")
Back to Journals » Drug Design, Development and Therapy » Volume 19
SNAr Reactive Pyrazine Derivatives as p53-Y220C Cleft Binders with Diverse Binding Modes
Authors Klett T , Stahlecker J , Schwer M , Jaag SJ , Masberg B , Knappe C , Lämmerhofer M , Stehle T , Boeckler FM
Received 16 January 2025
Accepted for publication 14 April 2025
Published 3 June 2025 Volume 2025:19 Pages 4727—4753
DOI https://doi.org/10.2147/DDDT.S513792
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Tamer Ibrahim
Theresa Klett,1 Jason Stahlecker,1 Martin Schwer,1 Simon J Jaag,2 Benedikt Masberg,2 Cornelius Knappe,2 Michael Lämmerhofer,2 Thilo Stehle,3 Frank M Boeckler1,4
1Laboratory for Molecular Design & Pharmaceutical Biophysics, Institute of Pharmaceutical Sciences, Department of Pharmacy and Biochemistry, Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany; 2Pharmaceutical (Bio-) Analysis, Institute of Pharmaceutical Sciences, Department of Pharmacy and Biochemistry, Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany; 3Interfaculty Institute of Biochemistry, Department of Pharmacy and Biochemistry, Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany; 4Interfaculty Institute for Biomedical Informatics (IBMI), Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany
Correspondence: Frank M Boeckler, Laboratory for Molecular Design & Pharmaceutical Biophysics, Institute of Pharmaceutical Sciences, Department of Pharmacy and Biochemistry, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 8 (Haus B), Tübingen, D – 72076, Germany, Tel +4970712974567, Fax +497071295637, Email [email protected]
Purpose: The tumor suppressor p53 is most commonly mutated in human cancer. The structural mutant in the β-sandwich of the protein, p53-Y220C, is the ninth most common p53 mutant. The p53-Y220C mutant has a solvent-accessible hydrophobic pocket, leading to thermal destabilization of the protein. Screening of our covalent fragment library (CovLib) revealed the highly reactive pyrazine derivatives SN006 and SN007, which arylate among other cysteines in p53, the mutation-generated Cys220. Herein, comprehensive structure-activity relationship (SAR) studies of these intrinsically reactive CovLib hits were performed, aiming to identify improved stabilizers for p53-Y220C, with a more balanced reactivity profile, diverse binding modes and a better potential for chemical optimization.
Methods: The compounds were screened for enhanced stabilization of p53 wild type and its mutants using differential scanning fluorimetry (DSF). To confirm covalent modification, intact mass spectrometry was performed. Thiol reactivity profiles were determined using a standardized Glutathione-modifying (GSH) assay. The binding modes of the identified hits and covalent modification of Cys220 were elucidated by X-ray crystallography. Moreover, the influence of the hits on the DNA-binding affinity of full-length p53 was investigated employing a fluorescence polarization assay (FPA).
Results and Conclusion: The promising pyrazine derivatives SN006/7-3, SN006/7-8, and SN006/7-9 were identified, occupying different subsites of the Y220C binding pocket. The compound SN006/7-8 substantially stabilized the thermosensitive cancer mutant Y220C by up to 5.0 °C, representing a strong enhancement over SN006 (1.8 °C) and SN007 (2.0 °C).
Keywords: tumor suppressor p53 mutant stabilizers, covalent fragment-based drug discovery, structure-activity relationship, differential scanning fluorimetry, intact protein mass spectrometry, X-ray crystallography
Graphical Abstract:

Introduction
The tumor suppressor protein p53, also known as the “guardian of the genome”, plays an essential role in the cell cycle.1–4 Activation of p53 in response to various stress signals leads to the regulation of the transcription of many genes, involved in cell cycle arrest, apoptosis, DNA-repair, and cellular senescence.5–7 In addition to the aforementioned canonical functions, it has been discovered that p53 is also involved in various processes, including autophagy, antiangiogenesis, protection against oxidative stress, metabolic homeostasis, ferroptosis, immune microenvironment, and regulation of stemness of cells.3,8–10
In many tumors, a mutation of p53 is found, or p53 activity is reduced by disruption of associated signaling pathways such as the upregulation of the two negative regulators murine double minute 2 homolog (MDM2) and murine double minute 4 homolog (MDM4).10,11 The majority of the oncogenic p53 mutations are missense mutations located in the core domain of p53.7,12–15 In addition to the loss of function, many of these mutants have a dominant-negative effect on wild-type (WT) p53 by forming mixed tetramers.4,13,16 Moreover, some of the p53 mutants acquire other transcriptional regulatory functions, resulting in a gain-of-function phenotype.3,4,7 The missense core domain mutants can be categorized as DNA contact mutants (eg R273H) and structural mutants, which induce structural changes within the protein, folding variations or packing defects destabilizing the protein (eg R282W, Y220C).13,14,17 Mutations of p53 are related to rapid tumor progression, worse cancer prognosis, and chemoresistance.13,18,19 Therefore, p53 is an attractive yet challenging target for cancer therapy, and there is a great interest in identifying drugs that can rescue p53.
The structural Y220C β-sandwich mutant is the ninth most common p53 mutant and is estimated to account for130 000 annual cases of human cancer.13,20–22 The mutation from tyrosine to cysteine creates a solvent-accessible hydrophobic crevice.13,14,23 The mutant p53-Y220C is strongly thermodynamically destabilized, and its melting temperature (Tm) is reduced by about 8 °C, resulting in rapid unfolding, denaturation, and aggregation at body temperature.13,24 However, the overall structure of the core domain remains intact.14 Furthermore, the mutation-induced Y220C cleft is far from the DNA-binding site, the dimerization interface, and not involved in essential protein contacts. Therefore, the crevice represents a possible binding pocket for stabilizing small molecules.13,14,23 A number of compounds were discovered that target the mutational cleft and enhance the thermal stability of the mutant Y220C protein.21,23–27 The N-ethylcarbazole based compound PK083 (ΔTm= 0.8 °C at 125 µM PK083),23,25 the iodophenol derivative PK5196 (ΔTm= 3.6 °C at 250 µM PK5196),21 the pyrazole derivative PK7088 (ΔTm= 1.0 °C at 350 µM PK7088),26 the aminobenzothiazole derivate MB710 (ΔTm= 2.0 °C at 250 µM MB710),27 and the nanomolar cleft binder JC744 (ΔTm= 2.70 °C at 200 µM JC744)24 are examples of stabilizers for the Y220C mutant. In addition, PMV Pharma has developed the small molecule PC14586 (Rezatapopt), which specifically targets and stabilizes the Y220C mutant and is currently evaluated in clinical trials for patients with advanced solid tumors harboring the p53-Y220C mutation (NCT04585750).28,29 Moreover, a covalent reactive compound, KG13, has been identified. KG13 contains an acrylamide warhead and targets Cys220 in the mutational cleft, thereby increasing the Tm of the Y220C mutant to levels comparable to those of the WT protein.30
We recently employed a covalent fragment-based approach and screened a covalent fragment library (CovLib) with p53 and its mutants.31,32 The highly reactive pyrazines derivatives SN006 and SN007 were identified, reacting via nucleophilic aromatic substitution (SNAr) with cysteines in p53 and also with the Cys220 in the crevice. Thiol reactivity was determined with a standardized GSH assay, highlighting complete reaction with glutathione at the first possible analysis time (20 min). Thus, their reaction half-time was initially reported by us as t1/2≪0.33h.32 We estimate that at least 4–6 half-time durations need to pass, until none of the unreacted fragment is detectable by HPLC-UV. Consequently, a t1/2 of 0.06 to 0.08 h (or even significantly below) is quite plausible based on the experimental method.
Covalent modification of Cys220 by SN006 was confirmed by X-ray crystallography. Nevertheless, SN006 and SN007 showed in line with the GSH assay a high reactivity and arylated cysteines in p53 non-specifically, resulting in a destabilization of the T-p53C protein at high compound concentrations.31 Therefore, we conducted structure-activity relationship (SAR) studies with the objective of identifying fragments with a more balanced reactivity profile, higher selectivity for Cys220, and different binding pose in the hydrophobic pocket.
Materials and Methods
Materials
The compounds SN006/7-1 to SN006/7-6 were purchased from Aldrich Market Select (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). The SAR compounds derived from SN006/7-3 and SN006/7-8 were purchased from the suppliers listed in Table S1. Purity of the compounds was confirmed to be >90% by high-performance liquid chromatography (HPLC)-UV on an Ultimate 3000 HPLC-System (Thermo Fisher Scientific, Dreieich, Germany). Upon completion of the biophysical measurements, it was determined that the purity of fragments SN009 and SN001-15 was below the required threshold of 90%. The precise point at which this occurred could not be ascertained. Consequently, compounds SN009 and SN001-15 are not referenced in this paper. However, the corresponding results are included in the Supporting Information. The corresponding HPLC chromatograms are provided in Figure S1 to Figure S23. Table S2 summarizes the analytical results.
Glutathione Assay
The glutathione (GSH) assay was performed as previously described32 using a method established by Keeley et al.33 The following reaction conditions were applied: 250 µM fragment, 100 µM ketoprofen or ibuprofen as an internal standard, 5 mM GSH excess, 10% acetonitrile, and phosphate buffered saline (PBS) pH 7.4 at 37 °C. The samples were analyzed on an Ultimate 3000 HPLC-System (Thermo Fisher Scientific, Dreieich, Germany) with UV-detection after 0, 1, 2, 4, 8, 12, and 24 h. Highly reactive fragments (half-life (t1/2) <1 h) were analyzed every 20 min. The reaction of the compounds with GSH was detected by measuring the decreasing area under the curve (AUC) of the compounds relative to the internal standard. OriginPro2020 (OriginLab, Northampton, MA, USA) was used to fit the relative AUC to the integrated rate equation of pseudo-first order kinetics:

The half-life t1/2 was calculated from the pseudo-first order rate constant k according to the following equation:

The GSH measurements were performed in duplicates and multiple runs were averaged using error propagation. In addition, measurements were performed in PBS buffer without GSH to test the hydrolytic degradation. Contrary to the calculations of Keeley et al,33 the rate constants kGSH and the corresponding t1/2 were not corrected for the degradation reaction in pure buffer.
A complete overview of all GSH assay data is provided in Table S4 in the Supporting Information.
Molecular Biology
The expression and purification of T-p53C (94–312, M133L/V203A/N239Y/N268D), its mutants, and full-length (FL)-T-p53 (1–393, M133L/V203A/N239Y/N268D) were carried out as previously described.22,31,32 The lysate was loaded onto a Ni-NTA column (HisTrap FF, Cytiva) and eluted using a gradient of imidazole from 10–300 mM (Buffer: 50 mM KPi, pH = 8, 300 mM NaCl, 10/300 mM Imidazole, 2 mM TCEP). After cleaving the His-Tag using the tobacco etch virus main protease, the sample was diluted 5–10 fold with heparin buffer A (50 mM KPi, pH = 7.5, 5 mM DTT) and loaded onto a heparin column (HiTrap Heparin HP, Cytiva). The sample was eluted setting a salt concentration of 800 mM (Buffer: 50 mM KPi, pH = 7.5, 5 mM DTT, 2 M NaCl). Final purification was performed via a size exclusion chromatography. For the core domain, a HiLoad 26/60 Superdex 75 pg (GE Healthcare) was used, while for the full length constructs a HiLoad 26/60 Superdex 200 pg was used (Buffer: 25 mM KPi, pH = 7.2, 5 mM DTT, 150 mM NaCl). This quadruple mutant is a frequently utilized p53 core domain construct, facilitating easier handling of p53 under lab conditions by enhancing its stability.34,35 The purity of the expressed proteins was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and protein mass was verified by ultra-high-performance liquid chromatography electrospray ionization mass spectrometry (UHPLC-ESI-MS). The protein sequences of all used constructs are listed in Table S3. A construct with a deletion of the GGS overhang was utilized for X-ray crystallography.
Differential Scanning Fluorimetry (DSF)
DSF was used to investigate the impact of the compounds on the melting temperatures of T-p53C and its mutants. DSF measurements were performed as previously described22,31,32 using a Qiagen Rotor-Q Model-5-Plex HRM real-time PCR instrument (Qiagen, Hilden, Germany). SYPRO Orange (Life Technologies Corporation, Eugene, OR, USA) served as a fluorescent dye (5x final concentration). A constant heating rate of 270 °C/h23 was used, while the temperature was raised from 28 °C to 60–70 °C. The excitation and emission filters were set at 470 nm and 610 nm, respectively. The DSF measurements were performed with 8 µM protein in phosphate buffer (25 mM KPi pH 7.2, 150 mM NaCl, 1 mM TCEP, 5% [v/v] DMSO). For the primary screening, a final fragment concentration of 1 mM was used, corresponding to a protein-to-compound ratio of 1:125.36 For concentration-dependent DSF measurements, the compound concentration was varied by diluting the compounds in pure DMSO before adding to the protein solution. For time-dependent measurements, all samples were incubated at 20 °C while shaking. The Tm of T-p53C and its mutants was obtained by calculating the maxima of the first derivative of the melting curves using OriginPro2020 (OriginLab, Northampton, MA, USA). Finally, the Tm of the protein sample was subtracted from the Tm of the sample containing both protein and compound to calculate ΔTm. All measurements were performed at least in triplicate and multiple runs were averaged according to the rules of error propagation. All DSF runs are summarized as Table S5 – Table S23 in the Supporting Information. In addition, the full set of DSF curves are provided as Figure S26 – Figure S39 and Figure S41 – Figure S49.
Intact Protein Mass Spectrometry
Sample preparation was performed as previously described.31 Therefore, the proteins in phosphate buffer (25 mM KPi pH 7.2, 150 mM NaCl, 5 mM DTT) were incubated with fragments, which were dissolved in DSMO yielding a protein-to-compound ratio of 1:100 and a final DMSO concentration of 5% [v/v]. The fragment concentration was varied by diluting the compound stock solution in DMSO before adding to the protein solution, resulting in a final DMSO concentration of 5% [v/v] and various molar ratios. The mixtures were incubated at 20 °C for 24 h while shaking at 150 rpm prior to intact protein mass analysis. The subsequent UHPLC-ESI-MS measurements, data acquisition, and data analysis were performed in accordance with the previously described procedure.22,31 Additional deconvoluted mass spectra are provided as Figure S24 to Figure S25, Figure S40, and Figure S50, complemented by Table S3 in the Supporting Information.
Protein Crystallization
Crystallization of T-p53C-Y220C was carried out as previously described.35 The T-p53C-Y220C protein (5 mg/mL) was mixed with reservoir solution (100 mM HEPES pH 7.2, 19% PEG4K, 10 mM DTT) and crystallized via the sitting drop vapor-diffusion technique and streak seeding. To generate complexes, compounds were dissolved to 5 mM and soaked for 1 day in cryo-protectant buffer (additional 20% glycerol).
Data Collection, Reduction, and Structure Refinement
Data sets were collected at the Swiss Light Source (Villingen, Switzerland) at beam lines X06SA (PXI). XDS was utilized for data processing and reduction.37 Initial phases were obtained by molecular replacement using 4AGL21 or 8A9222 as a search model for PHASER38 as part of the CCP4 suite.39 Structure and phase improvements were performed by multiple cycles of manual model building in Coot40 and structure refinement using PHENIX.41 Covalently bound fragment restraints were generated using JLigand42 in ACEdrg mode.43 The unbiased omit-maps were generated by removing all covalently bound fragments and the entire Cys sidechain, followed by refinement with simulated annealing. All crystallographic and refinement statistics are provided as Table S24 – Table S26 in the Supporting Information.
Fluorescence Polarization Assay (FPA)
The FPA was performed in accordance with the previously described methodology.31 FL-T-p53 in phosphate buffer (25 mM KPi pH 7.2, 150 mM NaCl, 5 mM DTT, 5% [v/v] DMSO, 0.2 mg/mL BSA) was incubated with fragment (protein-to-compound ratio 1:200) for 24 h at 20 °C on a rotating shaker.36 5’-end fluorescein-labeled GADD45α DNA response element served as a probe (sequence of the forward strand: 5’-[6-FAM]-GTACAGAACATGTCTAAGCATGCTGGGGAC-3’).36,44,45 Fluorescence polarization (FP) data were recorded on a CLARIOstar plate reader (BMG Labtech, Ortenberg, Germany) at 25 °C. Black non-binding polystyrene 96-well microplates (GBO, Frickenhausen, Germany) were utilized. Excitation and emission filters were set to 482 nm and 530 nm, respectively. The dichroic mirror was adjusted to 504 nm. During the one-hour measurement period, FP data were acquired at 10 min intervals and subsequently averaged. The normalized fluorescence polarization data were analyzed with OriginPro2020 (OriginLab, Northampton, MA, USA) according to the cooperative binding mechanism and fitted to the Hill equation with a linear drift term.36,46–48 Measurements were performed at least in quadruplicate and averaged according to the rules of error propagation.
Results
First Generation: Decomposition of the Initial CovLib Hits SN006 and SN007
The CovLib compounds SN006 and SN007, differing only in their leaving groups (LG), are highly reactive fragments with two electron-withdrawing nitrile groups, covalently modifying Cys220 in the hydrophobic pocket. However, due to their high reactivity, multiple cysteines in p53 were arylated. Therefore, we tried to decompose these initial hits, aiming for a simplified chemical lead structure for further development, showing attenuated reactivity and better selectivity, while maintaining or improving stabilization. Basically, we wanted to understand, which parts of SN006/7 are important for the stabilizing effect and the reactivity. Figure 1 shows the tested SN006/7 SAR compounds and t1/2 measured via GSH assay. Fragments SN006/7-1 and SN006/7-6 lack a second electron-withdrawing nitrile group in the para or ortho position to the LG compared to the original compound SN006. In compounds SN006/7-2 to SN006/7-5, the electron-donating NH2 group was also removed.
 |
Figure 1 Overview of SN006 and SN007 SAR compounds and their t1/2 ± standard deviation (SD) measured via GSH assay. |
The results of the DSF screen of T-p53C, the β-sandwich mutant Y220C, the DNA contact mutant R273H, the structural mutant R282W in the C-terminal helix (H2), and of the cysteine control mutant T-p53C-C124/182/277S with SN006/7 SAR compounds (1 mM) after 30 min, 4 h, and 24 h incubation time are depicted in Tables S5–S7. The cysteine control mutant was designed to abolish three solvent-exposed, rather reactive cysteine residues, which are frequently found to be covalently modified.22,36,49,50 The DSF results of the identified hits with T-p53C and its mutants after 24 h of incubation are listed in Table 1 and detailed results of the β-sandwich mutant Y220C incubated with the hits are listed Table 2.
 |
Table 1 Identified Fragment Hits of the SN006/7 SAR DSF Screen and Their Impact on the Tm of T-p53C and Its Mutants After 24 h of Incubation. WT Refers to T-p53C; Mutant Labels (eg, Y220C) Indicate Mutants of T-p53C, Such as T-p53C-Y220C |
 |
Table 2 Effect of SN006/7 SAR Hit Compounds on the Tm of T-p53C-Y220C |
The tested fragments SN006/7-1 and SN006/7-6 had no considerable effect on the melting temperatures of T-p53C or T-p53C-Y220C even after 24 h of incubation. Hence, the compounds, lacking the additional electron-withdrawing nitrile group in the para or ortho position to the LG, are unreactive (t1/2 GSH >100 h) and, therefore, did not stabilize T-p53C-Y220C.
All of the pyrazine derivatives SN006/7-2 to SN006/7-5 were identified as stabilizers by DSF. SN006/7-2 and SN006/7-4 have a halogen (chlorine or bromine, respectively) in the ortho position to the electron-withdrawing group (EWG), reducing their reactivity. No stabilization of T-p53C and its mutants was detected after only 30 min of incubation (Table S5). However, with increasing incubation time and target occupancy, the stabilizing effect increased. As shown in Table 1, after 24 h of incubation, both compounds stabilized p53 and its mutants except for the triple cysteine control mutant. Nevertheless, it is unclear from the DSF results, whether covalent binding to Cys220 in the binding pocket of T-p53C-Y220C occurred, as all mutants were stabilized. The melting curves of T-p53C supplemented with both compounds after 24 h were biphasic. The first derivatives showed two maxima (Figure S29A), corresponding to different arylated protein species with different melting points or non-covalently bound fragments with distinct melting points.51–53 A reversible covalent Pinner-type addition of proteinogenic nucleophiles to the nitrile group of the compounds33,54 is also conceivable, which could lead to differently modified protein species. Table S8 and Table 2 show that as the compound concentration decreased, the stabilization of T-p53C and T-p53C-Y220C also decreased.
After only 30 min of incubation, fragments SN006/7-3 and SN006/7-5, which have a chlorine or bromine para to the EWG, stabilized T-p53C-Y220C by 2.75 °C and 1.65 °C, respectively (Table 2). Fragments SN006/7-3 (t1/2 GSH= 0.23 h) and SN006/7-5 (t1/2 GSH= 0.14 h) also exhibited a higher reactivity in the GSH assay, compared to SN006/7-2 (t1/2 GSH= 6.5 h) and SN006/7-4 (t1/2 GSH= 3.1 h) with the ortho-LG. This higher reactivity is consistent with the increasing stabilization of the σ-complex by EWGs in the order meta ≪ ortho < para.55,56 In addition, after 30 min of incubation, SN006/7-3 and SN006/7-5 exhibited a particularly pronounced stabilizing effect on the Y220C mutant (Table S5), indicating covalent modification of the mutation-induced cysteine in the crevice. Similar to the original compounds SN006 and SN007, the R282W mutant showed no suitable melting curves and first derivatives with clearly defined peaks after incubation with SN006/7-3 and SN006/7-5 (Figures S27B, S28E, and S30B). After 24 h of incubation, compound SN006/7-3, bearing a chlorine atom in para position to the EWG, strongly increased the melting temperature of T-p53C-Y220C by up to 3.62 °C (1 mM, 24 h). Moreover, SN006/7-3 also stabilized T-p53C. After 24 h of incubation, the first derivatives of the melting curves of T-p53C showed two maxima with a ∆Tm of 0.70 °C and of 2.55 °C. Additionally, the compound slightly stabilized T-p53C-C124/182/277S by 0.55 °C (1 mM, 24 h), indicating covalent modification of additional cysteines besides Cys124, Cys182, and Cys277. The bromine analog SN006/7-5 stabilized T-p53C-Y220C less effectively (∆Tm= 1.65 °C, 1 mM, 24 h) than SN006/7-3. However, after 24 h of incubation, both T-p53C (∆Tm= −0.60 °C and 0.30 °C) and T-p53C-R273H (∆Tm= −1.48 °C) were destabilized. The destabilization can be attributed to the higher reactivity of SN006/7-5 (t1/2 GSH= 0.14 h), determined in the GSH assay compared to SN006/7-3 (t1/2 GSH= 0.23 h), leading to hyperarylation and impairment of normal protein folding.10
Table 2 indicates that the overall stabilization of T-p53C-Y220C was reduced at 250 µM SN006/7-3. In contrast, the stabilization of T-p53C-Y220C increased as the concentration of the reactive fragment SN006/7-5 decreased. This concentration-dependent reaction behavior was already observed for the highly reactive CovLib compounds SN006 and SN007.31 The stabilization of T-p53C-Y220C by 2.65 °C after 24 h of incubation with 250 µM SN006/7-5 was comparable to the ∆Tm value of 2.87 °C measured with SN006/7-3 at the same concentration.
In summary, SN006/7-3 was identified as the best hit, primarily stabilizing the Y220C mutant. The best stabilization of T-p53C-Y220C by about 3.62 °C (1 mM, 24 h) was also more pronounced than the maximum stabilization by the original compounds SN006 (∆Tm= 1.82 °C, 500 µM, 30 min) and SN007 (∆Tm= 2.03 °C, 250 µM, 30 min).31
Intact Protein Mass Spectrometry of T-p53C-Y220C with SN006/7 SAR Hits
For all identified SN006/7 SAR DSF hits, a covalent modification of the protein was detected by intact protein mass spectrometry (MS). After 24 h of incubation, all DSF hits multiply arylated T-p53C-Y220C at a protein-to-compound ratio of 1:100, as shown in Figure 2.
 |
Figure 2 Deconvoluted MS spectra of T-p53C-Y220C with the identified SN006/7 SAR hits: SN006/7-2 (A), SN006/7-3 (B), SN006/7-4 (C), SN006/7-5 (D) at a protein-to-compound ratio of 1:100 after 24 h of incubation at 20 °C (theoretical mass of unmodified T-p53C-Y220C: 24,696.00 Da). |
The deconvoluted MS spectra of compounds SN006/7-2 (Figure 2A) and SN006/7-4 (Figure 2C) showed signals corresponding to the triple arylated protein. The highest signals were observed for the protein species labeled with two molecules of SN006/7-2 or SN006/7-4. Additionally, in the deconvoluted MS spectrum of T-p53C-Y220C incubated with SN006/7-2, there was a prominent signal for the protein species with one molecule of SN006/7-2 attached via SNAr. The presence of a large signal for the single arylated protein may explain the slightly lower stabilization of T-p53C-Y220C in the DSF experiment by compound SN006/7-2 (∆Tm= 1.38 °C, 1 mM, 24 h) compared to the bromine analog SN006/7-4 (∆Tm= 2.00 °C, 1 mM, 24 h). Moreover, the 2-bromopyrazine derivative SN006/7-4 (t1/2 GSH= 3.1 h) also had a shorter t1/2 in GHS assay than the 2-chloropyrazine derivative SN006/7-2 (t1/2 GSH= 6.5 h).
In the deconvoluted MS spectra of SN006/7-3 (Figure 2B) and SN006/7-5 (Figure 2D), triple arylated protein species were recognizable as minimum modification. The protein species with one or two attached molecules were not discernible in the spectra of the compounds with the nitrile group in the para position to the halogen. Hence, fragments SN006/7-3 and SN006/7-5 caused more modifications of T-p53C-Y220C than fragments SN006/7-2 and SN006/7-4. This is consistent with the higher reactivity of SN006/7-3 and SN006/7-5, which have the π-accepting EWG in the para position.56 Moreover, the spectrum of T-p53C-Y220C incubated with SN006/7-3 (Figure 2B) showed signals where SN006/7-3 was attached to T-p53C-Y220C three, four, or five times via SNAr. Additionally, signals were visible corresponding to T-p53C-Y220C modified with SN006/7-3 via both SNAr and Pinner-type addition to the nitrile. In contrast, the deconvoluted MS spectrum of the bromine analog SN006/7-5 (Figure 2D) only displayed signals corresponding to the three and four times arylated T-p53C-Y220C. No reversible covalent Pinner-type addition to the nitrile was detected. The reduced stabilization of T-p53C-Y220C by fragment SN006/7-5 compared to the chlorine analog SN006/7-3 may be attributed to the differences in the deconvoluted MS spectra of T-p53C-Y220C with the compounds.
DSF Studies of the Y220C Stabilizing Hit SN006/7-3
The concentration-dependent DSF measurement with compound SN006/7-3 after 30 min of incubation (Table 3 and Figure 3A) showed that the highest stabilization of T-p53C-Y220C was achieved with the highest tested compound concentration (1 mM, ∆Tm= 2.65 °C). In contrast to the original hits SN006 and SN007, a high compound concentration did not lead to a reduced stabilization of the protein. After 30 min of incubation, up to a lowest effective compound concentration of 125 µM (protein-to-compound ratio 1:15.63), a slight stabilization was detectable (∆Tm= 0.70 °C). For the initially identified highly reactive fragments SN006 and SN007, no evaluable melting curves of the T-p53C-Y220C protein could be measured after 24 h incubation time at 1 mM compound concentration. In contrast, the SAR compound SN006/7-3 showed a characteristic concentration-dependent stabilization of T-p53C-Y220C (Figure 3B). Compared to the DSF measurement after 30 min, the overall ∆Tm values increased with prolonged incubation time. After 24 h of incubation, the highest stabilization of T-p53C-Y220C by up to 3.55 °C was achieved at 1 mM SN006/7-3. Up to a lowest effective fragment concentration of 31.25 µM, which is equivalent to a protein-to-compound ratio of 1:3.91, there was a considerable increase in the melting temperature of T-p53C-Y220C, with a ∆Tm of 0.98 °C.
 |
Table 3 Concentration-Dependent Effects of Compound SN006/7-3 on the Tm of T-p53C-Y220C After 30 min and 24 h Incubation Time |
 |
Figure 3 Concentration-dependent DSF measurements of T-p53C-Y220C with SN006/7-3. The first derivatives of the melting curves and resulting ∆Tm values of T-p53C-Y220C (8 µM protein) with various concentrations of compound SN006/7-3 after 30 min (A) and 24 h (B) of incubation at 20 °C are shown. |
The detailed time-dependent DSF measurements were performed with 1 mM SN006/7-3, representing the concentration leading to the highest stabilization of T-p53C-Y220C. Figure S33 and Table S10 show that a substantial increase in the melting temperature of T-p53C-Y220C was achieved after only 10 min of incubation (∆Tm= 2.30 °C). This is consistent with the short t1/2 of 0.23 h for fragment SN006/7-3 in the GSH assay. After about 2 h of incubation at 20 °C, the increase in the ∆Tm values plateaued, and temperature-sensitive cancer mutant T-p53C-Y220C was maximally stabilized.
Evaluation of the Hit SN006/7-3 with T-p53C-Y220C-CL
A cysteine light (CL) construct of T-p53C-Y220C (T-p53C-Y220C-C124/182/229/275/277S, T-p53C-Y220C-CL) was employed as a control in the DSF and intact protein MS analysis of SN006/7-3. Given the limited number of competingly accessible cysteine residues in the cysteine light construct, we sought to conduct a more detailed investigation into the possible arylation of SN006/7-3 at Cys220 in the hydrophobic pocket.30
The DSF results with the Y220C cysteine light mutant are depicted in Figure S34 and Table 4. SN006/7-3 stabilized T-p53C-Y220C-CL by up to 2.40 °C (1 mM, 24 h). At a compound concentration of 250 µM, the stabilization decreased, as previously observed for T-p53C-Y220C. However, the stabilization of T-p53C-Y220C-CL was lower compared to T-p53C-Y220C (∆Tm= 3.62 °C, 1 mM, 24 h), indicating that the labeling of cysteines other than Cys220 also has a stabilizing effect. Additionally, SN006/7-3 caused a smaller increase in the Tm of T-p53C-Y220C-CL than original compound SN006 (∆Tm= 3.05 °C, 1 mM, 24 h).31 However, the deconvoluted MS spectrum of T-p53C-Y220-CL with SN006 showed peaks with more than one fragment attached to the protein.31 As depicted in Figure 4, in the deconvoluted MS spectrum of the Y220C cysteine light mutant with the SAR compound SN006/7-3, one signal corresponded to the protein species with one molecule attached via SNAr, while the higher signal corresponded to protein with one molecule SN006/7-3 attached via reversible covalent Pinner-type addition to the nitrile. Hence, it is unclear from the MS data whether SN006/7-3 exclusively labels the remaining cysteine 220 in the pocket of the Y220C cysteine light mutant and which reaction mechanism is responsible for stabilizing T-p53C-Y220C-CL.
 |
Table 4 Effect of Compound SN006/7-3 on the Tm of T-p53C-Y220C-CL |
 |
Figure 4 Deconvoluted MS spectrum of T-p53C-Y220C-CL with SN006/7-3 at a molar ratio of 1:100 after 24 h of incubation at 20 °C (theoretical mass of unmodified T-p53C-Y220C-CL: 24,615.68 Da). |
SN006/7-3 Adopts an Alternative Binding Pose in the Y220C Induced Pocket
The X-ray structure of T-p53C-Y220C soaked with 5 mM SN006/7-3 confirmed that this pyrazine derivative is another Cys220 arylating compound, displaying well-defined density in the binding pocket of the Y220C mutant, as shown in Figure 5. Additional modifications were found at Cys182 and Cys277 after soaking for one day with 5 mM SN006/7-3. In the X-ray structure only the modification of cysteines by SN006/7-3 via SNAr was detected. The Pinner-type addition of proteinogenic cysteines to the nitrile group of the compound observed in the MS experiment was not visible, possibly due to the reversibility of the reaction. The 5 mM-soaked structure indicated that the pyrazine derivative SN006/7-3 reacted with Cys220 via an SNAr reaction with displacement of the chlorine atom.
 |
Figure 5 Overview of the binding mode of SN006/7-3 (soaked at 5 mM, PDB: 9G5H) into the Y220C induced crevice. The pyrazine derivative SN006/7-3 reacted with Cys220 via an SNAr reaction. The compound adopts an alternative conformation, occupying subsite 2 of the binding pocket. The unbiased omit map is shown at a contour level of 3 σ. |
Interestingly, the crystal structure revealed an alternative binding pose of SN006/7-3 in the Y220C cleft compared to the further identified Cys220 arylating compounds SN001 and SN006. The CovLib hits SN001 and SN006 adopted a Tyr-like conformation, occupying the central cavity and mimicking the WT Tyr220. Their electron-withdrawing nitrile groups pointed towards Thr150 and subsite 1. The meta-fluorine (SN001) or meta-nitrile moiety (SN006) pointed towards subsite 3. However, subsite 2 of the Y220C binding pocket was not occupied by SN001 and SN006.31 In contrast, as shown in Figure 5, the pyrazine derivative SN006/7-3 was positioned deeply in subsite 2 of the Y220C binding pocket. Its electron-withdrawing nitrile group occupied subsite 2 of the mutational crevice. Nevertheless, subsite 1 and subsite 3 were not targeted by compound SN006/7-3.
Second Generation: Scaffold Hopping Based on SN006/7-3
The identified SNAr reactive compound SN006/7-3, arylating Cys220 and occupying subsite 2 of the Y220C pocket, is still very reactive. Therefore, additional SAR fragments, which contain electron-donating methyl groups at the heteroarenes (Figure 6) were screened by DSF and intact protein MS. Since the previously identified cleft binders SN001, SN006, and SN006/7-3 are pyridine or pyrazine derivatives, additional six-membered heterocycles such as pyrimidines and pyridazines were tested.
 |
Figure 6 Overview of SN006/7-3 SAR compounds and their t1/2 ± SD measured via GSH assay. |
Compounds SN006/7-7 and SN006/7-8 have an additional electron-donating methyl group in the meta or ortho position to the LG compared to the pyrazine SN006/7-3. In comparison to SN006/7-3 (t1/2 GSH= 0.23 h), the GSH reactivity of SN006/7-7 (t1/2 GSH= 0.45 h) slightly diminished, whereas that of SN006/7-8 (t1/2 GSH= 0.28 h) was only minimally affected. Both compounds particularly stabilized T-p53C-Y220C, indicating cleft binding (Table 5). After 30 min of incubation, SN006/7-8 already stabilized all tested mutants, while SN006/7-7 only slightly increased the Tm of T-p53C (Table S11). However, SN006/7-8 had a smaller stabilizing effect on T-p53C-Y220C, with a ∆Tm of 1.71 °C after 30 min (Table 6), compared to SN006/7-3, which lacks the methyl group, with a ∆Tm of 2.75 °C. Time-dependent effects were observed for both SN006/7-7 and SN006/7-8. After 4 h of incubation, T-p53C-Y220C was especially stabilized by SN006/7-7 (∆Tm= 1.28 °C, 1 mM) and SN006/7-8 (∆Tm= 4.43 °C, 1 mM) (Table S12). The first derivatives of the melting curves of T-p53C incubated with SN006/7-8 for 4 h or 24 h showed two distinct peaks similar to compound SN006/7-3 (Figure S36A, Figure S37A). Interestingly, compound SN006/7-8 substantially increased the melting temperature of the Y220C mutant by up to 5.02 °C (1 mM) after 24 h of incubation (Table 5 and Table 6). The introduction of a methyl group in the ortho position to the LG increased the stabilizing effect of this scaffold compared to SN006/7-3 (∆Tm= 3.62 °C, 1 mM, 24 h) without a methyl group and SN006/7-7 (∆Tm= 2.08 °C, 1 mM, 24 h) with a methyl group in the meta position. Through the introduction of the methyl group, the electron deficiency of the heteroarene is reduced compared to fragment SN006/7-3. However, increased stabilization by fragment SN006/7-8 suggests that the methyl group at position 6 of the pyrazine ring induces favorable interactions in the Y220C pocket. Compound SN006/7-7 with the methyl group in the meta position to the LG resulted in a considerably lower stabilization of Y220C compared to fragments SN006/7-8 and SN006/7-3. Additionally, the compounds were tested at a concentration of 250 µM with T-p53C and T-p53C-Y220C (Table S14 and Table 6). As the compound concentration decreased, the stabilization of T-p53C and T-p53C-Y220C also decreased. The highest increase in the melting temperatures was achieved with 1 mM SN006/7-7 and SN006/7-8 after a 24-hour incubation period. However, even at a compound concentration of 250 µM, SN006/7-8 showed a higher stabilization of T-p53C-Y220C (∆Tm= 4.45 °C, 24 h) compared to SN006/7-7 (∆Tm= 1.10 °C, 24 h) and SN006/7-3 (∆Tm= 2.87 °C, 24 h).
 |
Table 5 Impact of the SN006/7-3 SAR Compounds on the Tm of T-p53C and Its Mutants After 24 h of Incubation. WT Refers to T-p53C; Mutant Labels (eg, Y220C) Indicate Mutants of T-p53C, Such as T-p53C-Y220C |
 |
Table 6 Effect of SN006/7-3 SAR Compounds on the Tm of T-p53C-Y220C |
The 2-chloropyrimidine SN010 caused a thermal stabilization of p53 and its mutants in the DSF experiment. Notably, SN010, which has an electron-donating NH2 group in the ortho position to the CN moiety, stabilized p53 after 24 h of incubation, in contrast to its pyrazine analog SN006/7-6. A pronounced stabilization of T-p53C-Y220C and binding in the Y220C pocket cannot be deduced from the DSF data for the 2-chloropyrimidine SN010. This discovery aligns with previous findings on pyrimidines substituted at position 2, such as the 2-sulfonylpyrimidine PK1100036 or the 2-chloropyrimidine 4482.22 These compounds achieved general stabilization of p53, but did not bind into the Y220C pocket.
After 24 h of incubation, both pyridazine derivatives, SN011 and SN012, moderately stabilized all tested mutants within a similar range. Compared to the pyrazine analog SN006/7-3 (∆Tm= 3.62 °C, 1 mM, 24 h), the stabilization of T-p53C-Y220C by the pyridazine derivative SN011 was less pronounced, with a maximum of 1.10 °C (1 mM, 24 h).
Overall, the results showed that the introduction of a methyl group, in the case of SN006/7-8, increased the stabilization of the oncogenic mutant T-p53C-Y220C. Moreover, the DSF results indicated that altering the heteroarene did not result in a high stabilization of the cancer mutant Y220C and selective binding into the mutational cleft.
Intact protein MS confirmed covalent modification of T-p53C-Y220C by all tested SN006/7-3 SAR compounds (Figure S40). The deconvoluted MS spectra of T-p53C-Y220C with the pyrazine derivatives SN006/7-7 and SN006/7-8 are depicted in Figure 7. Besides the electron-donating methyl group in the meta or ortho position to the LG, both compounds are still reactive, resulting in multiple arylations of T-p53C-Y220C. In both deconvoluted MS spectra, the protein species with three attached molecules of SN006/7-7 or SN006/7-8 represented the highest signal, and up to four times arylated protein was detected. Moreover, in the deconvoluted MS spectrum of SN006/7-7 (Figure 7A) with T-p53C-Y220C, an additional small signal corresponding to the two times arylated protein was evident. The number of modifications was reduced compared to the original fragment SN006/7-3 (Figure 2B). Furthermore, the deconvoluted MS spectrum of SN006/7-3 showed numerous signals where the protein was modified by reversible covalent addition to the nitrile group. In contrast, only signals for the protein arylated via SNAr were detectable in the spectra with the methylated SAR compounds SN006/7-7 and SN006/7-8.
 |
Figure 7 Deconvoluted MS spectra of T-p53C-Y220C with SN006/7-7 (A) and SN006/7-8 (B) at a protein-to-compound ratio of 1:100 after 24 h of incubation at 20 °C (theoretical mass of unmodified T-p53C-Y220C: 24,696.00 Da). |
SN006/7-8 Induced a Time and Concentration-Dependent Pronounced Stabilization of the Cancer Mutant T-p53C-Y220C
SN006/7-8 showed a distinct stabilization of the Y220C mutant up to a lowest effective compound concentration of 250 µM after only 30 min of incubation (Figure 8 and Table 7), in contrast to its analog SN006/7-7 (Figure S41 and Table S16). Nevertheless, the maximum stabilization achieved by fragment SN006/7-8 (∆Tm= 1.83 °C, 1 mM) after 30 min was lower than that of the original fragment SN006/7-3 (∆Tm= 2.65 °C, 1 mM, Table 3). Additionally, SN006/7-3 stabilized up to a lowest effective compound concentration of 125 µM after 30 min of incubation. After 24 h incubation time, the stabilization of T-p53C-Y220C by compound SN006/7-8 with 4.98 °C (1mM) was much more pronounced than by fragments SN006/7-3 (∆Tm= 3.55 °C, 1 mM, Table 3) and SN006/7-7 (∆Tm= 2.15 °C, 1 mM, Table S16). Even at concentrations of 500 µM and 250 µM, compound SN006/7-8 achieved substantially higher ∆Tm values than SN006/7-3. Similar to SN006/7-3, T-p53C-Y220C was stabilized up to a lowest effective compound concentration of 31.25 µM after 24 h of incubation, corresponding to a molar ratio of 1:3.91. Overall, the introduction of a methyl group in the ortho position to the LG markedly enhanced the stabilization of T-p53C-Y220C, compared to SN006/7-3 without a methyl group and SN006/7-7 with a methyl group in the meta position to the LG.
 |
Figure 8 Concentration-dependent DSF measurements of T-p53C-Y220C with SN006/7-8. The first derivatives of the melting curves and resulting ∆Tm values of T-p53C-Y220C (8 µM protein) with various concentrations of compound SN006/7-8 after 30 min (A) and 24 h (B) of incubation at 20 °C are shown. |
 |
Table 7 Concentration-Dependent Effects of Compound SN006/7-8 on the Tm of T-p53C-Y220C After 30 min and 24 h Incubation Time |
As shown in Figure S43 and Table S18, SN006/7-8 stabilized T-p53C-Y220C by up to 1.43 °C after only 10 min of incubation. The ∆Tm values of T-p53C-Y220C, supplemented with 1 mM SN006/7-8, increased with increasing incubation time, tending towards a maximum after 3 h of incubation. This time dependence is comparable to that of its constitutional isomer SN006/7-7 (Figure S42 and Table S17), whereas with SN006/7-3 (Figure S33 and Table S10) a higher stabilization of T-p53C-Y220C and thus target occupancy was already achieved at shorter incubation times.
Evaluation of SN006/7-7 and SN006/7-8 with T-p53C-Y220C-CL
The identified T-p53C-Y220C stabilizing hits SN006/7-7 and SN006/7-8 were further investigated with the Y220C cysteine light mutant (Figure S44, Table 8 and Figure 9). SN006/7-8 increased the Tm of the Y220C cysteine light mutant by up to 3.68 °C (1 mM, 24 h). As previously noted for T-p53C-Y220C, the fragment SN006/7-8, which has a methyl group in the ortho position to the LG, resulted in a higher stabilization of T-p53C-Y220C-CL compared to its constitutional isomer SN006/7-7 (∆Tm= 1.50 °C, 1 mM, 24 h) and SN006/7-3 (∆Tm= 2.40 °C, 1 mM, 24h). In addition, a comparably high stabilization of T-p53C-Y220C-CL was measured after 24 h of incubation with 1 mM (∆Tm= 3.68 °C) and 250 µM (∆Tm= 3.58 °C) SN006/7-8. Furthermore, as already observed for the Y220C mutant, the stabilization of T-p53C-Y220C-CL was time-dependent. Similar to SN006/7-3 and SN006/7-7, the stabilization of T-p53C-Y220C-CL by fragment SN006/7-8 was lower than that of T-p53C-Y220C (∆Tm= 5.02 °C, 1 mM, 24 h), indicating that the modification of cysteines besides Cys220 also has a stabilizing effect.
 |
Figure 9 Deconvoluted MS spectra of T-p53C-Y220C-CL (theoretical mass of unmodified T-p53C-Y220C-CL: 24,615.68 Da) with SN006/7-7 (A) at a molar ratio of 1:100, and with SN006/7-8 at a molar ratio of 1:100 (B) and 1:31.25 (C) after 24 h of incubation at 20 °C. |
 |
Table 8 Effect of SN006/7-7 and SN006/7-8 on the Tm of T-p53C-Y220C-CL |
In contrast to the deconvoluted MS spectrum of T-p53C-Y220C-CL with the original fragment SN006/7-3 (Figure 4), in the spectra of SN006/7-7 and SN006/7-8 with the Y220C cysteine light mutant, only the SNAr-type reaction was visible (Figure 9). Only the protein species arylated with one molecule of SN006/7-7 and SN006/7-8 was detected, indicating an arylation of the mutation-induced cysteine 220. In the deconvoluted MS spectra of T-p53C-Y220C-CL incubated with SN006/7-8 at a molar ratio of 1:100 (Figure 9B), only one signal was visible, corresponding to the single arylated protein. At a ratio of 1:31.25, an additional small signal was present for the native Y220C cysteine light protein (Figure 9C).
In summary, the DSF and intact protein MS results with the Y220C cysteine light mutant indicated that SN006/7-8, with a methyl group ortho to the LG, provided a higher thermal stabilization compared to SN006/7-3 and SN006/7-7. These findings may be attributed to a more selective binding of SN006/7-8 into the Y220C pocket.
SN006/7-8 Adopts a Tyr-Like Conformation in the Y220C Induced Crevice
The covalent modification of Cys220 by compound SN006/7-8 was confirmed using X-ray crystallography, as depicted in Figure 10. Compared to fragment SN006/7-3, pointing into subsite 2 of the hydrophobic pocket, fragment SN006/7-8 with the additional methyl group adopted the same Tyr-like conformation as the pyridine derivative SN001 and the pyrazine derivative SN006.31 Fragment SN006/7-8 reacted with Cys220 via an SNAr reaction with the chlorine atom as the LG. It occupied most of the space of the central cavity, mimicking the WT Tyr220. The electron-withdrawing CN moiety pointed towards subsite 1 and Thr150. The methyl group at position 6 of the pyrazine ring pointed into the hydrophobic subsite 3. Favorable hydrophobic interactions of the methyl group in subsite 3 could lead to the enhanced stabilization of T-p53C-Y220C compared to SN006/7-3 and SN006/7-7. It is noteworthy that the non-covalent cleft binders PK708826 and MB71027 also exhibited favorable hydrophobic interactions in subsite 3, formed by a flip of the Cys220, upon the introduction of a pyrrole side chain.
In addition, other residues that were arylated in the 5 mM-soaked structure included Cys182 and Cys277.
 |
Figure 10 Overview of the binding mode of SN006/7-8 (soaked at 5 mM, PDB 9G6T) into the Y220C induced crevice. The compound binds similarly to SN001 and SN006 in a Tyr-like conformation, with the nitrile pointing towards subsite 1 and Thr150.31 The meta-methyl group points into the hydrophobic subsite 3. The unbiased omit map is shown at a contour level of 3 σ. |
Third Generation: Structure-Activity Relationship Screening of the Identified Hit SN006/7-8
The promising fragment SN006/7-8 with improved stabilization of T-p53C-Y220C covalently modifies Cys220, adopting a Tyr-like conformation in the mutational cleft with the methyl group pointing into subsite 3 of the binding pocket. This prompted us to test additional SAR compounds with larger hydrophobic groups ortho to the LG to increase the selectivity for the pocket through improved hydrophobic interactions in subsite 3. However, no commercially available SAR compounds of SN006/7-8 with eg, ethyl or isopropyl groups at position 6 of the heteroarene were found. Hence, only 3.5-dichloropyrazine (SN006/7-9) or pyridine (SN006/7-10 to SN006/7-19) derivatives were purchased. The structures of the acquired SAR compounds are depicted in Figure 11.
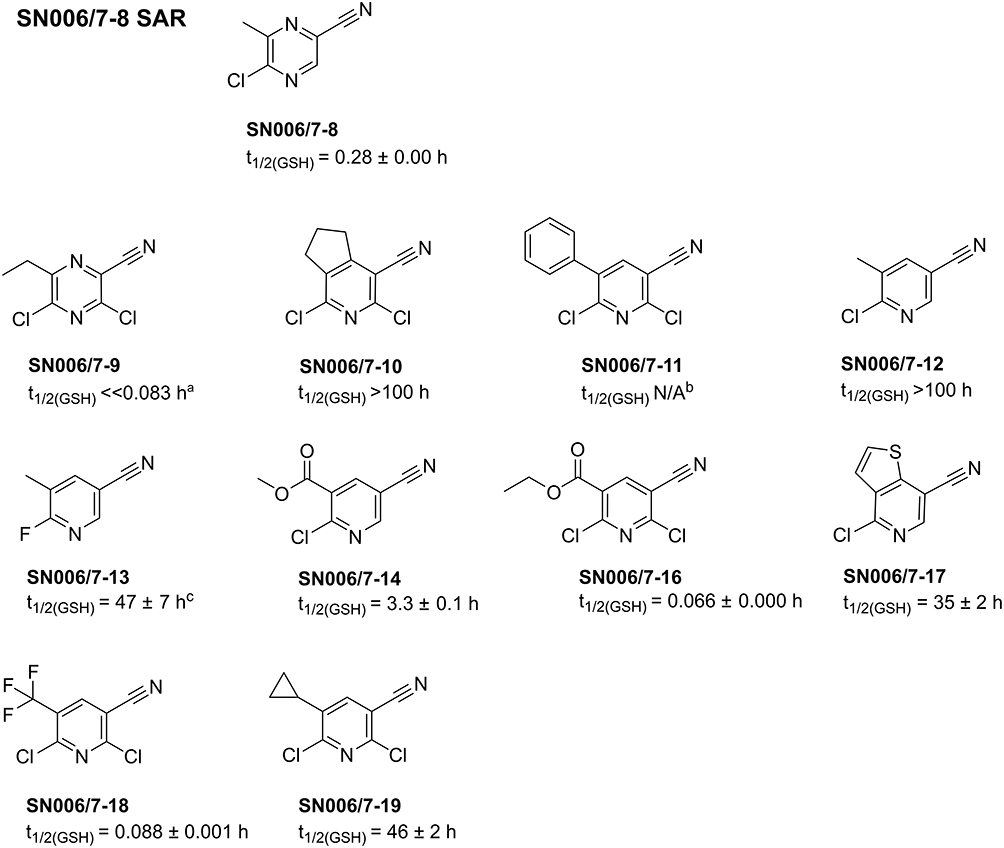 |
Figure 11 Overview of the tested SN006/7-8 SAR compounds and their t1/2 ± SD determined via GSH assay. aThe fragment was already degraded at the initial measurement point (0 h). The value of 0.083 h corresponds to the approximate time between adding the fragment to the reaction mixture and starting the HPLC run. bThe compound was not soluble in ACN buffer mixture at the required concentration. cThe fragment appeared to precipitate during the measurement process. |
The pyrazine fragment SN006/7-9 contains an ethyl group and an additional chlorine compared to the hit SN006/7-8. The introduction of a second electron-withdrawing chlorine in the ortho position to the EWG results in a highly reactive fragment with a t1/2 ≪ 0.083 h in the GSH assay. Incubation with 1 mM SN006/7-9 for 30 min markedly lowered the Tm of T-p53C (∆Tm= −4.85 °C), T-p53C-Y220C (∆Tm= −1.30 °C), and T-p53-C124/182/277S (∆Tm= −2.45 °C) (Table S19). However, the decrease in the Tm of T-p53C-Y220C was the least pronounced with a ∆Tm of −1.30 °C. After only 30 min incubation time, the first derivatives of the melting curves of the R273H and the R282W mutants did not display a peak (Figure S45). Furthermore, for all tested p53 mutants, no suitable melting curves were retained after 4 h and 24 h of incubation, or a destabilization of the triple cysteine control mutant occurred (Table 9). Fragment SN006/7-9 was also tested at a compound concentration of 250 µM with T-p53C and T-p53C-Y220C, as shown in Table S22 and Table 10. Nevertheless, at this concentration, fragment SN006/7-9 only stabilized the T-p53C-Y220C by up to 2.60 °C (1 mM, 24 h), indicating cleft binding. A higher stabilization of T-p53C-Y220C could be prevented by the high reactivity of the fragment, leading to unfavorable hyperarylation of the protein.
 |
Table 9 Impact of the Identified SN006/7-8 SAR Hits on the Tm of T-p53C and Its Mutants After 24 h of Incubation. WT Refers to T-p53C; Mutant Labels (eg, Y220C) Indicate Mutants of T-p53C, Such as T-p53C-Y220C |
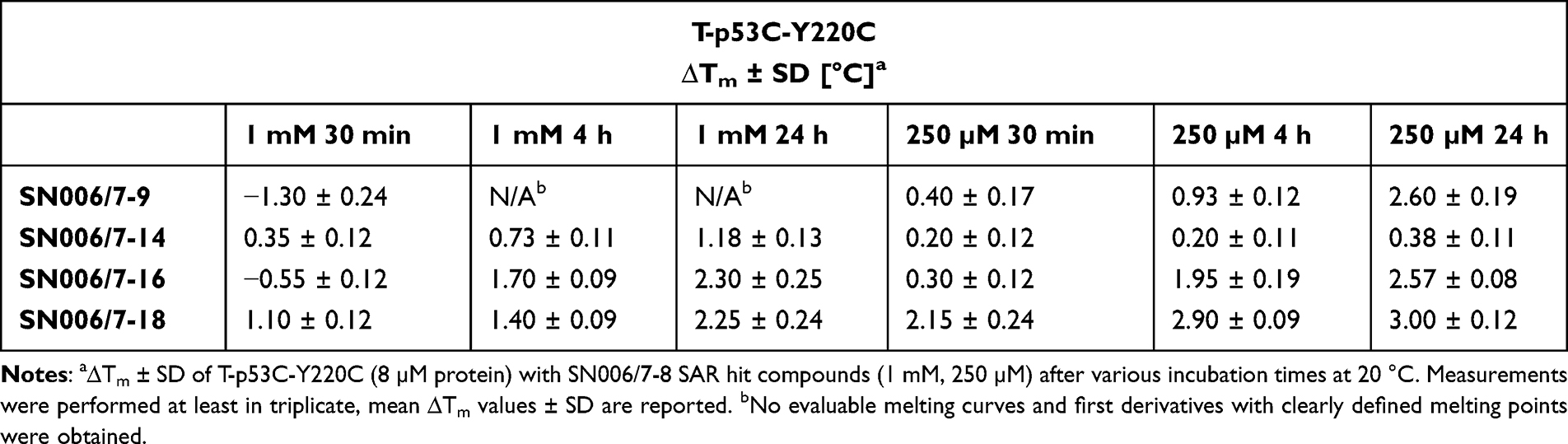 |
Table 10 Effect of SN006/7-8 SAR Hits on the Tm of T-p53C-Y220C |
SN006/7-12 and SN006/7-13 did not affect the Tm of T-p53C and its mutant, even after 24 h of incubation. The pyridines with an electron-withdrawing nitrile group and a halogen as the LG are less reactive or chemically stable and not suitable for covalent modification and stabilization of p53. Similarly, SN006/7-17 also showed no raise in the melting temperatures of p53 and its mutants after various incubation times. In addition, fragments SN006/7-10, SN006/7-11, and SN006/7-19 with two chlorine atoms, an electron-withdrawing CN group, and different electron-donating substituents at the pyridine ring, did not substantially influence the Tm of T-p53C, T-p53C-Y220C, and all tested mutants (Tables S19–S21).
Compound SN006/7-14 with an additional electron-withdrawing methoxycarbonyl group at the pyridine increased the Tm of all tested p53 mutants after 24 h incubation time at a concentration of 1 mM (Table 9). Nevertheless, as shown in Table 10, no substantial stabilization of T-p53C-Y220C was observed at a concentration of 250 µM. Compared to fragment SN006/7-12 (t1/2 GSH >100 h), the introduction of a second EWG in the ortho position to the LG in fragment SN006/7-14 (t1/2 GSH= 3.3 h) increases the reactivity, leading to a stabilization of p53 and its mutants. Fragment SN006/7-14 stabilized all mutants comparably, and no pronounced stabilization of the Y220C mutant was observed. Therefore, binding into the Y220C pocket is not expected.
The pyridine derivatives SN006/7-16 and SN006/7-18 with electron-withdrawing ethoxycarbonyl group or CF3 group and an additional chlorine atom are highly reactive. Both compounds particularly stabilized T-p53C-Y220C, suggesting a covalent modification of Cys220 (Table 9). After 24 h of incubation, the Tm of T-p53C-Y220C was increased by up to 2.57 °C with 250 µM SN006/7-16 and by up to 3.00 °C with 250 µM SN006/7-18.
The intact protein MS results emphasize that the hits SN006/7-9, SN006/7-14, SN006/7-16, and SN006/7-18 multiply modified T-p53C-Y220C after 24 h of incubation at a protein-to-compound ratio of 1:100 (Figure S50).
Overall, the SAR compound screen identified either less reactive or unreactive compounds (SN006/7-10,SN006/7-11, SN006/7-12, SN006/7-13, SN006/7-17, SN006/7-19) or highly reactive compounds (SN006/7-9, SN006/7-16, SN006/7-18). In total, no better hit was discovered than fragment SN006/7-8, which was more selective for the Y220C binding pocket and achieved a higher stabilization of T-p53C-Y220C. Although fragments SN006/7-9, SN006/7-16, and SN006/7-18 represent hits that suggest cleft binding, they are also highly reactive and destabilized other p53 mutants tested. The screen indicates that for the development of more T-p53C-Y220C selective SAR compounds, the scaffold of SN006/7-8 is necessary with electron-donating groups (EDGs) other than the methyl group ortho to the LG.
The Highly Reactive Fragment SN006/7-9 Also Adopts a Tyr-Like Conformation in the Mutational Cleft
The highly reactive fragment SN006/7-9 was identified as a cleft binder and Cys220 arylating compound using X-ray crystallography. As illustrated in Figure 12, compound SN006/7-9 reacted with Cys220 via an SNAr reaction with the displacement of the para-chlorine. In chain A (Figure 12A), similar to fragments SN001, SN006, and SN006/7-8, the compound SN006/7-9 also adopted a Tyr-like conformation, occupying the central cavity space.31 Its electron-withdrawing nitrile moiety pointed towards subsite 1 and Thr150, and the chlorine atom in ortho position pointed towards subsite 2. As anticipated, the ethyl group at position 6 of the pyrazine ring pointed into the hydrophobic subsite 3. In contrast to the pyrazine derivative SN006/7-3, subsite 2 of the binding pocket was not targeted.
 |
Figure 12 Overview of the binding mode of SN006/7-9 (soaked at 5 mM, PDB 9G6U) into the Y220C induced crevice. In chain A, the compound adopts a Tyr-like conformation, with the nitrile pointing towards subsite 1 and Thr150. The ortho-chlorine points towards subsite 2, and the meta-ethyl group points into the hydrophobic subsite 3 (A). In chain B, the binding pose of SN006/7-9 in the Y220C pocket is slightly altered, with SN006/7-9 oriented more towards subsite 2 (B). The unbiased omit maps are shown at a contour level of 3 σ. |
Additional modifications were found at Cys182 and Cys277 after soaking for one day with 5 mM SN006/7-9.
The Identified Fragment Hits SN006/7-3 and SN006/7-8 Did Not Alter the DNA Binding Capacity of Full-Length p53
The effect of the SN006/7-3 and its methylated analog SN006/7-8 on the DNA binding affinity of full-length (FL)-T-p53 to its non-apoptotic response element GADD45α was tested by FPA. Incubation of 2 h with the two original CovLib fragments SN006 and SN007 negatively affected the DNA binding of FL-T-p53 to GADD45α.31 However, after 24 h of incubation with SN006/7-3 and SN006/7-8 at a high molar ratio of 1:200, the DNA-binding of FL-T-p53 to GADD45α was maintained. The dissociation constant (Kd) for FL-T-p53 binding to GADD45α was only minimally increased from 83 nM without compound to 95 nM with SN006/7-3 and was perfectly conserved with a Kd of 84 nM after incubation with SN006/7-8 (Figure 13 and Table 11). The FPA results indicate that despite the binding of SN006/7-3 and SN006/7-8 to cysteines in p53 and especially to Cys277 in the DNA binding region of p53,4 regular DNA binding of FL-T-p53 is maintained. Moreover, the FPA outcomes suggest that normal transcription of p53 response elements is possible due to the thermal stabilization in cellular systems.
 |
Figure 13 Relative fluorescence polarization data [%] of FL-T-p53 with 20 nM 5’-carboxyfluorescein-labeled GADD45α response element and 5% DMSO.36 FL-T-p53 was incubated with compounds SN006/7-3 and SN006/7-8 at a protein-to-compound ratio of 1:200 for 24 h at 20 °C. |
 |
Table 11 Resulting Kd Values ± SD of the Direct Titrations of FL-T-p53 With or Without the Addition of SN006/7-3 or SN006/7-8 With GADD45α. The DMSO Column Indicates the Reference Measurement Without Adding Compound |
Conclusion
In summary, we herein employed an “electrophile first approach”,57 screening SAR fragments of the previously identified highly reactive CovLib hits SN006 and SN007 with the challenging target T-p53C-Y220C. We found a set of SNAr reactive fragments, targeting Cys220 in the hydrophobic pocket of T-p53C-Y220C and stabilizing the thermosensitive cancer mutant. Moreover, the discovered fragments showed different binding poses and occupied various subsites of the Y220C binding pocket. All identified fragments, covalently addressing Cys220 in the binding pocket of T-p53C-Y220C, are pyrazines with an electron-withdrawing CN group and a halogen as the LG in para position to the EWG. Variations in the heteroarene did not lead to more Cys220 selective hits. Compounds with ortho-EWG were also less reactive and not more selective for the Y220C pocket (SN006/7-2, SN006/7-4) than compounds with para-EWG (SN006/7-3, SN006/7-5).
The 5-chloro-pyrazine-2-carbonitrile (SN006/7-3) increased the Tm of T-p53C-Y220C by up to 3.62 °C. SN006/7-3 exhibited multiple modifications of the Y220C mutant in the intact protein MS experiment. However, arylation of the Cys220 in the binding pocket was confirmed by X-ray crystallography. The pyrazine derivative SN006/7-3 adopted a different conformation compared to the pyridine derivatives SN001 and the pyrazine CovLib hit SN006.31 The compound occupied subsite 2 of the binding pocket.
Interestingly, compound SN006/7-8, with an electron-donating methyl group in the ortho position to the LG of fragment SN006/7-3, substantially increased the melting temperature of the Y220C mutant by up to 5.02 °C (1 mM) after 24 h of incubation. The introduction of a methyl group in the ortho position to the LG increased the stabilizing effect of this scaffold compared to SN006/7-3 (∆Tm= 3.62 °C, 1 mM, 24 h) without a methyl group and SN006/7-7 (∆Tm= 2.08 °C, 1 mM, 24 h) with a methyl group in the meta position. Covalent modification of Cys220 by compound SN006/7-8 was confirmed by X-ray crystallography. Compared to fragment SN006/7-3, pointing into subsite 2 of the hydrophobic pocket, fragment SN006/7-8 with the additional methyl group adopted the same Tyr-like conformation as the pyridine derivative SN001 and the pyrazine derivative SN006.31 The methyl group at position 6 of the pyrazine ring of SN006/7-8 protruded into the hydrophobic subsite 3.
To increase the selectivity for the Y220C pocket through improved hydrophobic interactions in subsite 3, additional SAR compounds of SN006/7-8 with larger hydrophobic groups ortho to the LG were tested. The reactive fragment SN006/7-9 was identified as a Cys220 arylating fragment that also adopted a Tyr-like conformation in the Y220C pocket using X-ray crystallography. Overall, either unreactive compounds or highly reactive compounds, such as SN006/7-9, were identified, and no better hit was discovered than fragment SN006/7-8.
We have demonstrated that after incubation with SN006/7-3 and SN006/7-8 FL-T-p53 retains its potential to bind the GADD45α response element. However, in vivo experiments are essential to fully investigate the complexity of signaling and downstream effect activation.
In conclusion, all identified fragments are still highly reactive but represent a promising basis for optimization in a subsequent drug discovery process. To reduce the reactivity and increase the selectivity for the mutation-induced Y220C pocket with non-covalent interactions, the identified fragments can be merged, linked, and grown.58–60
Abbreviations
AUC, area under the curve; CL, cysteine light (C124/182/229/275/277S); CovLib, covalent fragment library; DSF, differential scanning fluorimetry; EDG, electron-donating group; EWG, electron-withdrawing group; FBDD, fragment-based drug discovery; FL, full-length; FP, fluorescence polarization; FPA, fluorescence polarization assay; GSH, glutathione; HPLC, high-performance liquid chromatography; Kd, dissociation constant; LG, leaving group; MDM2, mouse double minute 2 homolog; MDM4, mouse double minute 4 homolog; MS, mass spectrometry; N/A, not available; PBS, phosphate buffered saline; SAR, structure-activity relationship; SD, standard deviation; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SNAr, nucleophilic aromatic substitution; t1/2, half-life; Tm, melting temperature; T-p53C, quadrupole thermostable mutant of the p53 core-domain, 94–312, M133L/V203A/N239Y/N268D; UHPLC-ESI-MS, ultra-high-performance liquid chromatography electrospray ionization mass spectrometry; WT, wild-type.
Acknowledgments
We thank Dr. Georg Zocher and Dr. Michael Braun for their continuous advice in crystallography. We gratefully acknowledge the Swiss Light Source (SLS, Switzerland) for beam time.
Author Contributions
All authors made a significant contribution to the work reported: F.M.B. envisioned the research. T.K. and F.M.B. conceptualized the experiments and designed the study. T.K. prepared the proteins by heterologous expression and performed the DSF studies. TK prepared the MS samples. S.J.J., B.M., C.K, and M.L. performed and the UHPLC-ESI-MS experiments. S.J.J., B.M., C.K., T.K., and M.L. analyzed the UHPLC-ESI-MS experiments. Crystallographic data reduction and refinement was performed by J.S. and T.S. FPA experiments were performed by T.K. M.S. performed the Glutathione Assay. T.K. conducted data analysis and reprocessing of the experimental data. T.K., J.S., and F.M.B. wrote the manuscript. S.J.J., B.M., C.K., M.L., M.S., and T.S. critically reviewed the manuscript. All authors have agreed on the journal to which the article has been submitted. All authors agreed on all versions of the article at any stage of the submission, revision, publication and proofing process, particularly including the final version accepted for publication. All authors agree to take responsibility and be accountable for the contents of the article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi:10.1038/35042675
2. Lane DP. p53, guardian of the genome. Nature. 1992;358(6381):15–16. doi:10.1038/358015a0
3. Amin MN, Liu -Y-Y. Restoration of tumor suppression to cancer carrying p53 mutations. In: Piccaluga DPP, editor. Molecular Diagnostics of Cancer. IntechOpen; 2023. chap 0.
4. Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77(1):557–582. doi:10.1146/annurev.biochem.77.060806.091238
5. Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13(6):1027–1036. doi:10.1038/sj.cdd.4401910
6. el-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol. 1998;8(5):345–357. doi:10.1006/scbi.1998.0097
7. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9(12):862–873. doi:10.1038/nrc2763
8. Vousden KH, Prives C. Blinded by the light: the growing Complexity of p53. Cell. 2009;137(3):413–431. doi:10.1016/j.cell.2009.04.037
9. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370. doi:10.1038/nrc3711
10. Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. 2016;85(1):375–404. doi:10.1146/annurev-biochem-060815-014710
11. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6(12):909–923. doi:10.1038/nrc2012
12. Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19(10):1245–1256. doi:10.1038/sj.onc.1203434
13. Joerger AC, Fersht AR. Structure–function–rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26(15):2226–2242. doi:10.1038/sj.onc.1210291
14. Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci USA. 2006;103(41):15056–15061. doi:10.1073/pnas.0607286103
15. Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi:10.1016/s0065-230x(08)60785-x
16. Dearth LR, Qian H, Wang T, et al. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis. 2007;28(2):289–298. doi:10.1093/carcin/bgl132
17. Joerger AC, Ang HC, Veprintsev DB, Blair CM, Fersht AR. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J Biol Chem. 2005;280(16):16030–16037. doi:10.1074/jbc.M500179200
18. Zhou S, Chai D, Wang X, et al. AI-powered discovery of a novel p53-Y220C reactivator. Origin Res Front Oncol. 2023;13. doi:10.3389/fonc.2023.1229696
19. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170(6):1062–1078. doi:10.1016/j.cell.2017.08.028
20. Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28(6):622–629. doi:10.1002/humu.20495
21. Wilcken R, Liu X, Zimmermann MO, et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J Am Chem Soc. 2012;134(15):6810–6818. doi:10.1021/ja301056a
22. Stahlecker J, Klett T, Schwer M, et al. Revisiting a challenging p53 binding site: a diversity-optimized HEFLib reveals diverse binding modes in T-p53C-Y220C. 10.1039/D2MD00246A. RSC Med Chem. 2022;13(12):1575–1586. doi:10.1039/D2MD00246A
23. Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci USA. 2008;105(30):10360–10365. doi:10.1073/pnas.0805326105
24. Stephenson Clarke JR, Douglas LR, Duriez PJ, et al. Discovery of nanomolar-affinity pharmacological chaperones stabilizing the oncogenic p53 Mutant Y220C. ACS Pharmacol Transl Sci. 2022;5(11):1169–1180. doi:10.1021/acsptsci.2c00164
25. Bauer MR, Jones RN, Baud MGJ, et al. Harnessing Fluorine–Sulfur contacts and multipolar interactions for the design of p53 mutant Y220C rescue drugs. ACS Chem Biol. 2016;11(8):2265–2274. doi:10.1021/acschembio.6b00315
26. Liu X, Wilcken R, Joerger AC, et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013;41(12):6034–6044. doi:10.1093/nar/gkt305
27. Baud MGJ, Bauer MR, Verduci L, et al. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur J Med Chem. 2018;152:101–114. doi:10.1016/j.ejmech.2018.04.035
28. Vu BT, Dominique R, Fahr BJ, et al. Discovery of rezatapopt (PC14586), a first-in-class, small-molecule reactivator of p53 Y220C mutant in development. ACS Med Chem Lett. 2025;16(1):34–39. doi:10.1021/acsmedchemlett.4c00379
29. US National Library of Medicine. ClinicalTrials.gov. Available from: https://clinicaltrials.gov/study/NCT04585750.
30. Guiley KZ, Shokat KM. A small molecule reacts with the p53 somatic mutant Y220C to rescue wild-type thermal stability. Cancer Discov. 2023;13(1):56–69. doi:10.1158/2159-8290.Cd-22-0381
31. Klett T, Stahlecker J, Jaag S, et al. Covalent fragments acting as tyrosine mimics for mutant p53-Y220C rescue by nucleophilic aromatic substitution. ACS Pharmacol Transl Sci. 2024;7(12):3984–3999. doi:10.1021/acsptsci.4c00414
32. Klett T, Schwer M, Ernst LN, et al. Evaluation of a covalent library of diverse warheads (CovLib) binding to JNK3, USP7, or p53. Drug Des Devel Ther. 2024;18:2653–2679. doi:10.2147/DDDT.S466829
33. Keeley A, Ábrányi-Balogh P, Keserű GM. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. MedChemComm. 2019;10(2):263–267. doi:10.1039/C8MD00327K
34. Nikolova PV, Henckel J, Lane DP, Fersht AR. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc Natl Acad Sci USA. 1998;95(25):14675–14680. doi:10.1073/pnas.95.25.14675
35. Joerger AC, Allen MD, Fersht AR. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J Biol Chem. 2004;279(2):1291–1296. doi:10.1074/jbc.M309732200
36. Bauer MR, Joerger AC, Fersht AR. 2-Sulfonylpyrimidines: mild alkylating agents with anticancer activity toward p53-compromised cells. Proc Natl Acad Sci USA. 2016;113(36):E5271–E5280. doi:10.1073/pnas.1610421113
37. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(2):125–132. doi:10.1107/s0907444909047337
38. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(4):658–674. doi:10.1107/s0021889807021206
39. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi:10.1107/s0907444910045749
40. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi:10.1107/s0907444910007493
41. Liebschner D, Afonine PV, Baker ML, et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 2019;75(Pt 10):861–877. doi:10.1107/s2059798319011471
42. Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):431–440. doi:10.1107/s090744491200251x
43. Long F, Nicholls RA, Emsley P, et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr D Struct Biol. 2017;73(Pt 2):112–122. doi:10.1107/s2059798317000067
44. Ang HC, Joerger AC, Mayer S, Fersht AR. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J Biol Chem. 2006;281(31):21934–21941. doi:10.1074/jbc.M604209200
45. Tidow H, Veprintsev DB, Freund SMV, Fersht AR. Effects of oncogenic mutations and DNA response elements on the binding of p53 to p53-binding Protein 2 (53BP2). J Biol Chem. 2006;281(43):32526–32533. doi:10.1074/jbc.M604725200
46. Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements. J mol Biol. 2005;348(3):589–596. doi:10.1016/j.jmb.2005.03.014
47. Kaar JL, Basse N, Joerger AC, Stephens E, Rutherford TJ, Fersht AR. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010;19(12):2267–2278. doi:10.1002/pro.507
48. Arbely E, Natan E, Brandt T, et al. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc Natl Acad Sci USA. 2011;108(20):8251–8256. doi:10.1073/pnas.1105028108
49. Lambert JMR, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15(5):376–388. doi:10.1016/j.ccr.2009.03.003
50. Degtjarik O, Golovenko D, Diskin-Posner Y, Abrahmsén L, Rozenberg H, Shakked Z. Structural basis of reactivation of oncogenic p53 mutants by a small molecule: methylene quinuclidinone (MQ). Nat Commun. 2021;12(1):7057. doi:10.1038/s41467-021-27142-6
51. Sun C, Li Y, Yates EA, Fernig DG. SimpleDSFviewer: a tool to analyze and view differential scanning fluorimetry data for characterizing protein thermal stability and interactions. Protein Sci. 2020;29(1):19–27. doi:10.1002/pro3703
52. Gao K, Oerlemans R, Groves MR. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys Rev. 2020;12(1):85–104. doi:10.1007/s12551-020-00619-2
53. Kharenko OA, Patel RG, Brown SD, et al. Design and characterization of novel covalent Bromodomain and Extra-Terminal Domain (BET) inhibitors targeting a methionine. J Med Chem. 2018;61(18):8202–8211. doi:10.1021/acs.jmedchem.8b00666
54. Gehringer M, Laufer SA. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62(12):5673–5724. doi:10.1021/acs.jmedchem.8b01153
55. Birch AJ, Hinde AL, Radom L. A theoretical approach to the birch reduction. structures and stabilities of cyclohexadienyl anions. J Am Chem Soc. 1980;102(21):6430–6437. doi:10.1021/ja00541a009
56. Mayr H. Modern nucleophilic aromatic substitution. By Francois Terrier. Angew Chem Int Ed. 2014;53(28):7119. doi:10.1002/anie.201404183
57. Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022;21(12):881–898. doi:10.1038/s41573-022-00542-z
58. McAulay K, Bilsland A, Bon M. Reactivity of covalent fragments and their role in fragment based drug discovery. Pharmaceuticals. 2022;15(11):1366. doi:10.3390/ph15111366
59. Serafim RAM, Haarer L, Pedreira JGB, Gehringer M. Covalent chemical probes for protein kinases. Curr Res Chem Biol. 2023;3:100040. doi:10.1016/j.crchbi.2022.100040
60. Martin JS, MacKenzie CJ, Fletcher D, Gilbert IH. Characterising covalent warhead reactivity. Biorg Med Chem. 2019;27(10):2066–2074. doi:10.1016/j.bmc.2019.04.002
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.