")
Back to Journals » International Journal of Nanomedicine » Volume 20
Strategic Optimization of Nanoparticle Characteristics to Enhance Tumor Targeting and Doxorubicin Delivery
Authors Lee YJ, Hong J, Seo BY , Lee BH, Sarangthem V, Park RW
Received 19 December 2024
Accepted for publication 12 May 2025
Published 21 May 2025 Volume 2025:20 Pages 6357—6378
DOI https://doi.org/10.2147/IJN.S513336
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Misra
Young-Jin Lee, Jisan Hong, Bo-Yeon Seo, Byung-Heon Lee, Vijaya Sarangthem,* Rang-Woon Park*
Department of Biochemistry and Cell Biology, Cell & Matrix Research Institute, Kyungpook National University, School of Medicine, Daegu, 41944, Republic of Korea
*These authors contributed equally to this work
Correspondence: Rang-Woon Park, Email [email protected] Vijaya Sarangthem, Email [email protected]
Background: Doxorubicin (Dox) is a potent anticancer agent; however, its therapeutic efficacy is constrained by a narrow therapeutic index, resulting in nonselective cardiotoxicity and nephrotoxicity. To improve its specificity and therapeutic efficacy, multivalent targeting strategies are being developed.
Methods: A chimeric polypeptide consisting of an elastin-like polypeptides (ELP) copolymer with a repeating IL-4 receptor-specific targeting peptide, AP-1, and a (GGCGSCGSC)2 sequence encoding 6 cysteine residues (C6) at the carboxyl-terminus for Dox conjugation was designed. Several AP1-ELPs of varying molecular sizes and structures, ranging from unimers to micelle-forming polymers, were characterized to evaluate their influence on Dox delivery and tumor inhibition.
Results: Conjugating Dox to the C6 via an acid-labile linker induced self-assembly into micelle-like structures at body temperature. The size of these multivalent constructs significantly influenced their tumor penetration and overall therapeutic outcomes. High molecular weight, micelle-forming AP1-ELP constructs demonstrated faster tumor entry and enhanced inhibition compared to lower molecular weight linear AP1-ELPs. Tumor uptake of Dox was five times greater than that of free drug and twice that of low molecular weight, linear AP1-ELPs. Furthermore, systemic administration of these high molecular weight constructs effectively inhibited tumor growth in breast carcinoma xenograft models without inducing specific organ toxicity.
Conclusion: Outperforming free Dox, high molecular weight micelle-forming AP1-ELP constructs achieve superior tumor targeting and efficacy with minimal toxicity, highlighting their potential as safer and more promising carriers for targeted drug delivery.
Keywords: doxorubicin, tumor-targeting, elastin-like polypeptide, multivalent targeting, IL-4 receptor, AP1-ELPs, size dependency, tumor penetration, therapeutic efficacy, AP1-ELP-Dox
Introduction
Cancer remains a persistent global health challenge, requiring ongoing exploration of effective treatment strategies.1 Currently, various approaches are employed in cancer therapy, including surgery and radiotherapy for early-stage, localized cancers, as well as chemotherapy and immunotherapy for tumors resistant to surgery or radiation or those with metastases. Chemotherapeutics such as doxorubicin (Dox), epirubicin, paclitaxel, and sorafenib are commonly used in post-surgery and various cancer treatments.2 Dox is a potent anthracycline antibiotic widely used in cancer chemotherapy to treat various cancers, including breast cancer,3 lung cancer,4 sarcomas,5 lymphomas, and acute lymphocytic leukemia.6 However, the narrow therapeutic index of Dox limits its effectiveness, leading to off-target side effects, including nonselective cardiotoxicity, nephrotoxicity, alopecia, hematological toxicities, and cutaneous hyperpigmentation.4,7 Additionally, the efficacy of Dox is often reduced by intrinsic biological barriers and physiological processes in the body.7 Challenges such as hepatic metabolism and rapid filtration further reduce the therapeutic potential of these agents, complicating treatment outcomes. This necessitates exploring innovative approaches to enhance their efficacy. Polymeric nanocarriers emerge as promising platforms for the targeted delivery of chemotherapeutic agents, such as Dox, to tumor tissues. Polymeric nanocarriers with multivalent tumor-targeting strategies offer enhanced specificity and efficacy by harnessing the synergistic effects of multiple ligands.8 This polyvalent approach leverages the natural ability of cellular receptors to bind multiple ligands, significantly enhancing both binding affinity and internalization of targeted agents.9 Multivalent targeting enhances binding efficiency and improves selectivity for tumor cells over normal tissues, potentially reducing systemic toxicity.10 Various nanocarriers, including liposomes,11 polymeric nanoparticles,12 dendrimers,13 gold nanoparticles,14 silica nanoparticles,15 and carbon nanotubes16 are explored for encapsulating and delivering Dox to tumor sites. These nanocarriers can be engineered with precise control over physicochemical properties to optimize drug loading and release kinetics. Although polymeric nanocarriers-mediated multivalent tumor targeting of Dox offers several advantages, but also presents potential drawbacks. Designing and synthesizing polymeric nanocarriers with multivalent targeting functionality presents technical complexities and challenges. Achieving optimal ligand density, spacing, and orientation on the nanocarrier surface requires meticulous optimization, potentially increasing production costs and time.17 Moreover, some polymers or surface modifications utilized in nanocarrier synthesis may trigger undesirable immune reactions or exhibit poor biocompatibility, potentially resulting in adverse effects.18
In drug delivery, the size and molecular weight of biomaterials significantly influence the release kinetics of encapsulated drugs, thereby modulating their therapeutic efficacy.19 Moreover, these factors also shape interactions with biological barriers, such as the extracellular matrix and cellular membranes, further affecting drug delivery and tumor targeting.20 Nonetheless, they also play key roles in determining biodistribution, circulation time, tumor penetration, and cellular uptake, ultimately influencing therapeutic outcomes.21 Polymeric nanocarriers with larger molecular weights and sizes typically show prolonged circulation times and increased tumor accumulation through the enhanced permeability and retention (EPR) effect.22 However, excessively large biomaterials may have poor tumor penetration, potentially reducing their therapeutic efficacy.23 Conversely, smaller biomaterials may penetrate tumors more efficiently, but they are also rapidly cleared from circulation, which reduces their accumulation in the tumor.24 Thus, the size dependency of multivalent targeting strategies and their effect on tumor penetration and therapeutic efficacy remain active research areas.25 Large-scale production of polymeric nanocarriers with consistent quality and reproducibility presents significant manufacturing challenges. Complex synthesis processes, raw material variability, and regulatory requirements may limit the scalability and commercial viability of nanocarrier-based formulations.19,26–30
Elastin-like polypeptides (ELPs) emerge as a promising biomaterial for drug delivery, offering distinct advantages over other synthetic polymers. Since ELPs are derived from human tropoelastin, they are well-tolerated, biocompatible, and biodegradable, reducing the risk of long-term toxicity.31 ELPs can be produced in large quantities using recombinant DNA technology, ensuring scalability and reproducibility for clinical applications.32 They undergo a sharp phase transition with temperature changes, enabling controlled drug release. The physicochemical properties of ELPs, such as their molecular weight and hydrophobicity, can be precisely controlled by modifying the amino acid sequence. This tunability enables customization of the polymer to optimize drug loading and release kinetics, enhancing therapeutic outcomes.33 ELPs are engineered to incorporate targeting ligands, imaging agents, or other therapeutic molecules, forming multifunctional drug delivery platforms. This enables targeted delivery of Dox to specific tumor cells, minimizing off-target effects and enhancing drug efficiency.34 ELPs provide a stable environment for drug encapsulation, protecting Dox from premature degradation and enhancing its solubility a common challenge with hydrophobic drugs such as Dox.
Our previous study shows that fusion ELP-based carriers efficiently incorporate siRNA35 and therapeutic peptides,36 showing strong tumor-targeting potential in vivo. Additionally, we also demonstrate that the molecular weight and structural conformation of the ELP are crucial in determining tumor penetration, biodistribution, and retention time in tumor tissue. Furthermore, we assessed the biodistribution patterns of micelle-forming and non-micelle-forming targeting ELP polymers.37 Our in vivo evaluation in two different mouse models further confirms that high molecular weight micelle-forming targeting AP1-ELPs effectively localize to tumors and retain within them, demonstrating significant potential as carriers. Nevertheless, these carriers require further optimization to enhance the delivery of various therapeutic agents for clinical applications. Investigating the mechanisms of drug accumulation and release, as well as comparing the therapeutic effects of micelle-forming and non-micelle-forming AP1-ELPs with small molecule drugs, may offer insights into addressing current challenges in drug delivery.
Therefore, this study aims to examine the effectiveness of targeted delivery using AP1-ELPs and elucidate the antitumor effects of Dox conjugated to AP1-ELPs with varying molecular weights and structures. Furthermore, the inherent properties of AP1-ELP were assessed upon Dox conjugation, while the accumulation and anticancer effects of Dox across different sizes or molecular weights were evaluated. Additionally, leveraging the pH-responsive release properties of Dox, a novel drug delivery system that enhances the therapeutic efficacy of Dox was designed while reducing its adverse effects. Given the overexpression of IL-4R in various solid tumors, this study explores the potential of AP1-ELPs as a promising platform for targeted cancer therapy.
Materials and Methods
Materials
Synthetic oligonucleotide encoding (GGCGSCGSC)2 was obtained from Macrogen Inc., Seoul, South Korea. The essential restriction enzymes were sourced from New England Biolabs. For protein expression studies, competent BL21 (DE3) Escherichia coli cells and DH5α strains were obtained from Invitrogen (Carlsbad, CA, USA). Terrific Broth media, acquired from IBI Scientific, CA, USA, served as the growth medium, supplemented with 100 μg mL ampicillin (AMRESCO, LLC, OH, USA) and 1 mm IPTG (Carbosynth Limited, Berkshire, UK). Additionally, Dox-EMCH (the (6-maleimidocaproyl) hydrazone derivative of Dox was procured from ApexBIO (Houston, TX 77054, USA). In terms of cell lines, we obtained 4T1 murine breast cancer cells, MDA MB231 human breast cancer cells, H460 human lung cancer cells, and H226 murine lung cancer cells were obtained from the American Type Culture Collection (ATCC). 4T1, H460, and H226 cells were cultured in RPMI-1640 medium (Hyclone), while MDA-MB231-cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin, both from Hyclone. All cell cultures were maintained at 37°C in 5% CO2.
Dox Conjugation
The hydrazone derivative of Dox (DOXO-EMCH), covalently bound to the 6 cysteine residues (C6) on ELP, was synthesized. Briefly, 200 μM of E60 or AP1-ELPs were diluted in phosphate-buffered saline (PBS) and incubated with tris(2-carboxyethyl) phosphine (TCEP) at a 30-fold molar excess at 4°C for 30 min. Subsequently, the protein was precipitated using 1 M sodium chloride (NaCl) and centrifuged at 12000 rpm for 15 min. The resulting pellet was dissolved in cold PBS and conjugated to DOXO-EMCH via incubating with a 30-fold molar excess of DOXO-EMCH at room temperature for 16 h in the dark, with a final volume of 1 mL PBS. Most unreacted labels were removed through inverse thermal cycling. The resulting drug-protein conjugate was resuspended in PBS and filtered through a 3K molecular weight cutoff (MWCO) Amicon filter spin column (Thermo Scientific). The concentration of Dox was determined by measuring its absorbance at 495 nm, using an extinction coefficient of 9250 L/mol.cm.38 The absorbance of Dox at 280 nm was accounted for, and the protein concentration was calculated using its absorbance at 280 nm and a molar extinction coefficient of 7190 L/mol cm, according to the provided equation.

Thermal Characterization
The transition temperatures (Tt) of E60-Dox, A38-Dox, A60-Dox, and A86-Dox were determined by monitoring the turbidity profile of protein solutions as a function of temperature, using a UV-visible spectrophotometer (Agilent Technologies, CA, USA) at 350 nm. The protein solutions were prepared at a concentration of 25 μM, and measurements were taken over a temperature range of 20°C–55°C, with a 1°C per minute increment. The first derivative of the turbidity profile with respect to temperature was calculated, and Tt was defined as the temperature at which the turbidity reached 50% of its maximum value. The change in Tt after conjugation with Dox was compared to the Tt of the unmodified E60, A38, A60, and A86 proteins.
MALDI TOF/TOF MS Analysis
The molecular weights of E60-Dox, A38-Dox, A60-Dox, and A86-Dox were determined using an UltrafleXtreme mass spectrometer (Bruker). Protein samples were initially dissolved in acetonitrile and mixed with an equal volume of matrix solution in a 1:1 ratio. A 1-µL aliquot of the resulting mixture was then applied to a standard steel target and allowed to desiccate at room temperature. The prepared sample was then analyzed using mass spectrometry to determine the molecular weights of the protein-drug conjugates compared to those of the unmodified proteins.
Dynamic Light Scattering (DLS)
The E60-Dox, A38-Dox, A60-Dox, and A86-Dox solutions were diluted in PBS to a concentration of 25 μM. Particle size was measured at 24°C using a Malvern Zetasizer Nano-ZS ZEN 3600 instrument (Malvern P Analytical Ltd., United Kingdom). This advanced equipment enabled precise measurement of the particle size distribution using a dynamic light scattering (DLS) detector positioned at a 90-degree angle.
In vitro Drug Release Kinetic
To evaluate the in vitro release kinetics of Dox from E60-Dox, A38-Dox, A60-Dox, and A86-Dox nanoparticles, each suspended in PBS at a concentration of 0.1 mm, were loaded into Spectra/Por membrane tubes with a MWCO of 3500 Da. The tubes were then immersed in 50 mL of PBS solution adjusted to pH 5 and 7. The release system was maintained at a constant temperature of 37°C with gentle agitation at 100 rpm, protected from light. At predetermined time points, 50 µL was withdrawn from the 50 mL release medium and replaced with an equal volume of fresh medium. The concentration of Dox released into the medium was measured at 495 nm using a Cary UV-visible spectrophotometer. The cumulative release profile of Dox from the ELP nanoparticles was calculated relative to the initial concentration and plotted over time.
In vitro Cellular Uptake
Dox uptake was measured in various cancer cell lines, such as MDA-MB-231, 4T1, H226, and H460. Initially, 2×105 cells per well were seeded onto 4-well chamber slides and cultured until 80% confluency was reached. The cells were then exposed to ELP/AP1-ELP-Dox at a Dox equivalent concentration of 400 ng/mL for 1 h at 37°C. After incubation, the cells were carefully washed with cold PBS and fixed with paraformaldehyde (4%, v/v) at room temperature for 15 min. After three PBS rinses, the cell nuclei were stained with Hoechst dye and then examined for Dox fluorescence using a laser scanning confocal microscope (TS2 Leica).
In vitro Cell Cytotoxicity
Cancer cells (2 x 103 cells per well) were seeded in a 96-well plate and cultured for 24 h, then treated with free Dox, E60-Dox, A38-Dox, A60-Dox, and A86-Dox in a dose-dependent manner (0–400 ng/mL equivalents) for an additional 24 h. Subsequently, CCK solution and media were added to the cells, and the absorbance was measured at 450 nm using an automated microplate reader. Cell viability was expressed as a percentage normalized to untreated controls.
Apoptosis Assay
Cancer cells were seeded in 6-well plates at a density of 5×105 cells per well and incubated overnight at 37°C in a humidified 5% CO2 atmosphere to allow adherence and growth. The cells were then treated with free Dox, E60-Dox, A38-Dox, A60-Dox, and A86-Dox at a Dox-equivalent concentration of 400 ng/mL for 6 h. After treatment, the culture medium was replenished with fresh growth medium, and cells were incubated for an additional 24 h. After treatment, both adherent and nonadherent cells are harvested through trypsinization (0.25% trypsin-EDTA, Gibco, NY, USA). The harvested cells were washed with PBS and stained with Annexin V-FITC/PI (BD Life Sciences, USA). Apoptotic cells were analyzed using flow cytometry to determine the percentage of apoptotic cells in the population.
Hemolysis Tests
For hemolysis assay, blood samples were collected from 8-week female C57BL/6 mice in EDTA-coated container using standard protocol. Red blood cells were isolated from whole blood using centrifugation method for 3000 rpm, 10 min at 25 °C. RBCs were washed with PBS buffer saline (pH ~ 7.4) for three times at 3000 rpm for 5 min. For experiment purpose, 2% suspension of RBC were prepared with PBS as diluting reagent. All experiments performed with human RBC was in accordance with the rule of the institutional ethical committee. Different concentrations of AP1-ELP-Dox (eg, 200 and 400 µg/mL) were suspended with 0.8 mL of 2% RBC and incubate at 37 °C for 6 h with gentle shaking. After incubation, it was then centrifuge at 3000 rpm for 10 min and carefully transfer the supernatant to a new tube. Hemoglobin release was quantified by measuring the absorbance of the supernatant at 540 nm. The percentages of hemolysis were calculated using formula.

Tumor Inhibition Study
All studies were conducted in strict adherence to the National Institutes of Health (NIH) guidelines for the care and use of Laboratory Animals. The animal experiment protocol was approved by the Committee on the Ethics of Animal Experiments of the Kyungpook National University (Permit Number: KNU 2022-0328), ensuring compliance with ethical standards and animal welfare regulations. To evaluate the therapeutic efficacy of ELP/AP1-ELP-Dox, female BALB/c white mice were inoculated with 4T1 cells (1×106) until tumors were allowed to grow to a critical volume range of approximately 50–100 mm3, reached after 5 days of implantation. The mice were then randomly assigned to five groups, with 10 mice per group (n = 10). Dox (5 mg/kg), E60-Dox (20 mg/kg equivalent), A60-Dox (20 mg/kg equivalent), and A86-Dox (20 mg/kg) were administered intravenously via the tail vein on days 0, 3, and 6, respectively. PBS was used as the control in the experiments. Tumor dimensions were measured using a caliper every 3 days. Tumor growth inhibition was assessed by measuring tumor volume (V) (mm3) using the formula: Volume (mm3) = (W2 × L)/2, where L represents the longest diameter and W represents the shortest tumor diameter (mm). Body weight changes in all mice were also monitored throughout the treatment period. Mice were euthanized once tumor volume reached 2000 mm3 or upon the onset of ulceration.
In a separate experiment, the survival of tumor-bearing mice was assessed. Mice were monitored for up to 60 days posttransplantation of tumors or euthanized earlier under the following conditions: (1) if the mouse exhibited lethargy or illness affecting feeding, (2) a > 15% decrease in body weight from baseline, or (3) natural death. Survival rates were represented as Kaplan–Meier curves, and the median survival duration was calculated.
TUNEL Staining
For TUNEL staining, tumor tissues from each mouse group were harvested and fixed overnight in 4% paraformaldehyde at 4°C. Cryosections (8 μm thick) were prepared from frozen tumor tissues and subjected to TUNEL staining to evaluate apoptotic cell death in the tissue sections. Apoptotic cells were identified utilizing the deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay conjugated with FITC (TUNEL Apo-Green Detection Kit, Biotool.com, Seoul, Korea).
Systemic Toxicity Test
To evaluate off-target toxicity, blood samples were obtained, and hematological and biochemical parameters were analyzed after the therapeutic regimen was completed. Hematological indices, including white blood cell count (WBC), red blood cell count, platelet count (PLT), and hemoglobin concentration (HGB), were determined using whole blood specimens (Preclina Inc., Daegu, Korea). Serum samples were collected from each experimental group for biochemical profiling. This involved quantifying aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine (CRE), blood urea nitrogen (BUN), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK) levels to identify potential changes in biochemical parameters.
Statistical Analysis
Statistical differences between groups were evaluated using one-way analysis of variance (ANOVA) and Student’s t-test for pairwise comparisons. A significance level of p < 0.05 was considered statistically significant. Data are presented as the mean ± standard deviation.
Results
Synthesis and Characterization of ELPs of Varying Molecular Weights
Tailoring the size of ELPs has been shown to precisely modulate key parameters such as circulation half-life, cellular uptake efficiency, and payload capacity, thereby enhancing the overall efficacy and safety of the drug delivery system. The design of ELPs with varying molecular weights for Dox delivery requires a meticulous approach to optimize both the pharmacokinetic and pharmacodynamic properties of the drug carrier system. Studies show that high molecular weight, micelle-forming ELPs exhibit enhanced circulation in the bloodstream, improved accumulation at target sites, and prolonged retention times compared to lower molecular weight, non-micelle-forming AP1-ELPs.37 In this study, the coding sequences of ELPs with varying molecular weights, such as E60, A38, A60, and A86 (where the numerical values correspond to the molecular weights of the ELPs), were presented with the respective targeting peptides and ELP repeats (Figure S1A). Furthermore, all the ELPs were modified with a (GGCGSCGSC)2 sequence, encoding C6, at the carboxyl terminus to facilitate chemical conjugation with Dox (Supplementary information). The conjugation strategy involved the introduction of reactive groups, such as thiol functionalities, onto the ELP backbone to enable site-specific attachment of the drug molecule. The recombinant expression system used facilitated the successful purification of the proteins with sizes ranging from 38 kDa to 86 kDa (Supplementary information). The purified proteins were obtained to a high degree of purity, with each migrated at the expected molecular weight on SDS-PAGE (Figure S1B).
Synthesis and Characterization of ELP–Doxorubicin Conjugates and Characterization
The conjugation reaction between the cysteine-containing ELP and Dox was facilitated using an EMCH linker (Figure 1A). The maleimide group on EMCH reacted with the thiol group of the cysteine residue on the ELP, forming a stable thioether bond, while the hydrazide group was used to enable attachment to the keto group on Dox.38 Following conjugation with Dox via the EMCH linker, matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) analysis was conducted to assess the molecular weight of the conjugated product. Before conjugation, the MALDI-MS spectra for E60, A38, A60, and A86 exhibited molecular ion peaks at 54.3, 38.1, 54.3, and 89.4 (kDa), respectively, which closely corresponds to the expected molecular weight of the unmodified proteins (Figure 1B–E). After conjugation with Dox, the MALDI-MS spectrum revealed new molecular ion peaks at 55.6, 39.3, 56, and 90.5 kDa, indicating an increase in mass. This shift corresponds to the conjugation of Dox, with a molecular weight of approximately 750.75 Da per molecule. The observed mass increase of approximately 1100~1700 Da suggests the conjugation of 1~2 Dox molecules per protein molecule.
 |
Figure 1 Schematic Representation of Doxorubicin-N-ε-Maleimido-Caproic Acid Hydrazide Conjugation with AP1-ELP and Corresponding Molecular Weight Analysis (A) Dox, modified with N-ε-maleimido-caproic acid hydrazide (EMCH), was conjugated to AP1-ELP through a cysteine residue. The EMCH linker facilitates selective attachment of Dox to the cysteine thiol group on AP1-ELP, forming a stable thioether bond. (B–E) Mass spectrometry analysis further was used to confirm the molecular weight increase of AP1-ELP-Dox compared to unconjugated AP1-ELP, validating successful Dox conjugation. |
Following conjugation with Dox, the Tt and hydrodynamic radius (Rh) of ELP-Dox conjugates were determined and compared to unconjugated ELPs using turbidity assays and DLS. Conjugation with Dox did not affect the Tt of E60, while a slight increase in Tt was observed, ~ 2.81°C for A38 and ~ 4.05°C for A60. For A86, the Tt increased by ~ 7.30°C (Table S1 and Figure 2A–D). This observed increase in transition temperature may be due to the conjugation with Dox, which could reduce hydrophobic interactions. The presence of Dox, particularly its interaction with or disruption of the hydrophobic regions of the ELP, is expected to reduce the driving force for phase separation, thereby requiring a higher temperature to induce the transition.
 |
Figure 2 Thermal Characterization of ELP-Dox Conjugates. Turbidity profiles (OD350 nm) for (A) E60-Dox, (B) A38-Dox, (C) A60-Dox, and (D) A86-Dox were measured at a heating rate of 1°C/min with a 25-μM protein concentration. The transition temperature (Tt) was defined as the temperature at which the turbidity reached 50% of its maximum value. Thermal profiles of ELP – Dox conjugates were compared to those of the native ELP. The observed shift in Tt for the AP1-ELP-Dox conjugates highlights the effects of Dox conjugation on the thermoresponsive properties of AP1-ELP. |
The hydrodynamic particle size of ELP in aqueous solution was measured using DLS to evaluate the effects of conjugation with Dox on its self-assembly and aggregation behavior. The DLS analysis revealed no significant change in particle size for E60-Dox (74.92 nm) and A38-Dox (38.63 nm) compared to the native E60 (79.31 nm) and A38 (41.08 nm) at room temperatures, below the Tt (Figure 3A and B). In contrast, A60-Dox and A86-Dox exhibited a significant increase in particle size to 87.78 nm and 128.7 nm, respectively, compared to the native A60 (35.11 nm) and A86 (52.97 nm) (Figure 3C and D). This increase in particle size post-conjugation is due to the added mass of Dox and the modified self-assembly properties of the ELP upon drug conjugation.
 |
Figure 3 Size Determination via DLS. Hydrodynamic diameters of (A) E60–Dox, (B) A38-Dox, (C) A60-Dox, and (D) A86-Dox, along with native AP1-ELP, were measured using DLS at a 25-μM protein concentration at room temperature. Average hydrodynamic diameter of AP1-ELP-Dox demonstrated a slight increase compared to native AP1-ELP. Scale bar = 100 μm. |
Doxorubicin Release Kinetics
The release profile of Dox from ELP nanoparticles was assessed under two distinct pH conditions: pH 5 to simulate the acidic environment typical of lysosomes or tumor sites and pH 7.2 to represent the near-neutral physiological conditions of the bloodstream. A significantly faster release of Dox was observed at pH 5 than at pH 7.2 (Figure 4A–D). Within the first 24 h, approximately 94.7% of Dox was released from all ELP-Dox conjugates at pH 5. In contrast, at pH 7.2, the release rates were significantly slower, with E60, A38, A60, and A86 releasing 28.3%, 24.3%, 16.8%, and 15.7% of Dox, respectively, over the same 24-h period. The accelerated release observed at pH 5 is likely due to the protonation of the hydrazone linker (or other pH-sensitive linkages), which destabilizes the interaction between Dox and the nanoparticles, thereby promoting enhanced drug release. These results indicate that ELP-Dox conjugates are more likely to release their payload in acidic environments, such as endosomes or tumor microenvironments. In contrast, the release of Dox from the ELP nanoparticles at pH 7.2 was significantly slower, demonstrating their stability under neutral physiological conditions.
 |
Figure 4 Dox Release Profile from AP1-ELP-Dox. (A–D) Release kinetics of Dox from AP1-ELP-Dox conjugates were measured under physiological conditions (37°C) at pH 7.2 and pH 5. Dox release was monitored at regular intervals, demonstrating a sustained release profile over time. |
Cellular Binding and Cell Uptake of Micelles
Subsequently, the influence of different structures and sizes of ELP on the uptake and release of Dox in cancer cells was assessed. To evaluate the cellular association of ELP-Dox and AP1-ELP-Dox with tumor cell lines, H460, H226, 4T1, and MDA-MB231 cells were incubated with ELP/AP1-ELP-Dox at a concentration equivalent to 400 ng/mL of Dox, or with free Dox for 1 h. The cellular localization of Dox delivered using ELP/AP1-ELP or free Dox was examined using confocal fluorescence microscopy. Figure 5A displays that H460 cells, which exhibit low IL-4R expression, showed no significant Dox accumulation after 1 h of incubation. In contrast, in IL-4R-expressing 4T1 cells, Dox delivered by micelle-forming AP1-ELP (A86) was predominantly localized in the nucleus after 1 h, while no signal was detected for free Dox, E60, or A38. Only a weak signal was observed with A60 (Figure 5B). Similarly, in the H226 and MDA-MB231 cells, which exhibit high IL-4R expression, A60-Dox, and A86-Dox demonstrated predominant cytoplasmic distribution after 1 h of treatment (Figure 6A and B). In contrast, lower levels were detected in the E60 and A38-treated cells over the same exposure period. The distribution of free Dox shifted from the cytoplasm to exclusively nuclear localization by 4 h (Supplementary Information and Figures S2–S4). E60 showed no detectable Dox distribution in all cell lines, indicating that the presence of AP1 is crucial for effective Dox delivery into tumor cells.
 |
Figure 5 Intracellular Localization of AP1-ELP-Dox Conjugates in H460 and 4T1 cells. H460 (A) and 4T1 cells (B) were plated on a 0.2 μm coverslip, treated the following day with 20 μM AP1-ELP-Dox conjugates for 1 h at 37°C and subsequently washed and stained with Hoechst to label nuclei. After staining, they were mounted on slides and imaged under a confocal microscope. Scale bar = 100 μm. |
 |
Figure 6 Intracellular Localization of AP1-ELP-Dox Conjugates in H226 and MDA-MB231 cells. H226 (A) and MDA-MB231 (B) cells were seeded onto 0.2 μm coverslips and treated with AP1-ELP-Dox the next day at a 20-μM concentration for 1 h at 37°C. After treatment, cells were washed, stained with Hoechst to label nuclei, and mounted onto slides for imaging under a confocal microscope. Scale bar = 100 μm. |
Cytotoxicity of AP1-ELP-Dox Conjugates and Free Dox
To evaluate the effect of size on the cytotoxicity of ELP conjugates, in vitro toxicity assays were conducted using different ELP variants. ELPs of varying sizes (E60, A38, A60, and A86) were conjugated with Dox to assess their toxicity in cancer cells. The toxicity profiles of the ELP-Dox conjugates varied significantly based on the size and molecular weight of the ELP. However, in H460 cells, which exhibit low IL-4R expression, minimal toxicity was observed, potentially due to low internalization of Dox (Figure 7A). Larger, micelle-forming ELP constructs, such as A86, showed slightly enhanced toxicity in a dose-dependent manner compared to the smaller E60 and A38 constructs (Figure 7B–D). In 4T1, H226, and MDA-MB231 cells, treated with 400 ng of A86-Dox resulted in viability reductions to 40.47%, 39.83%, and 53.93%, respectively, compared to the untreated control (100%). This enhanced toxicity is potentially due to the increased ability of A86 to deliver payloads of Dox per molecule, leading to greater intracellular drug accumulation. In direct comparison, the micelle-forming A86-Dox conjugated exhibited the highest cytotoxicity across all tested cancer cell lines, followed by A60-Dox. The smaller, linear ELP-Dox conjugates, E60 and A38, exhibited comparatively lower cytotoxicity than their micelle-forming counterparts. This pattern suggests that micelle-forming ELP constructs deliver higher intracellular concentrations of Dox and prolong drug retention, thereby enhancing cytotoxic effects. Conversely, the smaller, linear ELP constructs release Dox more rapidly and less efficiently, leading to reduced cytotoxicity. These findings highlight the significance of optimizing the size and molecular weight of ELP constructs to enhance the therapeutic efficacy of ELP-based drug delivery systems.
 |
Figure 7 Cytotoxic Effects of Free Doxorubicin and AP1-ELP-Dox on Various Cancer Cell Lines. (A) H460, (B) 4T1 (C) H226 and (D) MDA-MB231 cells were exposed to different concentrations of E60-Dox, or AP1-ELP, or free Dox for 24 h at 37°C. Cell viability was assessed using the CCK-8 assay. Data represent the mean from at least three independents’ experiments, with error bars representing the SEM. ***p < 0.0001, **p < 0.001 *p < 0.05 between ELP-Dox or AP1-ELP-Dox compared to untreated (one-way ANOVA). |
Apoptosis Assay
The apoptotic efficacy of Dox-conjugated AP1-ELPs was evaluated with a focus on structural differences between linear and micelle-forming ELPs, alongside the influence of molecular weight on apoptotic potential. Apoptosis was assessed in multiple cancer cell lines (H460, 4T1, H226, and MDA-MB231) using Annexin V/PI staining followed by flow cytometry, examining ELP-Dox conjugates of varying molecular weights (E60, A38, A60, and A86). Cell death and apoptosis were quantified following a 4-h incubation with free Dox and ELP-Dox conjugates, followed by an additional 24-h culture period. The A86-Dox conjugate induced the highest levels of cell death in H226 (Figure 8B) and MDA-MB231 (Figure 8D) showing 82.33% and 81.01% cell death, respectively—both cell lines characterized by elevated IL-4R expression. In contrast, H460 (Figure 8A) and 4T1 (Figure 8C) cells demonstrated lower apoptosis levels, with 28.80% and 62.52% cell death, respectively. Among all tested conjugates, E60-Dox conjugates exhibited the lowest apoptotic activity across cell lines, potentially due to limited cellular internalization, highlighting the importance of molecular weight and targeted delivery mechanisms.
 |
Figure 8 Apoptosis Assay. Tumor cell lines, such as H460 (A), H226 (B), 4T1 (C), and MDA-MD231 (D) cells, were treated with 400 ng of Dox equivalent through AP1-ELP-Dox, E60-Dox, or free Dox. Apoptosis was analyzed 24 h posttreatment using an Annexin V/PI staining assay to assess cell death. Bar graphs represent the percentage of cell death cells as the average of three independent experiments. Statistical significance was evaluated using one-way ANOVA: ***p < 0.0001 and *p < 0.05 between AP1-ELP-Dox or E60-Dox compared to Free-Dox. (E) The hemocompatibility of the AP1-ELP-Dox conjugates were assessed through a hemolysis assay using red blood cells (RBCs) isolated from 8-week-old female C57BL/6 mice. RBCs were treated with AP1-ELP-Dox at two concentrations (Dox equivalent: 200 and 400 µg/mL) and incubated at 37 °C for 6 h. Hemolysis is observed depends on lysis of RBC. (F) Hemoglobin release was quantified by measuring the absorbance of the supernatant at 540 nm. The error bars represent the standard error of the mean (SEM) from three independent experiments (n=3). Abbreviations: PC, positive control; NC, negative control. |
In contrast, conjugation with AP1-ELPs significantly enhanced the apoptotic potential of Dox, with a clear dependence on the molecular weight of the AP1-ELPs. Larger AP1-ELPs, such as A86, induced a higher percentage of both early and late apoptotic cells than the smaller constructs, such as E60 and A38 (Figure S5). For example, in the 4T1, MDA-MB231, and H226 cell lines, the A86-Dox conjugate induced apoptosis in approximately 67.35%, 78.17%, and 58.74% of the cells, respectively, after 24 h. In contrast, the E60-Dox conjugate induced apoptosis in 25.36%, 50.17%, and 16.34% of the cells. These results suggest that higher molecular weight AP1-ELPs enhance drug delivery efficiency, leading to more pronounced apoptotic effects. For example, in the H460 cell line, the A86-Dox conjugate induced apoptosis in 23.17% of cells, while the E60-Dox induced apoptosis in only 19.30% after 24 h of treatment. This result demonstrates that Dox delivery occurs through an IL-4 receptor-mediated endocytosis mechanism.
Plasma Stability and Hemolysis Assay
Evaluation of nanoparticle stability in biologically relevant environments, such as plasma, is critical for determining carrier performance, biodistribution, and therapeutic outcomes. In this study, the colloidal stability of ELP-Dox and AP1-ELP-Dox constructs was assessed under physiologically conditions by measuring particle sizes using dynamic light scattering (Supplementary information). After 24 h of incubation at 37 °C, both ELP-Dox and AP1-ELP-Dox exhibited a slight increase in particle size, indicating good structural integrity with minor susceptibility to protein interactions or aggregation (Figure S6). In contrast, A86-Dox showed a relatively greater increase in size, suggesting that it may be more prone to micelle fusion, aggregation, or protein adsorption—common outcomes when nanocarriers interact with complex biological matrices like plasma. Interestingly, A38-Dox demonstrated a slight decrease in particle size, which may reflect improved resistance to plasma-induced destabilization. This enhanced stability could be attributed to its unique sequence composition or physicochemical properties that reduce unfavorable interactions with plasma proteins.
Simultaneously, the hemocompatibility of the AP1-ELP-Dox conjugates were assessed through a hemolysis assay using red blood cells (RBCs) isolated from 8-week-old female C57BL/6 mice. RBCs were treated with AP1-ELP-Dox at two concentrations (200 µg/mL and 400 µg/mL) and incubated at 37 °C for 6 h. Hemoglobin release was quantified by measuring the absorbance of the supernatant at 540 nm. The results showed that AP1-ELP-Dox induced minimal hemolysis at both tested concentrations (Figure 8E). At 200 µg/mL, the hemolysis percentage was approximately 2%, while at 400 µg/mL, it slightly increased to around 2.9% (Figure 8F). Both values were well below the generally accepted safety threshold of 5%, indicating that the formulation is hemocompatible under the experimental conditions. Overall, these findings suggest that AP1-ELP-Dox has good blood compatibility and does not cause significant damage to erythrocytes at the tested concentrations.
Tumor Growth Inhibition as a Function of ELP Size
To assess the sustained therapeutic potential of AP1-ELP-Dox constructs, their intratumoral retention was evaluated in 4T1 tumor-bearing mice. Once tumors reached a volume of approximately 100 mm³, the mice were intravenously injected with free Dox, E60-Dox, A60-Dox, or A86-Dox at a dose of 20 mg/kg (n = 4). In vivo fluorescence imaging using the IVIS system at 24- and 48-h post-administration revealed that A86-Dox exhibited markedly higher tumor accumulation, which persisted through 48 h (Figure S7). In contrast, constructs with lower molecular weights, E60-Dox and A60-Dox, demonstrated limited tumor localization. These results highlight the superior tumor-retention capability of the micelle-forming, high molecular weight AP1-ELP construct A86-Dox, underscoring its potential for enhanced therapeutic efficacy compared to non-micellar or lower molecular weight formulations.
Furthermore, the in vivo efficacy of AP1-ELPs-Dox conjugates with different molecular weights and structures (E60, A38, A60, and A86) was evaluated using a 4T1 allograft mouse model. Tumor-bearing mice were treated with these conjugates, and tumor volumes were monitored over time (Figure 9A). Tumor growth inhibition correlated directly with the size and molecular weight of the AP1-ELPs. Mice treated with larger, micelle-forming AP1-ELPs, such as A86-Dox, exhibited the most significant reduction in tumor volume, with 3.3-fold smaller tumors by day 21 than those in the control group (p =0.0001, Figure 9B). Mice treated with the intermediate A60-Dox conjugate showed a 2.1-fold decrease in tumor volume (p = 0.001), while those treated with the nontargeted E60-Dox exhibited a modest 1.2-fold reduction compared to those in the control (p ˃0.0038). Mice treated with free Dox demonstrated intermediate tumor inhibition; however, tumor regrowth occurred rapidly after treatment cessation (p =0.1446). In contrast, PBS-treated control mice exhibited the largest tumor growth.
 |
Figure 9 In vivo Assessment of Antitumor Effects of AP1-ELP-Dox Conjugates. (A) 4T1 cells were subcutaneously injected into the mammary fat pad of Balb/c wild-type mice. Once the tumors reached a volume of ∼150 mm3, treatment with PBS, free Dox, or AP1-ELP–Dox conjugates were initiated on day 0. Arrows indicate treatment days on days 0, 3, 6, and 9. (B) Tumor volume was monitored every other 3 days using caliper measurements. *** indicates statistical significance between the A86 group and PBS (p < 0.0001), **p < 0.001 between A60-Dox and PBS (C) At the end of the treatment, mice were sacrificed, and the tumors excised and weighed. (D) Average tumor weight after therapy. Statistical significance was evaluated using one-way ANOVA: ***p < 0.0001 between A86-Dox and PBS, **p < 0.001 between A60-Dox and PBS, *p < 0.05 between E60-Dox and PBS. (E) Average body weight of mice measured every 3 days throughout the treatment period (n = 5). (F) Kaplan–Meier survival curve illustrating the survival rates of tumor-bearing mice treated with AP1-ELP-Dox, E60-Dox, or free Dox. Data are presented as mean ± SEM, highlighting the enhanced therapeutic efficacy and survival advantage of AP1-ELP-mediated Dox delivery. **p < 0.001 indicates statistical significance between A86-Dox and PBS, *p < 0.05 between A60-Dox and PBS. |
Following the completion of the in vivo treatment study, excised tumors were immediately weighed to assess the effect of different molecular weights and sizes of AP1-ELPs on tumor burden. The results showed a significant correlation between tumor weight and the molecular size of the AP1-ELPs (Figure 9C). Tumors from mice treated with A86-Dox exhibited the most significant reduction in weight, with an average tumor mass of 0.29 g, representing a 2.8-fold decrease compared to those of PBS-treated controls (p = 0.0001, Figure 9D). Treatment with the intermediate-sized A60-Dox significantly decreased tumor weight to 0.43 g, a 1.9-fold decrease compared to controls (p=0.0013), though less pronounced than the reduction observed with A86-Dox. In contrast, tumors from mice treated with the E60-Dox construct exhibited only modest reductions in mass (0.58 g, p=0.0302), which were not statistically different from those of the free Dox group (0.79 g, p=0.9938). Tumors in the PBS-treated control group were the largest (0.82 g), confirming that the therapeutic effect is strongly influenced by the molecular size and drug delivery efficiency of the AP1-ELPs. Body weight was monitored throughout the treatment period to assess systemic toxicity and overall health in the mice. Mice treated with AP1-ELP-Dox conjugates of different molecular weights (E60, A38, A60, A86), free Dox, or PBS (control) were weighed at regular intervals to evaluate the treatment-related weight loss, a typical side effect of chemotherapy (Figure 9E). The results indicated that mice treated with AP1-ELP-Dox conjugates maintained relatively stable body weights compared to those receiving free Dox, regardless of conjugate size. Mice treated with free Dox exhibited only minor fluctuations in body weight, reaching a maximum weight loss of 16% by the end of the treatment period, which was not statistically significant (p > 0.05) compared to the PBS-treated control group. The control group maintained stable body weights throughout the experiment, serving as a baseline for comparison. These findings suggest that AP1-ELP-Dox conjugates, particularly micelle-forming constructs such as A86, reduce systemic toxicity compared to free Dox, as indicated by the maintenance of stable body weight throughout treatment. This reduced toxicity, along with enhanced therapeutic efficacy, supports the potential of larger AP1-ELP-based drug delivery systems as safer and more effective alternatives to conventional chemotherapy.
The effect of AP1-ELP-Dox conjugate size on overall survival was evaluated using Kaplan–Meier survival analysis in tumor-bearing mice treated with AP1-ELPs of difference molecular weights (E60, A38, A60, and A86), free Dox, or PBS (control). Mice were monitored over 45 days, with survival curves generated to assess the size-dependent effect on survival rates. The Kaplan–Meier survival curves demonstrated a clear size-dependent improvement in survival outcomes. Mice treated with the largest micelle-forming AP1-ELP construct, A86-Dox, showed a significantly prolonged median survival time of 45 days, with 50% of the mice surviving beyond 45 days (p < 0.001), compared to those in the control group (Figure 9F). Additionally, the A60-Dox conjugate produced a statistically significant increase in survival, with a median survival of 28 days (p < 0.05), while this effect was less pronounced than that observed with A86-Dox. In contrast, the E60-Dox conjugate demonstrated only a modest survival benefit, achieving a median survival of 24 days, similar to that observed with free Dox treatment. Mice in the PBS-treated control group exhibited the shortest median survival of 15 days, highlighting the poor prognosis of untreated tumors. The survival benefit associated with the larger AP1-ELP-Dox conjugates, particularly A86, suggests enhanced therapeutic efficacy, potentially due to increased drug accumulation, prolonged circulation, and improved tumor targeting. This size-dependent increase in survival further supports the hypothesis that higher molecular weight ELPs enhance therapeutic outcomes by improving drug delivery efficiency and reducing systemic clearance.
Histological examination of tumor tissue and major organs (heart, liver, kidneys, spleen, and lungs) using Hematoxylin and eosin (H&E) staining facilitated the evaluation of both therapeutic efficacy on the tumor and potential off-target toxicity in healthy organs during treatment (Supplementary Information and Figure 10). Tumor sections from mice treated with larger AP1-ELP-Dox conjugates, particularly A86, exhibited significant necrosis and apoptosis within the tumor mass, characterized by widespread cell death, nuclear fragmentation, and disrupted tissue architecture. In contrast, tumor tissues from mice treated with smaller E60-Dox conjugates or free Dox exhibited only focal areas of necrosis, with viable tumor cells remaining intact, indicating lower therapeutic efficacy. H&E staining of heart tissues revealed no evidence of cardiotoxicity in the A86-ELP-Dox and A60-ELP-Dox treatment groups, with normal myocardial structure observed. Conversely, free Dox-treated mice exhibited early signs of cardiomyopathy, including vacuolization and muscle fiber disarray, indicative of Dox-induced toxicity. The liver, kidneys, spleen, and lungs of mice treated with AP1-ELP-Dox conjugates, regardless of molecular size, exhibited normal histological architecture with no indications of toxicity, demonstrating the safety of the ELP-based delivery system. TUNEL assay results revealed a high degree of TUNEL-positive cells in tumor tissues, with a lower staining intensity observed in the heart, particularly in the A86-Dox treatment group (Figure S8). The A86 carrier enhances selective Dox accumulation within the tumor microenvironment, promoting increased apoptosis in cancer cells while potentially reducing cardiac toxicity. In contrast, mice treated with free Dox exhibited fewer TUNEL-positive cells in tumor tissue but higher TUNEL intensity in heart tissue, indicating reduced tumor-targeting efficiency and increased cardiac damage.
 |
Figure 10 Analysis of Cellular Morphology and Tissue Structure. Representative images of H&E-stained tumor and organ sections (liver, heart, spleen, lung and kidney) after the treatment. Scale bar = 50 μm. |
After completing the therapeutic regimen with AP1-ELP-Dox conjugates of different molecular weights, a comprehensive evaluation of hematological and biochemical parameters was conducted to assess the overall health of the mice and identify any potential treatment-related adverse effects. Hematological analysis of blood samples indicated that mice treated with the larger AP1-ELP-Dox conjugates (A86 and A60) maintained normal profiles, with WBC, HGB, and platelet counts within the physiological range compared to the PBS-control group (Figure 11A–C). Biochemical analysis of serum samples—conducted to evaluate liver, kidney, and heart function, alongside the overall metabolic health—demonstrated that mice treated with AP1-ELP-Dox conjugates exhibited normal biochemical profiles. Furthermore, ALT and AST levels were within normal limits, indicating no signs of hepatotoxicity (Figure 11D and E). Similarly, BUN and creatinine levels remained within the normal range, suggesting that the larger AP1-ELP constructs did not induce nephrotoxicity (Figure 11F and G). In contrast, free Dox-treated mice exhibited significantly elevated ALT and AST levels (p < 0.001), indicating liver damage. Additionally, higher molecular weight AP1-ELP-Dox conjugates demonstrated lower serum LDH and CPK levels than those of both free DOX and conjugates of lower molecular weights, indicating reduced cardiotoxicity (Figure 11H and I). In contrast, free Dox treatment led to elevated serum CPK levels, a specific marker for cardiac muscle injury, indicating DOX-induced cardiotoxicity.
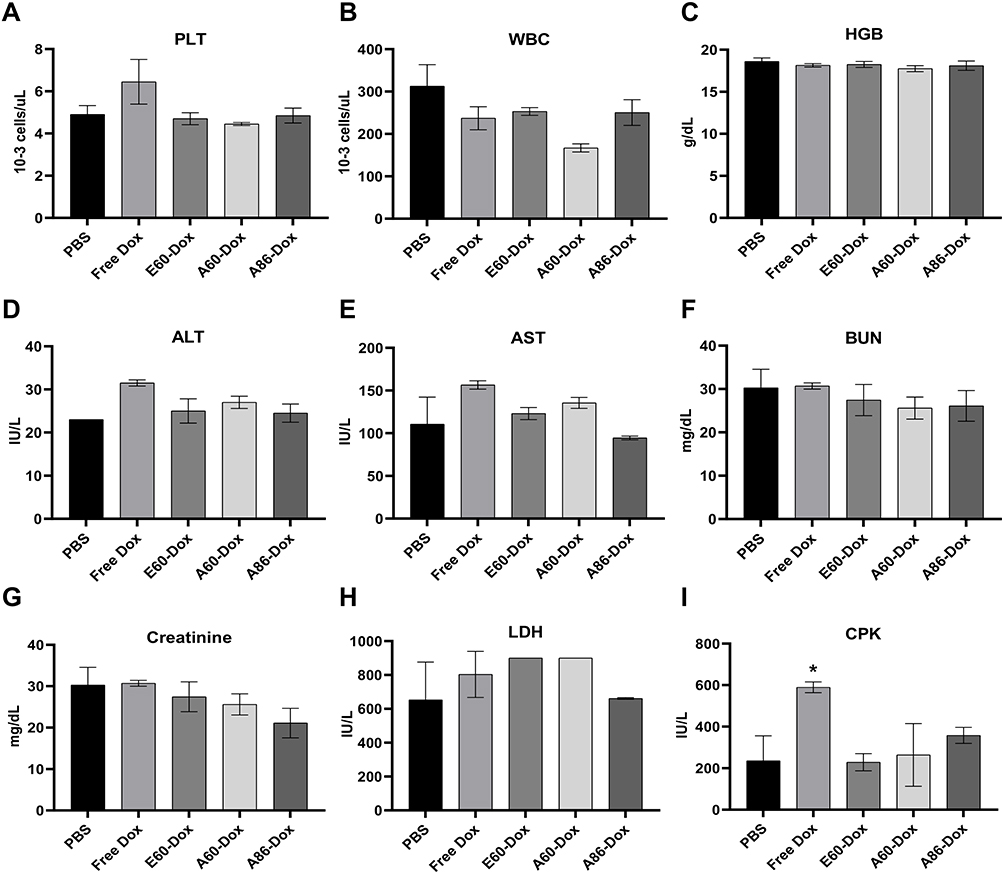 |
Figure 11 Blood and Serum Biochemistry for Systemic Toxicity. (A–I) Blood and serum samples were collected to evaluate liver, heart and kidney function markers, including ALT, AST, BUN, creatinine, LDH and CPK levels. Biomarker levels in AP1-ELP-Dox treated groups were consistent with baseline values, indicating low systemic toxicity. Data are presented as mean ± SEM from three independent experiments (n = 3). *p < 0.05 between free Dox and PBS. |
Discussion
The multivalent display of AP1 on ELP scaffolds improves receptor engagement through avidity effects, increasing the probability of binding to IL-4R-expressing tumor cells. This enhanced binding may improve the internalization and retention of therapeutic agents within tumors, resulting in higher localized drug concentrations and prolonged therapeutic efficacy. However, as particle size increases or as structural conformation changes due to thermal transition, steric hindrance may occur, limiting the ability of tumor-targeting AP1-ELP polymer receptors simultaneously on densely packed cell surfaces. Larger nanoparticles, while exhibiting poor tissue penetration, may accumulate in the liver and spleen, leading to off-target effects and potential toxicity. In contrast, smaller nanoparticles, despite their efficient tumor penetration, may be rapidly cleared from circulation, thereby reducing their overall therapeutic efficacy. Optimizing the size of AP1-ELPs to an intermediate range can maximize tumor retention while minimizing off-target effects and systemic toxicity.
Therefore, the size, molecular weight, and structure of the ELPs were optimized through genetic engineering to exhibit similar thermal transitions at physiological body temperature. The performance of these ELP-based Dox delivery systems was evaluated through both in vitro and in vivo studies, assessing parameters such as drug loading efficiency, release kinetics, cytotoxicity, and therapeutic efficacy. The Dox conjugation to the protein was confirmed through MALDI-MS based on the shift in the molecular peak compared to the native protein peak (Figure 1B–E). The differential release profiles observed under the two-pH conditions demonstrate the pH-sensitive nature of the ELP-Dox nanoparticles. At pH 5, the release rate is significantly higher due to the acidic environment that facilitates faster cleavage of the conjugation linkages. In contrast, at pH 7.2, the release is slower, resulting in a more sustained release under neutral physiological conditions (Figure 4A–D). Therefore, the design of AP1-ELPs is well-suited for targeted drug delivery, facilitating a faster release of Dox in acidic environments, such as tumor tissues or intracellular compartments, while maintaining stability in the bloodstream at neutral pH. Additionally, this controlled release profile further indicates that AP1-ELP-Dox nanoparticles have the potential to selectively deliver Dox to tumor sites, thereby minimizing off-target effects in healthy tissues.
The viability and apoptosis assay results highlight the significant influence of ELP size and molecular weight on the cytotoxicity of ELP-Dox conjugates. Larger, micelle-forming ELP constructs, such as A86, exhibit enhanced cellular uptake and increased toxicity, potentially due to their ability to deliver a higher drug load per nanoparticle (Figure 7A–D). In contrast, smaller ELPs, such as E60 and A38, demonstrate lower toxicity, correlating with their reduced drug delivery efficiency. This can be attributed to the temperature-sensitive transition to micellar structures, which enhances endocytosis and improves the exposure of targeting ligands, resulting in more efficient drug delivery into the cells.
Apoptosis assay results consistently demonstrated that both the size and molecular weight of ELP conjugates significantly affect their ability to induce apoptosis. Larger, micelle-forming ELPs, such as A86, were more efficient at delivering Dox and inducing apoptosis in cancer cells than the smaller, linear ELPs across all tested cancer cell lines (Figure 8A–D). This effect may be due to enhanced cellular uptake, prolonged drug retention, and increased activation of both intrinsic and extrinsic apoptotic pathways. The micellar structures may facilitate enhanced cellular uptake and prolonged retention of Dox, leading to increased DNA damage and activation of apoptosis pathways.
Subsequently, the influence of the size and structure of AP1-functionalized ELPs (AP1-ELPs) on the targeting and therapeutic performance of Dox conjugates was evaluated in vivo. The tumor-targeting efficacy of AP1-ELPs was assessed by evaluating their ability to inhibit tumor growth in the 4T1 allograft mouse model. The size-dependent efficacy of AP1-ELP-Dox conjugates is attributed to several factors, including their ability to exploit the EPR effect, enhanced tumor targeting via IL-4 receptor-mediated endocytosis, and improved pharmacokinetics associated with their larger size or nanoparticle retention due to temperature-triggered phase transition with the 100–200 nm range. This synergy improves the localization of Dox within the tumor, leading to significant tumor growth inhibition, as demonstrated in the 4T1 allograft model (Figure 9B–D). The optimal-sized AP1-ELP nanoparticles, such as A86, exhibit significantly improved tumor accumulation, increased cellular uptake, and enhanced therapeutic response compared to their smaller counterparts. A86 facilitates a more controlled release of Dox within the tumor microenvironment, contributing to the observed increase in apoptotic cells and overall tumor regression. This finding is consistent with those of previous studies indicating that macromolecular carriers of larger size exhibit better tumor targeting and less off-target distribution. In contrast, smaller ELPs, such as E60, may be more rapidly cleared from circulation, reducing their therapeutic efficacy.
Furthermore, Kaplan–Meier survival analysis revealed a clear size-dependent improvement in therapeutic outcomes for mice treated with AP1-ELP-Dox conjugates, with larger ELP constructs demonstrating significantly enhanced survival benefits. Specifically, the A86-Dox conjugate provided the greatest survival benefit, showing a significant increase in median survival and a higher percentage of long-term survivors compared to smaller constructs (E60) and free Dox treatment (Figure 9F). The superior efficacy of larger ELPs, particularly A86, can be attributed to several key factors. First, the increased molecular size and micelle-forming ability of these larger ELPs may contribute to improved pharmacokinetics, including prolonged circulation times and reduced renal clearance. In contrast, smaller constructs, such as A38 and A60, demonstrated only modest survival benefits, potentially due to their rapid clearance from circulation and reduced tumor-targeting efficiency. These findings are consistent with those of previous studies indicating that smaller drug delivery systems typically exhibit faster systemic clearance, leading to lower drug concentrations at the tumor site and reduced therapeutic outcomes. The limited efficacy of free Dox, as observed in the survival analysis, highlights the challenges of using free drugs, which are rapidly cleared and prone to off-target toxicity, thereby limiting their therapeutic window.
Evaluation of potential toxicity associated with AP1-ELP-Dox conjugates showed no significant differences in systemic toxicity among the various ELP-Dox and AP1-ELP-Dox formulations, as demonstrated via body weight and histopathological analyses of major organs (liver, kidneys, and lung) (Figure 10). However, free Dox-treated mice exhibited significant signs of cardiotoxicity, while AP1-ELP-Dox-treated mice exhibited no significant adverse effects, regardless of ELP size. Larger constructs, such as A86, improved tumor cell necrosis and significantly reduced systemic toxicity compared to the free Dox. Subsequent assessment of hematological and biochemical parameters after the therapeutic regimen indicated that larger molecular weight ELP constructs, such as A86, preserve normal blood profiles and biochemical markers, thereby reducing the risk of systemic toxicity typically associated with conventional Dox treatment (Figure 11). In contrast, the administration of free Dox was associated with significant biochemical disturbances, highlighting the benefits of utilizing ELP-based drug delivery systems to enhance therapeutic efficacy while minimizing adverse effects. These findings suggest that larger ELP-based drug carriers, with enhanced tumor targeting and reduced systemic exposure, can achieve a more favorable therapeutic index by selectively delivering the drug to the tumor while minimizing toxicity to healthy tissues.
Overall, these findings highlight the potential of higher molecular weight ELPs in cancer therapy, particularly when paired with targeted drug delivery strategies. Larger ELP constructs with micelle-forming capabilities may offer superior therapeutic efficacy by enhancing stability and facilitating controlled drug release within the tumor microenvironment (TME). Additionally, these findings highlight the potential of A86 as a safer alternative for cancer treatment, necessitating further investigation into its clinical applications. Future studies could build upon these findings by investigating the effects of even larger ELP constructs or by integrating AP1-ELP-Dox conjugates with additional targeting ligands or combination therapies to improve survival outcomes. Additionally, optimizing ELP size drug loading efficiency and exploring combination treatments could further enhance antitumor effects. Expanding this approach to include metastatic or aggressive cancer models would provide insight into the broader therapeutic potential of AP1-ELP-Dox conjugates.
Conclusions
The efficacy of therapeutic agents targeting solid tumors depends largely on the interplay between nanoparticle size, structure, and their ability to penetrate the TME. This study shows that delivering Dox using AP1-ELPs of varying molecular weight, from linear to micelle-forming structures, highlights the significant role of nanoparticle architecture in drug delivery efficiency, tumor penetration, and therapeutic outcomes. Compared with nontargeted E60, all AP1-ELP constructs exhibited phase transition changes within a clinically applicable temperature range, with enhanced internalization observed in a time-dependent manner. The micelle-forming AP1-ELP, A86, self-assembles into more stable structures at physiological temperature, with faster tumor accumulation and uptake compared to its linear counterpart. Overall, these findings indicate that micelle-forming A86, particularly those with higher molecular weights, enhances therapeutic efficacy and survival in allograft mice while reducing systemic toxicity. These findings suggest the potential of micelle-forming AP1-ELPs as a more effective and safer nanocarrier for cancer therapy, advancing their development for clinical applications focused on optimizing tumor-targeted drug delivery and minimizing adverse side effects.
Abbreviations
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CPK, creatine phosphokinase; CRE, creatinine; C6, 6 cysteine residues; DLS, dynamic light scattering; Dox, doxorubicin; ELPs, Elastin-like polypeptides; EPR, enhanced permeability and retention; H&E, Hematoxylin and Eosin; HGB, hemoglobin concentration; LDH, lactate dehydrogenase; MWCO, molecular weight cutoff; PBS, phosphate-buffered saline; PLT, platelet count; Tt, transition temperatures; WBC, white blood cell count.
Data Sharing Statement
The dataset generated during this study is available from the corresponding author upon request.
Ethics Approval and Consent to Participate
Animal experiments were reviewed and approved by the Committee on the Ethics of Animal Experiments of the Kyungpook National University (Permit Number: KNU 2022-0328).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2021-NR060094).
Disclosure
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Xu M, Han X, Xiong H, et al. Cancer nanomedicine: emerging strategies and therapeutic potentials. Molecules. 2023;28(13):5145. doi:10.3390/molecules28135145
2. Anand U, Dey A, Chandel AKS, et al. Cancer chemotherapy and beyond: current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023;10(4):1367–1401. doi:10.1016/j.gendis.2022.02.007
3. Gao J, Deng F, Jia W. Inhibition of indoleamine 2,3-dioxygenase enhances the therapeutic efficacy of immunogenic chemotherapeutics in breast cancer. J Breast Cancer. 2019;22(2):196–209. doi:10.4048/jbc.2019.22.e23
4. Thorn CF, Oshiro C, Marsh S, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21(7):440–446. doi:10.1097/FPC.0b013e32833ffb56
5. Cortes-Funes H, Coronado C. Role of anthracyclines in the era of targeted therapy. Cardiovasc Toxicol. 2007;7(2):56–60. doi:10.1007/s12012-007-0015-3
6. Escherich G, Zimmermann M, Janka-Schaub G; CoALL Study Group. Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr Blood Cancer. 2013;60(2):254–257. doi:10.1002/pbc.24273
7. Basak D, Arrighi S, Darwiche Y, Deb S. Comparison of anticancer drug toxicities: paradigm shift in adverse effect profile. Life. 2021;12(1):48. doi:10.3390/life12010048
8. Porębska N, Ciura K, Chorążewska A, Zakrzewska M, Otlewski J, Opaliński Ł. Multivalent protein-drug conjugates–an emerging strategy for the upgraded precision and efficiency of drug delivery to cancer cells. Biotechnol Adv. 2023;67:108213. doi:10.1016/j.biotechadv.2023.108213
9. Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chemie Int Ed Engl. 2006;45(15):2348–2368. doi:10.1002/anie.200502794
10. Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed Engl. 1998;37(20):2754–2794. doi:10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3
11. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi:10.1016/j.addr.2012.09.037
12. Langer R. Drug delivery and targeting. Nature. 1998;392(6679 Suppl):5–10.
13. Svenson S, Tomalia DA. Dendrimers in biomedical applications—reflections on the field. Adv Drug Deliv Rev. 2012;64:102–115. doi:10.1016/j.addr.2005.09.018
14. Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA. The golden age: gold nanoparticles for biomedicine. Chem Soc Rev. 2012;41(7):2740–2779. doi:10.1039/c1cs15237h
15. Slowing II, Vivero-Escoto JL, Wu C-W, Lin VS-Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv Drug Deliv Rev. 2008;60(11):1278–1288. doi:10.1016/j.addr.2008.03.012
16. Liu Z, Chen K, Davis C, et al. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008;68(16):6652–6660. doi:10.1158/0008-5472
17. Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine. 2011;6(4):715–728. doi:10.2217/nnm.11.19
18. Nel AE, Mädler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano–bio interface. Nat Mater. 2009;8(7):543–557. doi:10.1038/nmat2442
19. Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. doi:10.1021/mp800051m
20. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. doi:10.1016/j.jconrel.2012.01.043
21. Park K, Kim JH, Nam YS, et al. Effect of polymer molecular weight on the tumor targeting characteristics of self-assembled glycol chitosan nanoparticles. J Control Release. 2007;122(3):305–314. doi:10.1016/j.jconrel.2007.04.009
22. Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem. 2010;21(5):797–802. doi:10.1021/bc100070g
23. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nature Rev Mater. 2016;1(5):1–12. doi:10.1038/natrevmats.2016.14
24. Gratton SE, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci. 2008;105(33):11613–11618. doi:10.1073/pnas.0801763105
25. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. doi:10.1016/s0168-3659(99)00248-5
26. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi:10.1038/nrd1632
27. Malik N, Wiwattanapatapee R, Klopsch R, et al. Dendrimers:: relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J Control Release. 2000;65(1–2):133–148. doi:10.1016/s0168-3659(99)00246-1
28. Zhang X-D, Wu D, Shen X, et al. Size-dependent in vivo toxicity of PEG-coated gold nanoparticles. Int J Nanomed. 2011:2071–2081. doi:10.2147/IJN.S21657
29. Lu S, Bennett WF, Ding Y, et al. Design and characterization of a multifunctional pH-triggered peptide C8 for selective anticancer activity. Adv Healthc Mater. 2015;4(17):2709–2718. doi:10.1002/adhm.201500636
30. Atabaev TS. Chapter 8 - Multimodal inorganic nanoparticles for biomedical applications. In: Grumezescu AM, editor. Nanobiomaterials in Medical Imaging. William Andrew Publishing; 2016:253–278.
31. Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat Biotechnol. 1999;17(11):1112–1115. doi:10.1038/15100
32. Meyer DE, Chilkoti A. Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules. 2004;5(3):846–851. doi:10.1021/bm034215n
33. MacEwan SR, Chilkoti A. Applications of elastin-like polypeptides in drug delivery. J Control Release. 2014;190:314–330. doi:10.1016/j.jconrel.2014.06.028
34. Liu W, Dreher MR, Furgeson DY, et al. Tumor accumulation, degradation and pharmacokinetics of elastin-like polypeptides in nude mice. J Control Release. 2006;116(2):170–178. doi:10.1016/j.jconrel.2006.06.026
35. Yi A, Sim D, Lee Y-J, Sarangthem V, Park R-W. Development of elastin-like polypeptide for targeted specific gene delivery in vivo. J Nanobiotechnology. 2020;18:1–14. doi:10.1186/s12951-020-0574-z
36. Lee B-HL, Park R-W, et al. Multivalent targeting based delivery of therapeutic peptide using AP1-ELP carrier for effective cancer therapy. Theranostics. 2016;6(12):2235–2249. doi:10.7150/thno.16425
37. Sarangthem V, Seo B-Y, Yi A, et al. Effects of molecular weight and structural conformation of multivalent-based elastin-like polypeptides on tumor accumulation and tissue biodistribution. Nanotheranostics. 2020;4(2):57. doi:10.7150/ntno.39804
38. Moktan S, Perkins E, Kratz F, Raucher D. Thermal targeting of an acid-sensitive doxorubicin conjugate of elastin-like polypeptide enhances the therapeutic efficacy compared with the parent compound in vivo. Mol Cancer Ther. 2012;11(7):1547–1556. doi:10.1158/1535-7163.MCT-11-0998
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.