVs in activated CFs fibrosis. To determine the concentration of TGF-β1 that induces fibrosis in CFs, we treated CFs with different concentrations of TGF-β1 for 24h, 48h, and 72h, respectively. As shown in Figure S2, TGF-β1 significantly increased the viability of CFs in a time- and dose-dependent manner and promoted the expression of fibrotic proteins in a dose-dependent manner. Therefore, in this study, we used 10 ng/mL of TGF-β1 to treat CFs for 24 hours to evaluate cell proliferation, migration, and the expression levels of fibrotic proteins and mRNA. To investigate the effect of Young-sEVs on the fibrosis of CFs, we added 100 μg/mL of Young-sEVs or Old-sEVs into TGF-β1-induced CFs, with the dosage based on previous studies.26 Through cell proliferation assays (EdU immunofluorescence staining and CCK-8 assays) and migration assays (Transwell and Wound healing assays), we observed that Young-sEVs effectively reversed the proliferation and migration of TGF-β1-induced CFs (Figure 3A–D). Furthermore, Western blotting and qRT-PCR analysis demonstrated that Young-sEVs significantly inhibited the upregulation of Col-I, Col-III and α-SMA mRNA and protein expression in TGF-β1-induced CFs (Figure 3E and F). Consistent with the in vivo results, the Old-sEVs group did not achieve the aforementioned therapeutic effects. The above results indicated that Young-sEVs had an inhibitory effect on fibrosis in CFs.
 |
Figure 3 Young-sEVs inhibit fibrosis of TGF-β1-induced CFs in vitro. (A-C) Proliferation of CFs was assessed using the EdU assay and CCK-8 assay. For the EdU experiment, red fluorescence indicated proliferating cells, while blue fluorescence denoted the cell nuclei. The proportion of EdU-positive CFs and results of CCK-8 assay were shown in panel (scale bar = 100 μm). (D) The Wound healing assay and Transwell assay were used to determine the migration of CFs. The two methods were evaluated after incubation for 24 hours and the relative migration speed were measured (scale bar = 100 μm). (E) Western blotting analysis and quantification of Col-I, Col-III and α-SMA protein levels in CFs. (F) qRT-PCR analysis of the mRNA expressions of Col-I, Col-III and α-SMA in CFs. Data are presented from three independent experiments. Data were presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the negative control (NC) group. #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the TGF-β1 group. &P < 0.05, &&P < 0.01, &&&P < 0.001, compared with the TGF-β1 + Old-sEVs group. Abbreviations: α-SMA, alpha smooth muscle actin; CCK-8, cell counting kit-8; CFs, cardiac fibroblasts; Col-I, collagen I; Col-III, collagen III; EdU, 5-ethynyl-20-deoxyuridine; Old-sEVs, small extracellular vesicles from old healthy human plasma; Young-sEVs, small extracellular vesicles from young healthy human plasma; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; ns, no statistical difference; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation; TGF-β1, transforming growth factor beta 1. |
Differential Expression of miRNAs in Young-sEVs
It is known that sEVs can transfer various bioactive molecules, such as miRNA, to recipient cells. To analyze the molecular basis of sEVs-mediated inhibition of cardiac fibrosis, this study used miRNA-sequencing to screen the differential miRNA expression profiles (Figure 4A). At the same time, to effectively identify potential active microRNAs, we isolated sEVs from the plasma of old healthy human to serve as a control. Based on the sequencing results, the top fifteen differentially expressed miRNAs derived from sEVs are miR-1304-3p, miR-6885-5p, miR-183-3p, miR-183-5p, miR-3188, miR-6750-5p, miR-1914-5p, miR-664a-3p, miR-1323, miR-150-3p, miR-2116-3p, miR-3130-3p, miR-483-3p, miR-877-3p and miR-182-5p. We again verified the expression of the above 15 miRNAs in plasma-derived sEVs from 10 young healthy individuals and 10 old healthy individuals using qRT-PCR. The results showed that miR-6750-5p, miR-664a-3p, miR-1323, and miR-3130-3p were abundantly present in the Young-sEVs, whereas their levels were significantly reduced in the plasma sEVs of the old control group (Figure 4B; The expression of other miRNAs was shown in Figure S3). We selected these four miRNAs for further investigation. Expression levels of the four miRNAs were analyzed using qRT-PCR in both TGF-β1-induced fibrotic cell models and in cardiac tissues from mice post-MI. The results indicated that only miR-664a-3p was significantly downregulated in TGF-β1-induced CFs and cardiac tissues post-MI. However, its expression significantly increased after treatment with Young-sEVs (Figure 4C and D). Therefore, we hypothesized that miR-664a-3p played a crucial role in the development of cardiac fibrosis and warranted further investigation. Additionally, the data indicated that Young-sEVs were enriched with miR-664a-3p, which could potentially be transferred to the heart via intramyocardial injection.
 |
Figure 4 Identification of miR-664a-3p as the main effector in Young-sEVs against cardiac fibrosis. (A) Heatmap of upregulated and downregulated miRNAs in Young-sEVs compared to Old-sEVs. Select differentially expressed miRNAs (marked in red) for further validation. (B) Further qRT-PCR validation experiments comparing 10 young healthy individuals with 10 old healthy individuals revealed that 4 out of the top 15 highly expressed miRNAs identified in sequencing showed significantly higher expression levels (n = 10). (C) qRT-PCR verified the expression of the above 4 miRNAs in TGF-β1-induced CFs (n = 3). (D) qRT-PCR was further utilized to confirm the expression of the above 4 miRNAs in cardiac tissue of mice after MI (n = 3). Data were presented as mean ± SD. ***P < 0.001, compared with the negative control (NC) group or sham group or old group. ##P < 0.01, compared with the TGF-β1 group or MI group. Abbreviations: CFs, cardiac fibroblasts; Old-sEVs, small extracellular vesicles from old healthy human plasma; Young-sEVs, small extracellular vesicles from young healthy human plasma; MI, myocardial infarction; miR-Seq, microRNA sequencing; ns, no statistical difference; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation; TGF-β1, transforming growth factor beta 1. |
MiRNA-664a-3p Inhibits Proliferation, Migration and Fibrosis of TGF-β1-Induced CFs in vitro
To further investigate the specific effects of miR-664a-3p, which was abundantly present in Young-sEVs, gain-of-function and loss-of-function studies were conducted. CFs were successfully transfected with miR-664a-3p mimic, miR-664a-3p inhibitor, and their NCs and were validated using qRT-PCR after 48 hours (Figure 5A). By performing EdU immunofluorescence staining and CCK-8 assays, we observed that TGF-β1 significantly promoted the proliferation of CFs, while miR-664a-3p mimic (miR-664a-3p) effectively reversed the TGF-β1-induced upregulation of proliferation (Figure 5B-D). Furthermore, transwell migration assays and wound healing assays demonstrated that TGF-β1 significantly promoted the migration of CFs, while miR-664a-3p efficiently reversed the upregulation of migration induced by TGF-β1 (Figure 5E). Similarly, Western blotting and qRT-PCR analysis revealed that miR-664a-3p effectively inhibited the upregulation of Col-I, Col-III and α-SMA expression in TGF-β1-induced CFs (Figure 5F and G). Conversely, transfection of CFs with the miR-664a-3p inhibitor further enhanced TGF-β1-induced proliferation and migration of CFs (Figure 6A-D) and further increased the expression of TGF-β1-induced Col-I, Col-III and α-SMA (Figure 6E and F). Overall, these results suggested that miR-664a-3p played a crucial role in the fibrotic process of CFs induced by TGF-β1.
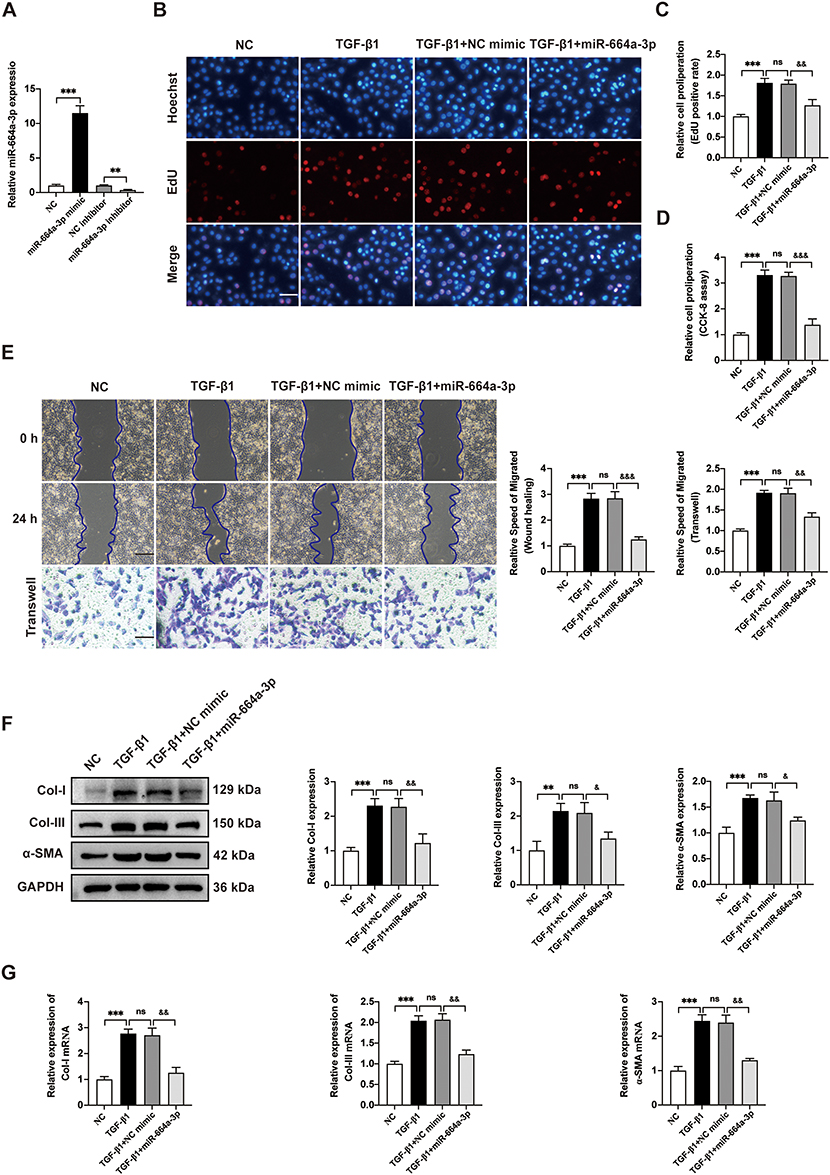 |
Figure 5 The function of miR-664a-3p mimic in inhibiting fibrosis of TGF-β1-induced CFs. (A) The expression of miR-664a-3p in CFs transfected with miR-664a-3p mimic, inhibitor or negative control (NC) was detected by qRT-PCR. (B-D) The proliferation of CFs transfected with the miR-664a-3p mimic was evaluated using EdU and CCK-8 cell proliferation assays. The proportion of EdU-positive CFs and results of CCK-8 assay were shown in panel (scale bar = 100 μm). (E) The migration of CFs transfected with the miR-664a-3p mimic was assessed using the Wound healing assay and Transwell migration assay. The two methods were evaluated after incubation for 24 hours and the relative migration speed were measured (scale bar = 100 μm). (F) Western blotting analysis and quantification of Col-I, Col-III and α-SMA protein levels in CFs after transfection with miR-664a-3p mimic. (G) qRT-PCR analysis of the mRNA expressions of Col-I, Col-III and α-SMA in CFs transfected with miR-664a-3p mimic. Data are presented from three independent experiments. Data were presented as mean ± SD. **P < 0.01, ***P < 0.001, compared with the negative control (NC) group. &P < 0.05, &&P < 0.01, &&&P < 0.001, compared with the TGF-β1 + NC mimic group. Abbreviations: α-SMA, alpha smooth muscle actin; CCK-8, cell counting kit-8; CFs, cardiac fibroblasts; Col-I, collagen I; Col-III, collagen III; EdU, 5-ethynyl-20-deoxyuridine; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; ns, no statistical difference; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation; TGF-β1, transforming growth factor beta 1. |
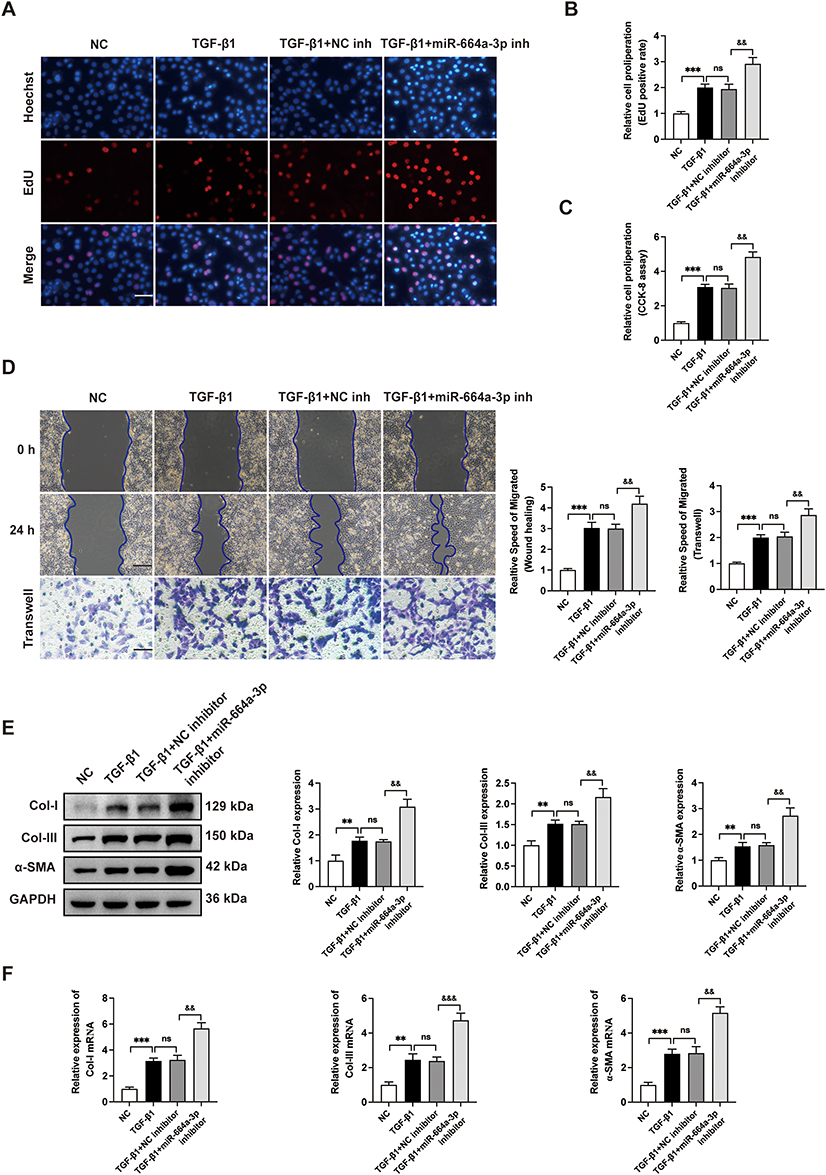 |
Figure 6 The miR-664a-3p inhibitor further aggravates the fibrosis of TGF-β1-induced CFs. (A-C) The proliferation of CFs transfected with the miR-664a-3p inhibitor was evaluated using EdU and CCK-8 cell proliferation assays. The proportion of EdU-positive CFs and results of CCK-8 assay were shown in panel (scale bar = 100 μm). (D) The migration of CFs transfected with the miR-664a-3p inhibitor was assessed using the Wound healing assay and Transwell migration assay. The two methods were evaluated after incubation for 24 hours and the relative migration speed were measured (scale bar = 100 μm). (E) Western blotting analysis and quantification of Col-I, Col-III and α-SMA protein levels in CFs after transfection with miR-664a-3p inhibitor. (F) qRT-PCR analysis of the mRNA expressions of Col-I, Col-III and α-SMA in CFs transfected with miR-664a-3p inhibitor. Data are presented from three independent experiments. Data were presented as mean ± SD. **P < 0.01, ***P < 0.001, compared with the negative control (NC) group. &&P < 0.01, &&&P < 0.001, compared with the TGF-β1 + NC inhibitor group. Abbreviations: α-SMA, alpha smooth muscle actin; CCK-8, cell counting kit-8; CFs, cardiac fibroblasts; Col-I, collagen I; Col-III, collagen III; EdU, 5-ethynyl-20-deoxyuridine; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; inh, inhibitor; ns, no statistical difference; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation; TGF-β1, transforming growth factor beta 1. |
The Impact of miR-664a-3p Inhibition on the Anti-Cardiac Fibrosis Effects of Young-sEVs
To verify the role of miR-664a-3p in mediating the anti-cardiac fibrotic effects of Young-sEVs, we transfected Young-sEVs with the miR-664a-3p inhibitor or NC inhibitor. We then evaluated the therapeutic efficacy of NC inhibitor-Young-sEVs and miR-664a-3p inhibitor-Young-sEVs in myocardial fibrosis post-MI in mice. Echocardiography and Masson’s trichrome staining at 28 days post-injection showed that treatment with NC inhibitor-Young-sEVs significantly improved cardiac function (Figure 7A and B) and reduced fibrotic area (Figure 7C), while treatment with miR-664a-3p inhibitor-Young-sEVs reversed these improvements. Further analysis of the expression of fibrosis-related proteins and mRNA revealed that compared to the NC inhibitor-Young-sEVs group, the miR-664a-3p inhibitor-Young-sEVs abolished the suppression of Col-I, Col-III and α-SMA protein and mRNA expression by Young-sEVs (Figure 7D and E). These data indicated that in the mouse MI model, inhibition of miR-664a-3p expression abolished the cardioprotective effects mediated by Young-sEVs. This also confirmed that miR-664a-3p derived from Young-sEVs could inhibit the progression of cardiac fibrosis in mice.
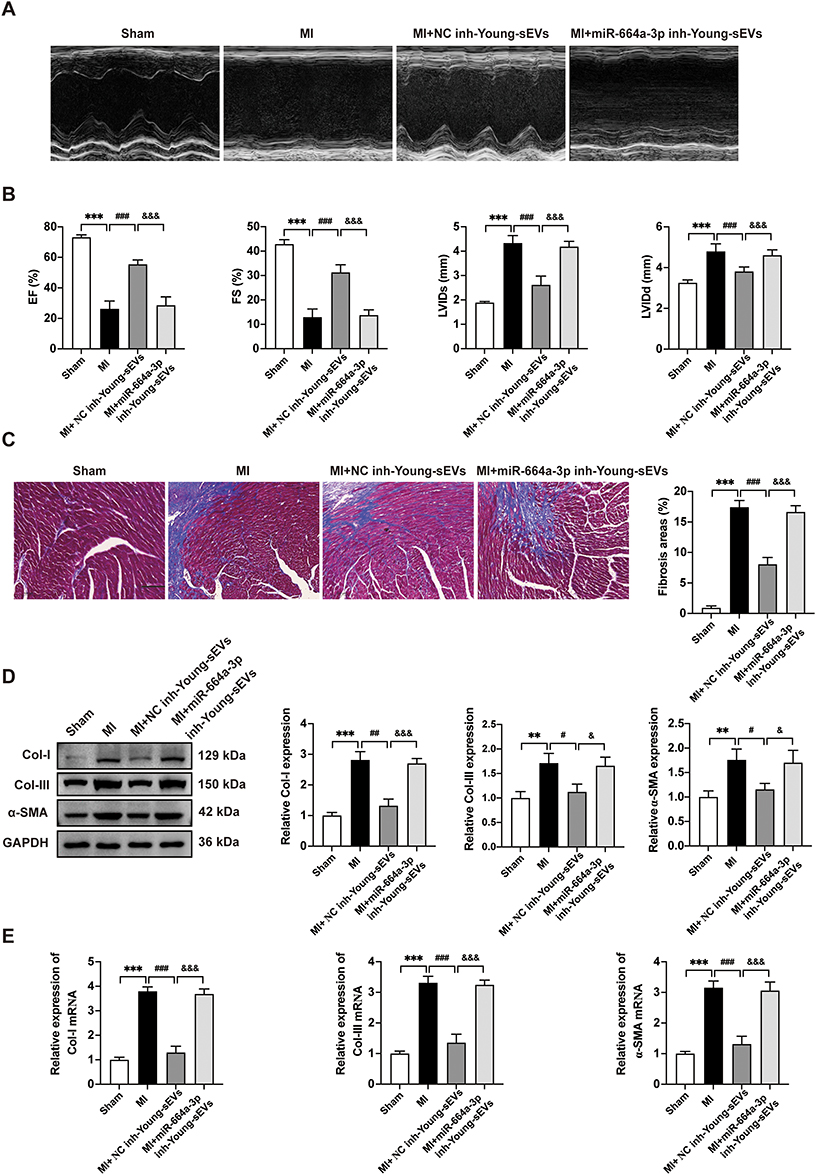 |
Figure 7 The suppression of miR-664a-3p expression in Young-sEVs abolishes its function in inhibiting cardiac fibrosis in vivo. (A) Representative echocardiograms of mouse hearts in each group at 28 days post-MI. (B) Echocardiographic measurements of EF, FS, LVIDs and LVIDd (n = 6). (C) Masson’s trichrome staining of representative cardiac cross-sections and the collagen volume fraction (n = 6, magnification: 20 ×; scale bar = 100 μm). (D) Western blotting analysis and quantification of Col-I, Col-III and α-SMA protein levels in cardiac tissues (n = 3). (E) qRT-PCR analysis of the mRNA expressions of Col-I, Col-III and α-SMA in cardiac tissues (n = 3). Data were presented as mean ± SD. **P < 0.01, ***P < 0.001, compared with the sham group. #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the MI group. &P < 0.05, &&&P < 0.001, compared with the MI + NC inhibitor-Young-sEVs group. Abbreviations: α-SMA, alpha smooth muscle actin; Col-I, collagen I; Col-III, collagen III; Young-sEVs, small extracellular vesicles from young healthy human plasma; EF, ejection fraction; FS, fractional shortening; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; inh, inhibitor; LVIDd, left ventricular internal dimension in diastole; LVIDs, left ventricular internal dimension in systole; MI, myocardial infarction; NC, negative control; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation. |
SMAD4 is Identified as the Target of miR-664a-3p to Mediate Its Antifibrotic Effects
To explore potential signaling pathways associated with biological activity of miR-664a-3p, we conducted a combined analysis using miRWalk (http://mirwalk.umm.uni-heidelberg.de/), TargetScanHuman 8.0 (http://www.targetscan.org/) and miRBase (http://www.mirbase.org/) to predict the target genes of miR-664a-3p, resulting in the identification of 24 candidate target genes (Figure 8A; Table S2). To gain further insight into the biological functions of these target genes, we performed a GO analysis on the predicted gene transcripts, finding involvement in various biological processes, including cellular and metabolic processes, regulation of biological processes, cellular functions, and regulation of protein binding, among others (Figure S4). The KEGG analysis revealed the top 15 KEGG pathways with significant p-values, among which included the TGF-β signaling pathway closely associated with fibrosis (Figure S5). Combined with previous literature,27,28 among the 24 candidate genes, SMAD4 was a critical component of the classical TGF-β signaling pathway. Furthermore, in bioinformatics analysis, it exhibited high target prediction scores in the above three miRNA target gene prediction databases. The TargetScanHuman database revealed sequence consistency at the binding site between miR-664a-3p and SMAD4 in both humans and mice, demonstrating a high degree of species conservation in the critical functional regions of miR-664a-3p. Additionally, SMAD4 could be enriched in both GO and KEGG pathways, making it worthy of further study. To confirm this hypothesis, the expression of SMAD4 mRNA in CFs transfected with miR-664a-3p mimic, inhibitor and their corresponding NCs was assessed using qRT-PCR. As shown in Figure 8B and C, the results demonstrated that transfection with miR-664a-3p inhibitor significantly increased the protein expression levels of SMAD4 in CFs, while transfection with miR-664a-3p mimic led to a marked decrease in SMAD4 protein expression in CFs. However, there was no significant difference in the expression levels of SMAD4 mRNA in CFs. This suggested that miR-664a-3p might regulate the expression of SMAD4 in CFs at the post-transcriptional level.
Figure 8 Continued. Figure 8 SMAD4 is identified as the target of miR-664a-3p. (A) Potential targets of miR-664a-3p in the TargetScan 8.0, miRBase, and miRWalk databases. (B) The expression of SMAD4 mRNA in CFs after transfection with miR-664a-3p mimic, inhibitor, or their respective controls. (C) The protein expression levels of SMAD4 in CFs after transfection with miR-664a-3p mimic, inhibitor, or their respective controls. (D) Diagram showing the predicted binding sites of miR-664a-3p on SMAD4 3′-UTR, with the sequences marked in red indicating mutations in the miR-664a-3p matching sequence. (E) After co-transfecting CF cells with mimic-NC, miR-664a-3p mimic, inhibitor-NC, or miR-664a-3p inhibitor and plasmids containing either wild-type or mutant-type 3′-UTR of SMAD4, luciferase activity was detected using a dual-luciferase reporter assay system. (F) Western blotting analysis and quantification of SMAD4 protein levels in TGF-β1-induced CFs treated with Young-sEVs (n = 3). (G) The expression of SMAD4 mRNA in TGF-β1-induced CFs treated with Young-sEVs by qRT-PCR analysis (n = 3). (H) Western blotting analysis and quantification of SMAD4 protein levels in mouse cardiac tissue of post-MI treated with Young-sEVs (n = 3). (I) The expression of SMAD4 mRNA in mouse cardiac tissue of post-MI treated with Young-sEVs by qRT-PCR analysis (n = 3). Data were presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the negative control (NC) group or NC mimic/inhibitor group or sham group. ##P < 0.01, ###P < 0.001, compared with the TGF-β1 group or MI group. &P < 0.05, &&P < 0.01, &&&P < 0.001, compared with the TGF-β1+ Old-sEVs group or MI + Old-sEVs group. Abbreviations: CFs, cardiac fibroblasts; Old-sEVs, small extracellular vesicles from old healthy human plasma; Young-sEVs, small extracellular vesicles from young healthy human plasma; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; MI, myocardial infarction; MUT, mutant type; ns, no statistical difference; qRT-PCR, quantitative real-time polymerase chain reaction; SMAD4, SMAD family member 4; SD, standard deviation; TGF-β1, transforming growth factor beta 1; UTR, untranslated region; WT, wild type.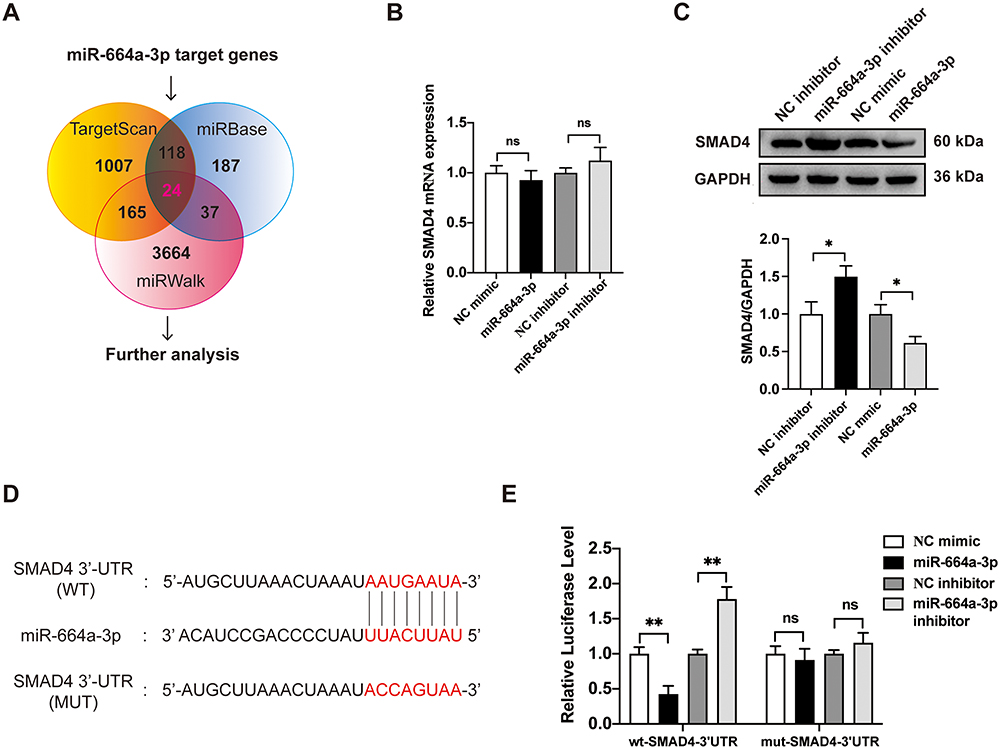

To further ascertain whether miR-664a-3p directly binded to the 3′-UTR region of SMAD4, we performed chimeric constructs containing either the luciferase mutant-type 3′-UTR sequence (mut-SMAD4-3′-UTR) or the wild-type 3′-UTR sequence (wt-SMAD4-3′-UTR) (Figure 8D). Compared to the control group, the overexpression of miR-664a-3p significantly suppressed the luciferase activity of the reporter gene in the wild-type construct, while inhibition of miR-664a-3p enhanced the luciferase activity (Figure 8E). However, the mutant miR-664a-3p binding site in the SMAD4 3′-UTR could reverse these effects. In addition, we measured the protein and mRNA expression of SMAD4 in CFs and observed that SMAD4 expression was significantly increased in TGF-β1-induced CFs but significantly decreased after treatment with plasma sEVs from young healthy human (Figure 8F and G). Similarly, the expression of SMAD4 in mouse cardiac tissue was also consistent with the aforementioned trends (Figure 8H and I). Overall, these results suggested that miR-664a-3p could directly target SMAD4 in CFs.
The Overexpression SMAD4 Protein Blocks the Anti-Fibrotic Effect of miR-664a-3p
To investigate the role of SMAD4 in miR-664a-3p-mediated CFs fibrosis, overexpressed SMAD4 and control vector plasmids (control vectors) were transfected into CFs. The upregulation of SMAD4 plasmid was confirmed using Western blotting. As shown in Figure 9A, the expression of SMAD4 protein significantly increased in a dose-dependent manner. Subsequently, qRT-PCR and Western blotting analysis were performed to assess the expression levels of fibrotic markers in the presence or absence of TGF-β1, miR-664a-3p mimic, and SMAD4 overexpression plasmids. As illustrated in Figure 9B–E, overexpression of SMAD4 reversed the inhibitory effects of miR-664a-3p on TGF-β1-induced proliferation and migration of CFs. Furthermore, SMAD4 overexpression also counteracted the miR-664a-3p-mediated suppression of protein and mRNA expression levels of Col-I, Col-III, and α-SMA in TGF-β1-stimulated CFs (Figure 9F and G). These results indicated that miR-664a-3p directly targeted SMAD4 to inhibit the development of cardiac fibrosis.
Figure 9 Continued. Figure 9 The SMAD4 protein can block the anti-fibrotic effect of miR-664a-3p. (A) Western blotting analysis for the SMAD4 protein expression using different amounts of transfected plasmid DNA. (B-D) The proliferation of CFs transfected with the SMAD4 overexpression plasmid was evaluated using EdU and CCK-8 cell proliferation assays. The proportion of EdU-positive CFs and results of CCK-8 assay were shown in panel (scale bar = 100 μm). (E) The migration of CFs transfected with the SMAD4 overexpression plasmid was assessed using the Wound healing assay and Transwell migration assay. The two methods were evaluated after incubation for 24 hours and the relative migration speed were measured (scale bar = 100 μm). (F) Western blotting analysis and quantification of Col-I, Col-III and α-SMA protein levels in CFs after transfection with the SMAD4 overexpression plasmid. (G) qRT-PCR analysis of the mRNA expressions of Col-I, Col-III and α-SMA in CFs transfected with the SMAD4 overexpression plasmid. Data are presented from three independent experiments. Data were presented as mean ± SD. **P < 0.01, ***P < 0.001, compared with the negative control (NC) group. #P < 0.05, ##P < 0.01, ###P < 0.001, compared with the TGF-β1 group or 1 μg group. &P < 0.05, &&P < 0.01, &&&P < 0.001, compared with the TGF-β1 + miR-664a-3p group or 2 μg group. Abbreviations: 664a-3p, miR-664a-3p; α-SMA, alpha smooth muscle actin; CCK-8, cell counting kit-8; CFs, cardiac fibroblasts; Col-I, collagen I; Col-III, collagen III; EdU, 5-ethynyl-20-deoxyuridine; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; qRT-PCR, quantitative real-time polymerase chain reaction; SD, standard deviation; SMAD4, SMAD family member 4; TGF-β1, transforming growth factor beta 1.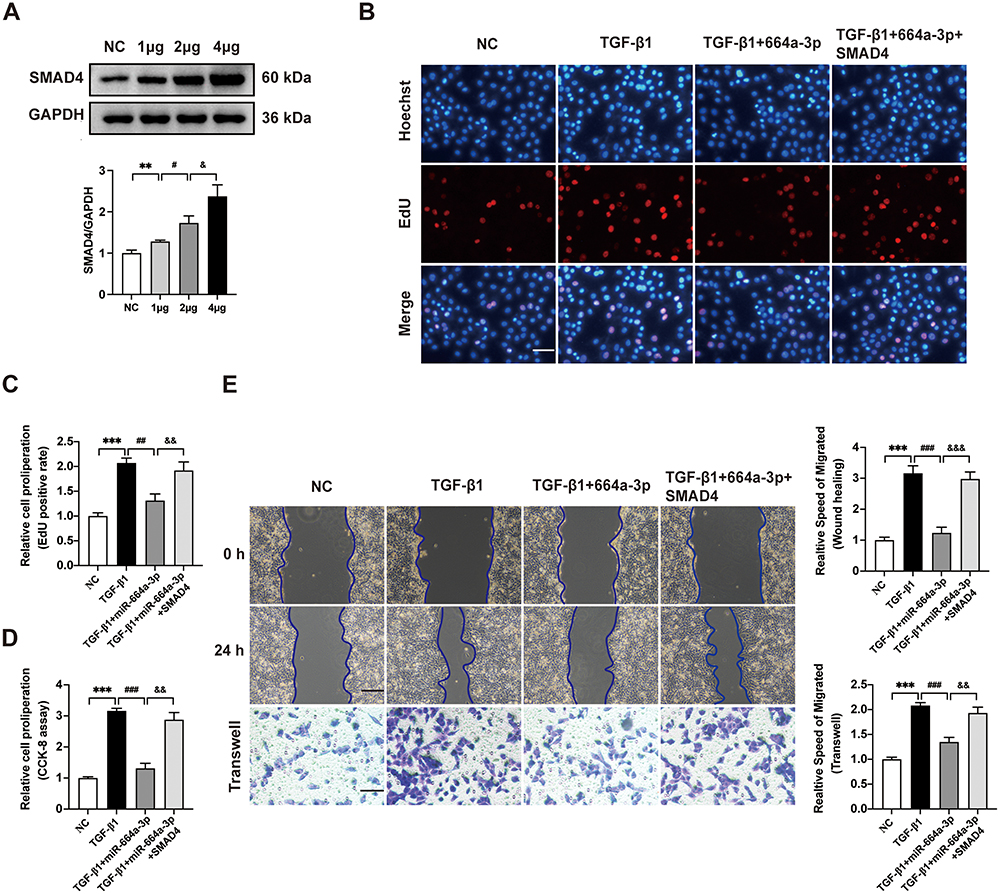

Discussion
Cardiac fibrosis occurs in the progression of many cardiovascular diseases, and its global incidence and healthcare burden are increasing dramatically every year.1 However, current clinical treatments fail to slow down or reverse the progression of fibrosis towards end-stage organ failure. Therefore, there is an urgent need for advanced anti-fibrotic therapies. As summarized in graphical abstract, we have reported that Young-sEVs significantly improved cardiac function in mice after MI and inhibited the progression of cardiac fibrosis both in vivo and in vitro. Further investigating the underlying mechanisms, we demonstrated that Young-sEVs inhibited cardiac fibrosis via miR-664a-3p. Moreover, through functional gain and loss studies, we identified SMAD4 as a direct target of miR-664a-3p, and downregulation of SMAD4 prevented excessive proliferation and deposition of the extracellular matrix. To the best of our knowledge, this is the first study to demonstrate that miR-664a-3p delivered through intramyocardial injection of Young-sEVs can effectively inhibit cardiac fibrosis. This study further demonstrates that the delivery system based on sEVs from young healthy human plasma is a promising therapeutic approach for heart failure patients, with considerable translational application value in clinic.
It had been found that transferring young blood into old mice could enhance memory and learning abilities in elderly rodents, and it could also induce rejuvenation and regenerative effects, effectively reversing the aging process.29–32 These studies provided evidence supporting the hypothesis that youthful factors in the bloodstream could rejuvenate aging biological systems. However, further exploration is needed to identify the reparative components present in young blood and elucidate their mechanisms of action. A recent study found that exosomes from young healthy human plasma could promote functional recovery after cerebral hemorrhage through miR-25-3p.12 Furthermore, there have been research reports indicating that plasma exosomes from healthy individuals can protect the myocardium from ischemia-reperfusion injury.15 These studies further demonstrate that sEVs (including exosomes) present in young plasma are one of the primary reparative components in young blood. However, the aforementioned studies mainly focused on disease models related to improving functional recovery in the brain or ischemia-reperfusion injury in the heart. Nonetheless, the impact and underlying mechanisms of Young-sEVs on cardiac fibrosis remain unclear. This study utilized Young-sEVs to treat cardiac fibrosis after MI, and achieved unexpected positive outcomes. The findings not only revealed the potential benefits of Young-sEVs but also introduced a novel pathway for the treatment of cardiac fibrosis.
Increasing evidence suggests that miRNAs are one the most significant molecules through which sEVs modulate the functions of recipient cells.33,34 In the field of cardiovascular research, miRNAs within sEVs often act as communicators between cardiovascular cells. Almost all types of cardiovascular cells have been demonstrated to release and receive miRNAs from sEVs, suggesting their potential regulatory role in cardiac fibrosis.6 For example, various sources of exosomal miRNAs, including embryonic stem cells, induced pluripotent stem cells (iPSCs), MSCs and cardiac stem/progenitor cells, had been shown to possess cardiac protective effects by enhancing the survival and function of cardiomyocytes.35 In addition to cellular sources, miRNAs from sEVs present in serum or plasma had also been demonstrated to possess cardiac protective effects. For instance, exosomes in serum and plasma derived from long-term exercise have been shown to protect the heart from myocardial ischemia/reperfusion injury through the action of miR-342-5p.36 Overall, these findings suggest that natural sEVs derived from various cell types and circulation can protect the heart by modulating miRNAs, but research on miRNAs in circulating sEVs in the context of cardiac fibrosis is relatively limited. Thus, in this study, we utilized Young-sEVs to treat cardiac fibrosis and demonstrated their efficacy through in vivo and in vitro experiments. Further analysis using high-throughput miRNA sequencing and qRT-PCR identified reparative miRNA-664a-3p in Young-sEVs and confirmed its crucial role in mediating the therapeutic effects against TGF-β1-induced CFs fibrosis and post-MI cardiac fibrosis in mice. Furthermore, the study by Ronan et al37 demonstrated that sEVs derived from the left ventricular tissue of young humans and mouse models could inhibit cardiac fibrosis in vitro through the synergistic action of four miRNAs. This finding also supports our research results, indicating that sEVs from young individuals exhibit significant cardioprotective effects due to their unique miRNA content. The development of this therapeutic approach could offer more precise and effective treatment options for patients with cardiac fibrosis. As a natural and biocompatible delivery system, sEVs make the application of miRNA-based gene therapy more feasible, while reducing potential immunogenicity and other safety risks.
MicroRNAs are approximately 22-nucleotide-long non-coding single-stranded RNA molecules encoded by endogenous genes. They can inhibit mRNA translation or induce mRNA degradation, thereby controlling the pattern of gene expression.38 Through sequencing and qRT-PCR, we have confirmed the abundant presence of miR-664a-3p in Young-sEVs. Previous studies on miR-664a-3p have been primarily concentrated in the fields of oncology, chronic obstructive pulmonary disease (COPD), and polycystic ovary syndrome (PCOS).39–42 However, there is almost no research on the association between miR-664a-3p and cardiac fibrosis, and to the best of our knowledge, this is the first study to elucidate the therapeutic potential of miR-664a-3p in the context of cardiac fibrosis. The pathological mechanism of cardiac fibrosis is complex, involving multiple signaling pathways, among which the TGF-β pathway is the most extensively studied fibrosis pathway.27,43 In our study, bioinformatics analysis and further validation experiments indicated that SMAD4 was a critical downstream target of miR-664a-3p. SMAD4 is crucial for TGF-β signal transduction, maintaining the activated phenotype of fibroblasts, and promoting the synthesis of collagen.44 The results demonstrated that during treatment with Young-sEVs, the expression level of SMAD4 was significantly downregulated in both post-MI cardiac tissue and TGF-β1-induced CFs. In vitro, overexpression of SMAD4 could eliminate the therapeutic effects of miR-664a-3p on cardiac fibrosis. In addition, luciferase assays revealed that miR-664a-3p would bind to the 3′-UTR region of SMAD4, reducing its expression at the post-transcriptional level. In summary, Young-sEVs enriched with miR-664a-3p, could inhibit the progression of cardiac fibrosis by modulating TGF-β signaling through SMAD4.
We acknowledged that there were some limitations to this study. Firstly, we only sequenced the plasma samples from three young healthy individuals to identify highly expressed miRNAs. However, a larger scale database was needed to further validate our primary findings. Secondly, the involvement of other molecules in the progression of cardiac fibrosis remained uncertain and required further investigation. Thirdly, in our study, we used loss/gain-of-function assays with mimics or inhibitors transfection to evaluate the therapeutic effects of sEVs. However, transgenic animal models would provide better information for further understanding the underlying mechanisms.16 Fourthly, the local myocardial injection of sEVs was employed for treating cardiac fibrosis, yet the distribution of sEVs in the mouse heart remained unclear. Although studies have demonstrated that myocardial injection of sEVs resulted in longer retention time in the heart compared to coronary artery and tail vein injections, which mainly accumulated in the liver and spleen,10,45 further research was still needed regarding the choice of injection site, injection frequency, and sEVs dosage. Future research focusing on loading these engineered sEVs with bioactive materials and optimizing therapeutic systems for heart failure would be highly meaningful. Lastly but not least, it would be valuable to elucidate whether plasma sEVs from young healthy individuals can improve the progression of human cardiac fibrosis.16
Conclusions
This study demonstrated that Young-sEVs promoted cardiac function recovery by inhibiting fibrosis, thereby potentially serving as a treatment for heart failure. Mechanistically, sEVs-derived miR-664a-3p inhibited the expression of SMAD4 by binding to its 3′-UTR, subsequently inhibiting the TGF-β/SMAD4 signaling pathway to protect heart from fibrosis. To the best of our knowledge, this is the first study revealing the role of Young-sEVs in suppressing cardiac fibrosis via the miR-664a-3p/SMAD4 axis, providing a novel avenue for the treatment of heart failure patients in the future.
Data Sharing Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was set in compliance with Helsinki Declaration and was approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University (2023-SRFA-401). Non-opposition to participate in the study from the patient or his/her next of kin was collected prior to inclusion.
Acknowledgments
This study was funded by the Jiangsu Province Key Medical Talent Program (grant number ZDRCA2016016), the Young Scholars Fostering Fund of the First Affiliated Hospital of Nanjing Medical University (grant number PY2022024) and Jiangsu Funding Program for Excellent Postdoctoral Talent (grant number 2022ZB736).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation. 2022;145(8):e153–e639. doi:10.1161/CIR.0000000000001052
2. Krumholz HM, Chen YT, Wang Y, Vaccarino V, Radford MJ, Horwitz RI. Predictors of readmission among elderly survivors of admission with heart failure. Am Heart J. 2000;139(1 Pt 1):72–77. doi:10.1016/s0002-8703(00)90311-9
3. Su M, Li W, Yuan Y, et al. Epididymal white adipose tissue promotes angiotensin II-induced cardiac fibrosis in an exosome-dependent manner. Transl Res. 2022;248:51–67. doi:10.1016/j.trsl.2022.05.004
4. Wei T, Du Y, Shan T, et al. The crystallin alpha B (HSPB5)-tripartite motif containing 33 (TRIM33) axis mediates myocardial fibrosis induced by angiotensinogen II through transforming growth factor-beta (TGF-beta1)-Smad3/4 signaling. Bioengineered. 2022;13(4):8836–8849. doi:10.1080/21655979.2022.2054913
5. Ranjan P, Kumari R, Verma SK. Cardiac fibroblasts and cardiac fibrosis: precise role of exosomes. Front Cell Develop Biol. 2019;7:318. doi:10.3389/fcell.2019.00318
6. Xue R, Tan W, Wu Y, et al. Role of exosomal miRNAs in heart failure. Front Cardiovasc Med. 2020;7:592412. doi:10.3389/fcvm.2020.592412
7. Pu Y, Li C, Qi X, et al. Extracellular vesicles from NMN preconditioned mesenchymal stem cells ameliorated myocardial infarction via miR-210-3p promoted angiogenesis. Stem Cell Rev Rep. 2023;19(4):1051–1066. doi:10.1007/s12015-022-10499-6
8. Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Mol Aspect Med. 2019;65:2–15. doi:10.1016/j.mam.2018.06.003
9. Fan J, Ren M, He Y. Diagnostic and therapeutic properties of exosomes in cardiac fibrosis. Front Cell Develop Biol. 2022;10:931082. doi:10.3389/fcell.2022.931082
10. Gallet R, Dawkins J, Valle J, et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J. 2017;38(3):201–211. doi:10.1093/eurheartj/ehw240
11. Chen F, Li X, Zhao J, Geng J, Xie J, Xu B. Bone marrow mesenchymal stem cell-derived exosomes attenuate cardiac hypertrophy and fibrosis in pressure overload induced remodeling. Vitro Cell Dev Biol Anim. 2020;56(7):567–576. doi:10.1007/s11626-020-00481-2
12. Yang W, Ding N, Luo R, et al. Exosomes from young healthy human plasma promote functional recovery from intracerebral hemorrhage via counteracting ferroptotic injury. Bioact Mater. 2023;27:1–14. doi:10.1016/j.bioactmat.2023.03.007
13. Li X, Lian Y, Wu Y, et al. Neonatal plasma exosomes contribute to endothelial cell-mediated angiogenesis and cardiac repair after acute myocardial infarction. Int J Mol Sci. 2023;24(4). doi:10.3390/ijms24043196
14. Luo Z, Hu X, Wu C, et al. Plasma exosomes generated by ischaemic preconditioning are cardioprotective in a rat heart failure model. Br J Anaesth. 2023;130(1):29–38. doi:10.1016/j.bja.2022.08.040
15. Vicencio JM, Yellon DM, Sivaraman V, et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. Journal of the American College of Cardiology. 2015;65(15):1525–1536. doi:10.1016/j.jacc.2015.02.026
16. Ma C, Qi X, Wei YF, et al. Amelioration of ligamentum flavum hypertrophy using umbilical cord mesenchymal stromal cell-derived extracellular vesicles. Bioact Mater. 2023;19:139–154. doi:10.1016/j.bioactmat.2022.03.042
17. Nagpal V, Rai R, Place AT, et al. MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation. 2016;133(3):291–301. doi:10.1161/CIRCULATIONAHA.115.018174
18. Tao L, Bei Y, Chen P, et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. 2016;6(12):2068–2083. doi:10.7150/thno.15007
19. Cerasi E, Luft R. The plasma insulin response to glucose infusion in healthy subjects and in diabetes mellitus. Acta Endocrinol. 1967;55(2):278–304. doi:10.1530/acta.0.0550278
20. Horder K, Horder M. Plasma haptoglobin and physical exercise: changes in healthy individuals concomitant with a strenuous march. Clinica Chimica Act. 1970;30(2):369–372. doi:10.1016/0009-8981(70)90128-2
21. Horder M, Toft H, Christensen NC, Simonsen EE. Plasma pre-beta lipoprotein in healthy adults. Atherosclerosis. 1971;14(1):31–37. doi:10.1016/0021-9150(71)90036-0
22. Hemmingsen L, Skaarup P. The 24-hour excretion of plasma proteins in the urine of apparently healthy subjects. Scand J Clin Lab Invest. 1975;35(4):347–353. doi:10.1080/00365517509095751
23. Li H, Ding J, Liu W, et al. Plasma exosomes from patients with acute myocardial infarction alleviate myocardial injury by inhibiting ferroptosis through miR-26b-5p/SLC7A11 axis. Life Sci. 2023;322:121649. doi:10.1016/j.lfs.2023.121649
24. Wang K, Li Z, Ding Y, et al. Klotho improves cardiac fibrosis, inflammatory cytokines, ferroptosis, and oxidative stress in mice with myocardial infarction. J Physiol Biochem. 2023. doi:10.1007/s13105-023-00945-5
25. Chen G, Xu H, Xu T, et al. Calycosin reduces myocardial fibrosis and improves cardiac function in post-myocardial infarction mice by suppressing TGFBR1 signaling pathways. Phytomedicine. 2022;104:154277. doi:10.1016/j.phymed.2022.154277
26. Xiao YY, Xia LX, Jiang WJ, et al. Cardiopulmonary progenitors facilitate cardiac repair via exosomal transfer of miR-27b-3p targeting the SIK1-CREB1 axis. Cell proliferation. 2024;57(5):e13593. doi:10.1111/cpr.13593
27. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022;21(1):104. doi:10.1186/s12943-022-01569-x
28. Dees C, Chakraborty D, Distler JHW. Cellular and molecular mechanisms in fibrosis. Exp Dermatol. 2021;30(1):121–131. doi:10.1111/exd.14193
29. Kaiser JA. ‘Rejuvenation factor’ in blood turns back the clock in old mice. Science. 2014;344(6184):570–571. doi:10.1126/science.344.6184.570
30. Scudellari M. Ageing research: blood to blood. Nature. 2015;517(7535):426–429. doi:10.1038/517426a
31. Zhang K, Mizuma H, Nakatani Y, et al. Neural correlates of beneficial effects of young plasma treatment in aged mice: PET-SPM analyses and neuro-behavioural/molecular biological studies. Eur J Nucl Med Mol Imaging. 2022;49(5):1456–1469. doi:10.1007/s00259-021-05598-4
32. Zhang W, Qu J, Liu GH, Belmonte JCI. The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol. 2020;21(3):137–150. doi:10.1038/s41580-019-0204-5
33. Stahl PD, Raposo G. Exosomes and extracellular vesicles: the path forward. Essays Biochem. 2018;62(2):119–124. doi:10.1042/EBC20170088
34. Han C, Yang J, Sun J, Qin G. Extracellular vesicles in cardiovascular disease: biological functions and therapeutic implications. Pharmacol Ther. 2022;233:108025. doi:10.1016/j.pharmthera.2021.108025
35. Moghaddam AS, Afshari JT, Esmaeili SA, Saburi E, Joneidi Z, Momtazi-Borojeni AA. Cardioprotective microRNAs: lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis. 2019;285:1–9. doi:10.1016/j.atherosclerosis.2019.03.016
36. Hou Z, Qin X, Hu Y, et al. Longterm exercise-derived exosomal miR-342-5p: a novel exerkine for cardioprotection. Cir Res. 2019;124(9):1386–1400. doi:10.1161/circresaha.118.314635
37. Ronan G, Bahcecioglu G, Yang J, Zorlutuna P. Cardiac tissue-resident vesicles differentially modulate anti-fibrotic phenotype by age and sex through synergistic miRNA effects. Biomaterials. 2024;311:122671. doi:10.1016/j.biomaterials.2024.122671
38. Qiao L, Hu S, Liu S, et al. microRNA-21-5p dysregulation in exosomes derived from heart failure patients impairs regenerative potential. J Clin Invest. 2019;129(6):2237–2250. doi:10.1172/JCI123135
39. Pu X, Zhang C, Ding G, et al. Diagnostic plasma small extracellular vesicles miRNA signatures for pancreatic cancer using machine learning methods. Transl Oncol. 2024;40:101847. doi:10.1016/j.tranon.2023.101847
40. He M, Mao G, Xiang Y, et al. MicroRNA-664a-3p inhibits the proliferation of ovarian granulosa cells in polycystic ovary syndrome and promotes apoptosis by targeting BCL2A1. Ann translat Med. 2021;9(10):852. doi:10.21037/atm-21-1614
41. Zhong S, Chen C, Liu N, et al. Overexpression of hsa-miR-664a-3p is associated with cigarette smoke-induced chronic obstructive pulmonary disease via targeting FHL1. Int J Chron Obstruct Pulmon Dis. 2019;14:2319–2329. doi:10.2147/COPD.S224763
42. Wang L, Li B, Zhang L, et al. miR-664a-3p functions as an oncogene by targeting Hippo pathway in the development of gastric cancer. Cell proliferation. 2019;52(3):e12567. doi:10.1111/cpr.12567
43. Shi Y, Liu C, Xiong S, et al. Ling-Gui-Qi-Hua formula alleviates left ventricular myocardial fibrosis in rats with heart failure with preserved ejection fraction by blocking the transforming growth factor-beta1 /Smads signaling pathway. J Ethnopharmacol. 2023;317:116849. doi:10.1016/j.jep.2023.116849
44. Xu P, Yi Y, Xiong L, et al. Oncostatin M/Oncostatin M receptor signal induces radiation-induced heart fibrosis by regulating SMAD4 in fibroblast. Int J Radiat Oncol Biol Phys. 2024;118(1):203–217. doi:10.1016/j.ijrobp.2023.07.033
45. Roefs MT, Heusermann W, Brans MAD, et al. Evaluation and manipulation of tissue and cellular distribution of cardiac progenitor cell-derived extracellular vesicles. Front Pharmacol. 2022;13:1052091. doi:10.3389/fphar.2022.1052091
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.