")
Back to Journals » International Journal of Nanomedicine » Volume 19
Strategies for Non-Covalent Attachment of Antibodies to PEGylated Nanoparticles for Targeted Drug Delivery
Authors Ho KW , Liu YL, Liao TY, Liu ES, Cheng TL
Received 29 May 2024
Accepted for publication 5 September 2024
Published 1 October 2024 Volume 2024:19 Pages 10045—10064
DOI https://doi.org/10.2147/IJN.S479270
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dongwoo Khang
Kai-Wen Ho,1,2 Yen-Ling Liu,2,3 Tzu-Yi Liao,2,3 En-Shuo Liu,2,3 Tian-Lu Cheng1– 3
1Department of Biomedical Science and Environmental Biology, Kaohsiung Medical University, Kaohsiung, Taiwan; 2Drug Development and Value Creation Research Center, Kaohsiung Medical University, Kaohsiung, Taiwan; 3Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
Correspondence: Tian-Lu Cheng, Department of Biomedical Science and Environmental Biology, Kaohsiung Medical University, No. 100 Shih-Chuan 1st Road, Kaohsiung, 80708, Taiwan, Tel +886 7 3121101 2697, Fax +886 7 3227508, Email [email protected]
Abstract: Polyethylene glycol (PEG)-modified nanoparticles (NPs) often struggle with reduced effectiveness against metastasis and liquid tumors due to limited tumor cell uptake and therapeutic efficacy. To address this, actively targeted liposomes with enhanced tumor selectivity and internalization are being developed to improve uptake and treatment outcomes. Using bi-functional proteins to functionalize PEGylated NPs and enhance targeted drug delivery through non-covalent attachment methods has emerged as a promising approach. Among these, the one-step and two-step targeting strategies stand out for their simplicity, efficiency, and versatility. The one-step strategy integrates streptavidin-tagged antibodies or bispecific antibodies (bsAbs: PEG/DIG × marker) directly into PEGylated NPs. This method uses the natural interactions between antibodies and PEG for stable, specific binding, allowing the modification of biotin/Fc-binding molecules like protein A, G, or anti-Fc peptide. Simply mixing bsAbs with PEGylated NPs improves tumor targeting and internalization. The two-step strategy involves first accumulating bsAbs (PEG/biotin × tumor marker) on the tumor cell surface, triggering an initial attack via antibody-dependent and complement-dependent cytotoxicity. These bsAbs then capture PEGylated NPs, initiating a second wave of internalization and cytotoxicity. Both strategies aim to enhance the targeting capabilities of PEGylated NPs by enabling specific recognition and binding to disease-specific markers or receptors. This review provides potential pathways for accelerating clinical translation in the development of targeted nanomedicine.
Keywords: PEGylated nanoparticle, monoclonal antibody, antibody-containing nanodrugs, functionalization, targeted cancer therapy
Introduction
Polyethylene glycol (PEG)-based nanoparticles (NPs) are diverse and promising drug delivery vehicles. PEG is water-soluble, non-toxic, and used as an excipient in medication formulations to improve pharmacokinetics, biodistribution, solubility, and stability, reducing side effects.1–3 PEG polymers coupled to a lipid anchor inhibit the reticuloendothelial system (RES). The PEGylated NPs escape from the RES system and are stable within PEGylated liposomes, extending their half-life and enhancing therapeutic efficacy.4–8 The use of nanotechnology to develop these systems has been well-established over the past decade, both in pharmaceutical research and the clinical setting. For instance, Doxil and Caelyx, which are PEGylated liposomal doxorubicin (PLD)9–11 have been utilized in treating AIDS-related Kaposi’s sarcoma and solid cancers.10 The pharmacokinetic profile of Doxil at 50 mg/m² in humans shows a 300-fold increase in the area under the curve (AUC) compared to the free drug.10,12 Metastatic pancreatic cancer patients can receive Onivyde, a PEGylated liposomal irinotecan.13,14 After administering Onivyde 60–180 mg/m2 to cancer patients, the overall AUC increased by 46.2 times compared to nonliposomal irinotecan.15,16 Tumor accumulation of PEGylated NPs can boost treatment efficiency due to the enhanced permeability and retention (EPR) effect.17–19 Although non-targeted PEGylated NPs exhibit improved pharmacokinetics by evading the RES, EPR effects-lacking metastases and liquid tumors present difficulties. PEGylated NPs may internalize less in metastatic or hematopoietic cancers. PEGylated NPs had one-third of the intracellular absorption of non-PEGylated NPs.20 PEGylated NPs are less effective against metastasis or liquid tumors because the EPR effect is weaker, reducing tumor cell uptake and therapeutic efficacy. Thus, actively targeted liposomes with tumor selectivity and internalization augmentation are needed to promote tumor uptake and therapeutic efficacy.
The significance of active targeted PEGylated NP internalization lies in its pivotal role in optimizing drug delivery to solve hematologic malignancies, metastasis, and drug resistance.21–25 Hematologic malignancies like leukemia lack the EPR effect. Chen et al developed a bispecific antibody (bsAb: mPEG × CD20) that simultaneously binds PEGylated NPs and CD20.25 This yielded multivalent αCD20-armed liposomes, enhancing internalization and anticancer efficacy against CD20-expressing lymphoma cells. The one-step formulation of mPEG × CD20 modified PLD, resulting in αCD20/PLD specifically targeting Raji cells, demonstrating 56-fold increased internalization. In metastasis, the distribution of MM-302, an anti-human epidermal growth factor receptor 2 (HER2) Ab-targeted PEGylated liposomal doxorubicin, was also associated with better activity.26 Cu-MM-302 PET/CT data revealed its ability to access difficult metastatic locations, such as brain and lung metastasis.24 Cheng et al23 developed a bsAb (mPEG × HER2) to confer the PLD with tumor tropism to enhance PLD internalization. The αHER2/PLD specifically accumulated doxorubicin in tumor-bearing mice and produced significantly greater antitumor activity against doxorubicin-resistant MDA-MB-361 (P < 0.05) tumors compared to untargeted PLD. Various strategies can be employed to transform conventional NPs into targeted NPs, ensuring their precise delivery to specific cells or tissues and internalization enhancement. These methods include bsAbs, peptide conjugation, and ligand-receptor interactions. Further advancements in active targeting of NP techniques may revolutionize drug development, possibly ushering in the third generation of therapeutic interventions.
The chemical covalent conjugation of NPs is often used to incorporate Abs into cancer treatment. Chemical conjugation between Abs and NPs can occur via Ab amino acids like lysine and cysteine.27–29 The limitation is that reactive and abundant amine groups cause random conjugation and heterogeneity in Abs and NP mixtures. Many chemical conjugation techniques use site-specific conjugation to solve heterogeneity, such as thiol chemistry,30 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) chemistry,31 and Click chemistry.32 However, the modification of these strategies is complicated. To address these drawbacks, one-step formulation strategies have been proposed that involve the utilization of streptavidin-tagged Ab, Ab, or bsAb (PEG/DIG × marker), which possess the ability to attach to PEGylated NPs that will modify the biotin/Fc binding domain (such as protein A, G or anti-Fc peptide)/mPEG/digoxigenin (DIG) molecules separately, and these strategies can bind to tumor markers simultaneously. The simple one-step mixing of bsAb with these PEGylated NPs can confer tumor tropism and internalization enhancement. Moreover, in the two-step targeting strategy, pre-targeting is conducted using a bispecific adaptor that can accumulate on the tumor cell surface and capture the PEGylated NPs to induce a second attack by enhancing internalization and cytotoxicity against targeted tumor cells. The two-step targeting strategy maintains the original formulation and characteristics of PEGylated NPs with minimal modification. This review details these innovative approaches, the one-step formulation and the two-step targeting strategy (Figure 1), which improve PEGylated NP tumor targeting and cellular internalization. After tumor cells internalize NPs, they can release chemotherapeutic drugs, nucleic acids, or imaging agents, improving efficacy and reducing side effects. The one-step formulation and two-step targeting strategy of creating targeted NPs has great potential for use in precision medicine and personalized cancer therapy.
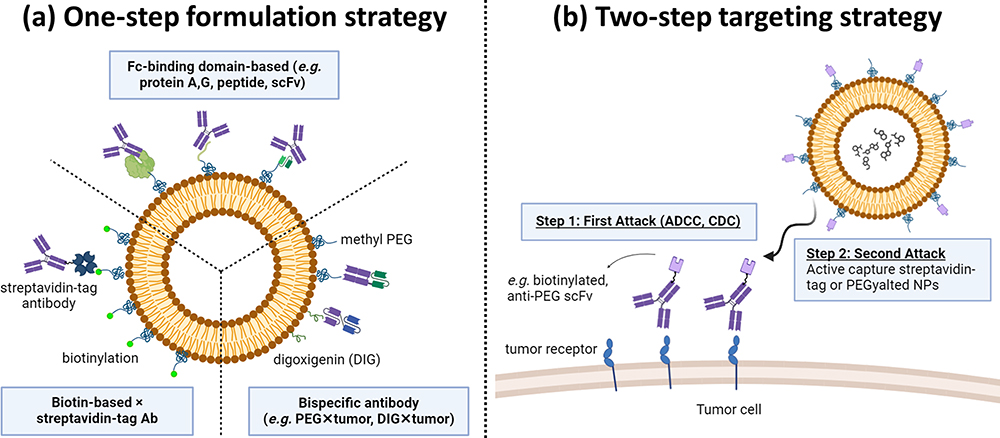 |
Figure 1 Schematic representation of one-step and two-step targeting nanoparticles (NPs) for enhanced drug delivery. (a) One-Step Formulation Strategy: The strategies use streptavidin-tagged antibody (Ab), Ab, or bispecific Ab (bsAb: PEG/DIG × marker) that can be mixed in one step with NPs which are modified with biotin/Fc binding domain (eg, protein A, G, or anti-Fc peptide)/polyethylene glycol (PEG)/digoxigenin (DIG) molecules separately. These strategies can confer the NPs with tumor tropism and enhance internalization and therapeutic efficacy. (b) Two-Step Targeting Strategy: The bsAb (PEG/biotin × tumor marker) accumulates on the tumor cell surface to activate the first attack of Ab-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) through Abs. Then, bsAbs can capture PEGylated/streptavidin-tagged NPs to trigger a second attack by increasing internalization and cytotoxicity. |
Content
One-Step Formulation Strategy for Active Targeting
Biotin-Based × Streptavidin-Tagged Formulation
Numerous Ab-NP systems have been developed using the biotin-streptavidin ligation technique.33,34 Biotinylated NPs were formed by activating the carboxylic group of biotin and covalently attaching it to the amine group of PEG-amine.35 Streptavidin interacts with biomaterials like Abs via thiol conjugation. The biotin-streptavidin ligation technique is employed to attach monoclonal Abs (mAbs) or other ligand types onto the surface of liposomes. This technique involves a one-step formulation process, creating targeted liposomes that can selectively bind to tumor receptors (such as HER2, EGFR, CD20, and others) and enhance drug internalization (Figure 2). For instance, by using streptavidin-tagged anti-TfR monoclonal Abs conjugating through the biotin group located at the distal end of the PEG spacer of PEGylated immuno-lipopolyplexes (PILP) for gene delivery. The targeted PILP transfection efficiency (67.5 ± 3.8%) increased by nearly 3-fold compared to non-targeted PILP (25 ± 4%).36 Attachment of anti-TfR monoclonal Abs to PEGylated NPs increases the evaluated in vitro and in vivo gene transfection efficiency, stability, and cytotoxicity without increasing toxicity.37,38 Also, Chen et al39 developed biotinylated anti-CD33 and biotinylated anti-CD7 Abs that were coupled to chemo-drug cytarabine encapsulated streptavidin-tagged liposomes (SALs) by using the biotin-streptavidin technique. The internalization efficacy was 93%, 74%, and 81% of targeted SALs compared to 63%, 27%, and 18% of non-targeted SALs in the three cell lines, respectively. The biotin-streptavidin technique has also been applied to specific protein assay systems for immobilizing Abs.40,41 Biotin-streptavidin ligation involves the specific binding of biotin to streptavidin, creating a stable and robust linkage widely used in various applications, such as biomolecule immobilization, targeted drug delivery, and diagnostic assays. In immobilizing Abs on NPs, the challenge is to prevent aggregation and ensure proper orientation, which is crucial for maintaining functionality and paratope accessibility.42 Therefore, careful optimization is essential in biotin-streptavidin ligation (Table 1).
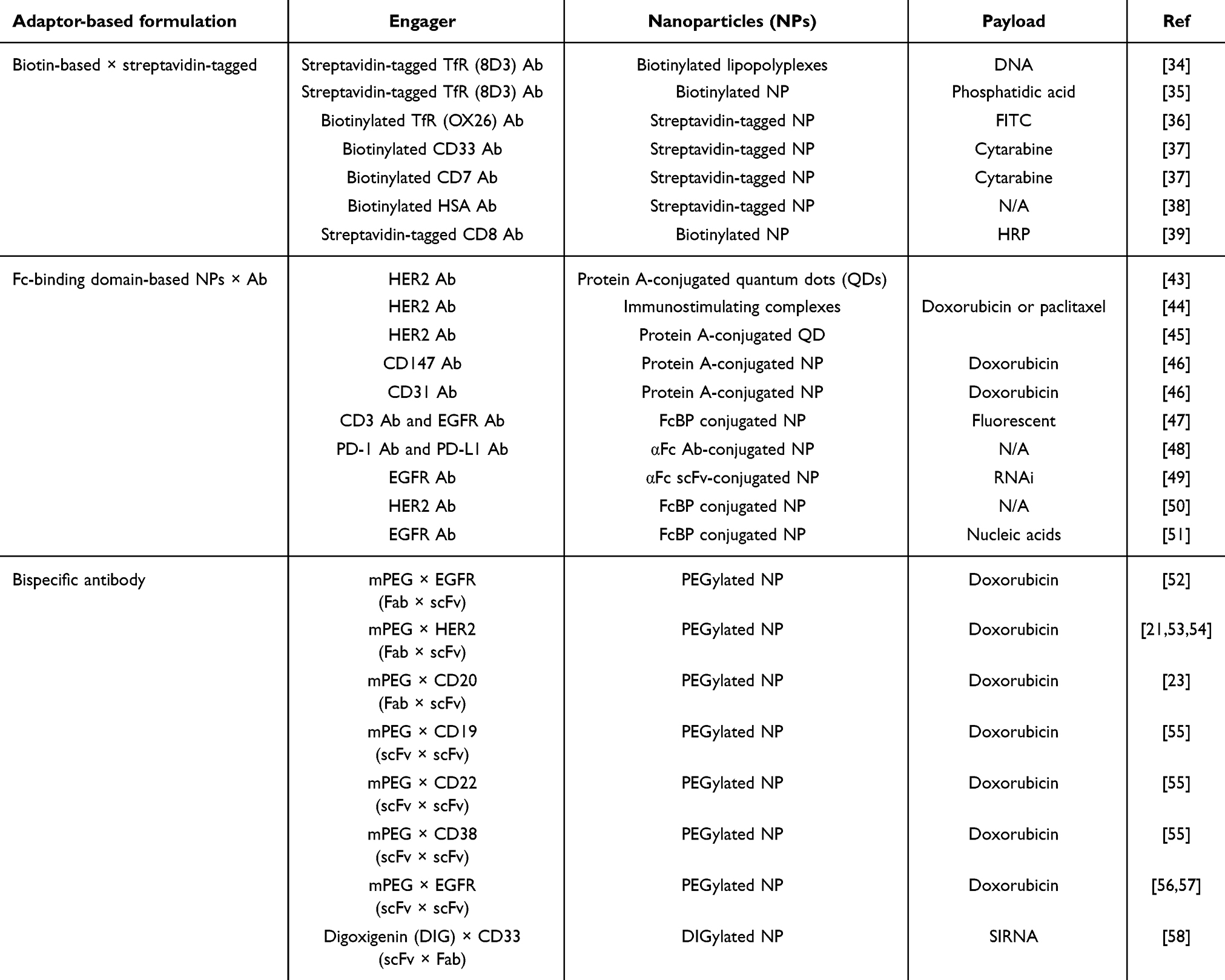 |
Table 1 One-Step Formulation Strategy for Active Targeting |
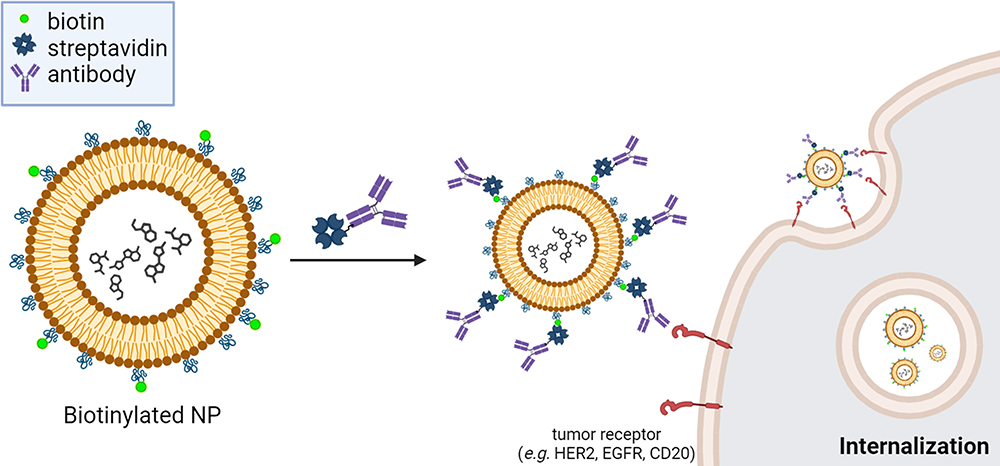 |
Figure 2 Schematic of one-step mixing of biotin-based NPs with streptavidin-tagged Abs for targeted drug delivery and internalization enhancement. Biotinylated NPs were formed by activating the carboxylic group of biotin and covalently attaching it to DSPE–PEG2000 using EDC/NHS chemistry. The biotinylated NPs one-step mix with streptavidin-tagged Abs to form targeted NPs. The antibody specifically binds to its target antigen such as transferrin receptor (TfR), CD33, CD7, etc. The targeted NPs allow for the specific delivery of therapeutic payloads to the desired tumor site, enhancing drug efficacy and minimizing off-target effects. |
The biotin-streptavidin interaction is well-known as one of the most robust non-covalent interactions (affinity: Kd ~10−15 M) found in nature. High affinity prevents the ligand from detaching from NPs, resulting in high stability and enabling the efficient operation of Abs.59 However, biotin-streptavidin ligation introduces foreign entities (biotinylated ligands) to which the immune system may respond. Streptavidin, a bacterial protein, may trigger an immune response leading to clearance by phagocytic cells in humans.60,61 This may result in the formation of anti-streptavidin Abs, impacting the pharmacokinetics and efficacy of the delivery system. The immune response generated in the context of biotin-streptavidin ligation might lack specificity and control, potentially leading to unintended immune reactions. Streptavidin’s tetrameric structure provides numerous binding sites,62 resulting in different biotin attachment orientations. This heterogeneity may impair biotin-streptavidin interactions and tests or therapeutic applications that rely on this binding affinity. To provide consistent and dependable results in biological and medicinal applications, conjugation methods must be carefully considered and optimized.63 This optimization process aims to achieve consistent and controlled binding interactions between biotinylated molecules and streptavidin, preventing the formation of varied or mixed conjugates. Proper conditions during ligation are essential for optimizing the effectiveness to maintain the integrity and specificity of Ab-NP conjugates.64
Fc-Binding Domain-Based NPs × Ab Formulation
In this section, three methods to improve the antigen-binding activity of liposome-tagging Abs for capturing Abs are discussed: (1) protein A and protein G, which are bacterial proteins known for their high affinity to the Fc region of Abs, (2) anti-Fc scFv, a single-chain variable fragment engineered to specifically bind to the Fc region of Abs, and (3) anti-Fc peptide, short peptide sequences designed to mimic the binding site of Abs’ Fc region. Each method offers unique advantages and applications to confer the PEGylated NPs with tumor tropism and internalization enhancement (Figure 3 and Table 1).
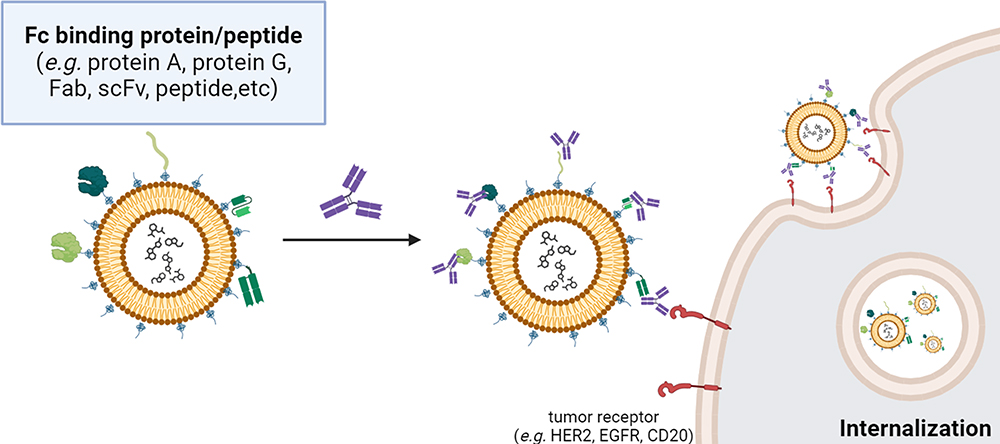 |
Figure 3 Schematic representation of one-step mixing of Fc-binding domain-based NPs with Abs for targeted drug delivery. The Fc-binding domain-based NPs are engineered with Fc-binding domains (eg, protein A, protein G, Fab, peptide) displayed on their surface. The Fc-binding domain, such as protein A/G, was covalently attached to NPs through N-hydroxysuccinimide ester groups at the PEG end (bis-NHS esters). On the other hand, the Ab fragments and Fc-binding peptides were synthesized by conjugating with maleimide-PEG-DSPE through amide bond formation. These domains exhibit a high affinity for the Fc region of Abs, facilitating specific binding (such as HER2, EGFR, CD147, CD31, etc.) and subsequent functionalization of the NPs with therapeutic payloads. Upon mixing, the Abs readily interact with the Fc-binding domains on the NPs, forming targeted NPs. This one-step mixing strategy offers a simple and efficient approach for the development of targeted NPs and holds significant promise for various biomedical applications, including targeted drug delivery and precision therapeutics. |
To improve the antigen-binding activity of liposome-tagging Abs, IgG Fc-binding proteins, like protein A or protein G, were conjugated to the surface of NPs using numerous chemical methods such as carbodiimide chemistry,65 maleimide chemistry43,65,66 or Click chemistry.43 This strategy can be used to couple Abs to NPs in an oriented manner, and the Fc binding affinity also allows the Ab orientation to face the antigen binding site outward.44 This non-covalent approach has emerged as a prevalent strategy for the facile attachment of diverse targeting or therapeutic Abs to NPs.45,46 As an example, protein A was conjugated to quantum dots (QDs) that were subsequently employed for the immobilization of anti-HER2 Abs.45 Delivery efficiency was assessed in breast cancer cells with both HER2 overexpression and HER2 non-overexpression evaluated by confocal microscopy. The findings showed that HER2-targeted QDs enhanced cellular uptake by endocytosis.47 This non-covalent coupling system could serve as an effective carrier for drug delivery targeting breast cancer cells with HER2 overexpression. Also, Hama et al demonstrated rapid Ab modification on high-affinity protein A-displaying liposomes (PAR28-PEG-lipo), allowing specific uptake by CD147- and CD31-positive cells. Liposomes modified with anti-CD147 Abs significantly increased cellular uptake of doxorubicin-capsuled PAR28-PEG-lipo and enhanced delivery compared to controls.67 Apart from proteins A and G, growing interest in site-specific Ab coupling has spurred research on affinity peptides derived from protein.68,69 However, the unexplored potential of protein A or protein G derivatives in the creation of conjugates between anticancer Abs and NPs remains an active area of investigation. In a different investigation, Gil-Garcia et al constructed a tripartite fusion protein, incorporating ZapB (for assembly), green fluorescent protein (GFP, for cell imaging), and the Z-domain (an engineered analog of the B-domain of Staphylococcus aureus protein A with high IgG affinity).70 The resulting ZapB-GFP-Z NPs were then selectively bound to the Fc regions of anti-EGFR and anti-CD3 Abs in a controllable ratio, forming a bsAb mimic capable of redirecting T lymphocytes to cancer cells.
Similar to biotin-streptavidin ligation, protein A or G ligation might provide limited control over immune activation, potentially causing non-specific immune reactions. Abs and smaller Ab fragments, such as Fab’ and scFv, are hypothesized to be less immunogenic.48,71 Jiang et al devised a versatile platform for Ab immobilization. They conjugated oxidized anti-IgG (Fc specific) Abs (αFc) onto amino-functionalized polystyrene NPs (αFc-NP). Subsequently, anti-programmed death-1 (anti-PD-1) and anti-PD-L1 monoclonal Abs (mAbs) were immobilized onto αFc-NPs through Fc-specific noncovalent interactions.49 These resulting immunomodulating nano-adaptors demonstrated the ability to co-engage T cells and tumor cells using two types of mAbs, showcasing their potential for efficient Ab-based cancer immunotherapy. Moreover, Kampel et al employed an Anchored Secondary scFv Enabling Targeting (ASSET) strategy50 that enables anchoring of anti-IgG Fc single chain Fv (scFv) to the LNP surface.51 The ASSET strategy allowed for the noncovalent and uniform attachment of anti-EGFR monoclonal Abs and facilitated targeted delivery using Cy5-labeled LNPs; they explored tumor localization and observed targeted LNPs in direct association with the cell surface of head and neck cancer.69 Specialized peptides have been utilized to immobilize Abs for the functionalization of NPs. In the synthesis of a peptide conjugate NP using maleimide chemistry, the NP surface is initially activated with maleimide groups, facilitating the subsequent conjugation reaction. For instance, Shim et al covalently attached an Fc-binding peptide (FcBP, amino acid: DCAWHLGELVWCT), selected from a cyclic peptide library and refined through monovalent phage display, to the lactic-co-glycolic acid-polyethylenimine (LGA-PEI) NP. The liposome can non-covalently target HER2-specific Ab, enabling targeted delivery of the Ab-conjugated LGA-PEI NPs to HER-expressing cells.26 HER2/FcBP-NPs showed 5.3-fold higher binding affinity to HER2 than isotype IgG Ab-modified NP and demonstrated significantly higher cellular uptake in HER2-positive cells compared to other formulations. Similar results were also reported by Lü et al, who covalently attached the anti-EGFR Ab cetuximab, enabling targeted delivery of therapeutic nucleic acids loaded in the Ab-conjugated LGA-PEI NPs to EGFR-expressing pancreatic cancer cells. Anti-EGFR Ab/FcBP-LGA-PEI/miR-198 mimic NPs showed enhanced internalization in Mia-MSLN cells compared to non-targeted NPs (P < 0.01).72 In the Fc-binding domain-based NP strategy, an Ab can be non-covalently attached to the surface of FcBP-expressing NPs with precise orientation.26
Protein A (or protein G) is a bacterial protein that may be recognized by the immune system, potentially triggering an immune response. This can lead to the formation of anti-protein A Abs. Similar to streptavidin, the immunogenicity of protein A may impact the stability and effectiveness of the delivery system. The presence of anti-protein A Abs could affect the system’s performance. Protein A and protein G display differing affinities for various Ab isotypes,73,74 resulting in variations in the binding specificity of Abs to NPs. The alignment of Abs attached to NPs impacts their function and effectiveness in therapy. In 1984, Uhlén et al discovered five locations where Protein A binds to IgG. Various binding locations and Ab conformations create difficulty in standardizing the orientation of NP Abs.75,76 Variability in protein A and protein G binding can introduce challenges in achieving stable and reproducible Ab-NP conjugates. Rigorous optimization and quality control measures are essential to address issues related to heterogeneity. The potential dissociation protein A (or protein G) and Abs can have a significant impact on the stability and targeting efficacy of NP formulation. The binding affinity (10−7 to 10−8 M)77 between protein A and Abs on NPs plays a crucial role in determining the specificity and stability of the conjugation. A higher binding affinity ensures strong and specific interactions, promoting a stable and durable association between protein A and Abs on the NP surface.78 The FcBPs have varying Kd values (10−9 M to 10−5 M),79–83 enabling selection for unique application needs. Drug administration uses affinity ligands because of their high selectivity, stability, low toxicity, affordability, and configurable affinity capacity. However, FcBPs may attract different Ab subclasses and species, resulting in inconsistent binding efficacy and stability of targeted NPs. FcBP sequences must be optimized and selected based on target Ab attributes for optimal binding affinity and performance.
Bispecific Ab Formulation
BsAbs are engineered proteins designed to simultaneously target two different antigens. According to this concept, one end of the bsAb recognizes a specific hapten such as PEG digoxigenin (DIG), while the other end targets a tumor marker such as HER2, EGFR, CD20, etc. This innovative bsAb is engineered to simultaneously target multiple antigens, offering unique therapeutic advantages in various applications, including cancer treatment, immunotherapy, and other medical interventions.52,53 It is essential to balance bsAb functionalization strategies with the stealth properties of PEGylated NPs when aiming to enhance in vivo tumor accumulation or increase NP uptake by tumor cells (Figure 4). The humanized bsAb platform can confer PEGylated NPs with tumor tropism in a simple one-step formulation. Several studies achieved the successful fabrication of methoxy PEGylated NPs through the straightforward mixing of PEGylated NPs with different formats of bsAbs such as Fab × scFv format23,25,54–57 or scFv × scFv format.58,84,85 These bsAbs comprised a humanized anti-mPEG (clone: h15-2b)86 Fab fragment along with an anti-tumor antigen (EGFR, HER2, CD19, CD20, CD22, or CD38) scFv. Take the bsAb strategy reported in Kao et al for example,54 cell imaging was conducted by combining bsAbs with PEG-imaging probes (Lipo/Rho, Qdot565nm, or FeOdots) and then assessing probe localization in cancer cells using confocal microscopy and magnetic resonance (MR) imaging. Confocal microscopy of αEGFR-Lipo/Rho added to SW480 (EGFR+) and SW620 (EGFR−) cancer cells revealed a red fluorescence signal exclusively on SW480 cells. In addition, the αHER2/PLD effectively increased doxorubicin accumulation and cytotoxicity in both HER2-amplified MCF-7/HER2 breast cancer cells and DOX-resistant MDA-MB-361 cells. PEGylated NPs lack efficacy for hematologic malignancies due to the absence of the EPR effect. To address this, a one-step formulation of mPEG × CD20 modified PLD was used, resulting in αCD20/PLD specifically targeting leukemic cells, which demonstrated a 56-fold increased internalization and 15.2-fold higher cytotoxicity compared to controls. mPEG × CD20 modification presents a convenient strategy to enhance the efficacy of PEG-NPs against hematologic malignancies.25 Similarly, Moles et al reported that bsAb-assisted enhancements in leukemia cell-targeting and cytotoxic potency correlated with receptor expression minimally affected normal peripheral blood mononuclear cells and hematopoietic progenitors in vitro and in vivo, and contributed to extended overall survival in patient-derived xenograft models of high-risk childhood leukemia, with reduced drug accumulation in the heart and kidneys (Table 1).
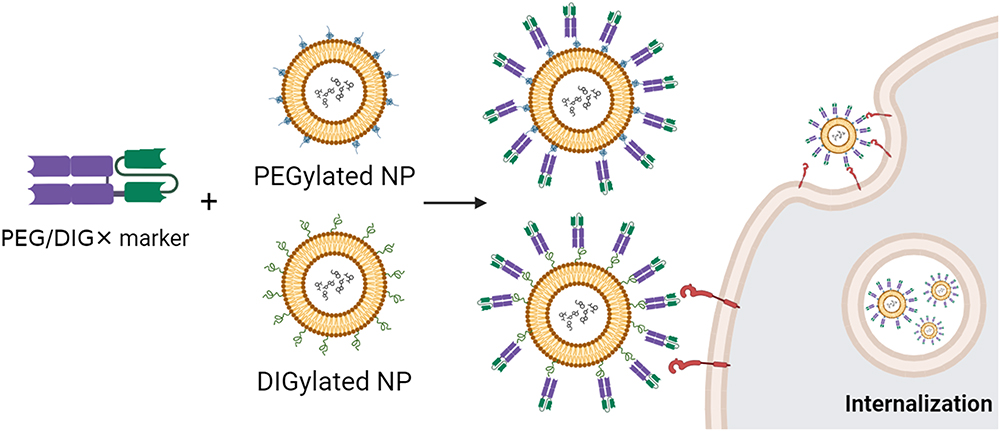 |
Figure 4 Schematic representation of one-step mixing of PEGylated or DIGylated (digoxigenin-modified) NPs with a bsAb for the formation of targeted NPs. DIGylated NPs are synthesized by chemically coupling NHS- or maleimide-DIG molecules to PEG. The bsAb is engineered to simultaneously recognize PEG or DIG and a specific tumor marker. Upon mixing, the bsAbs readily interact with the PEG/DIG moiety of NPs and confer NPs with tumor tropism. This one-step method streamlines NP functionalization and delivers therapeutic payloads to the tumor site efficiently and specifically, potentially improving treatment efficacy. |
In addition to PEG binding, small chemical haptens such as digoxigenin (DIG)87 incorporated into the lipid formulation, allow anti-DIG bsAbs to be recognized and confer the DIG-containing NPs with tumor tropism. Irmgard et al developed two bsAb formats for decorating siRNA-containing NPs with DIGs. DIG-binding scFv are fused at the C-terminal to the anti-CD33 IgG or Fab.88,89 Using such αCD33/NPs, they achieved a knockdown efficiency of up to 90% knockdown compared to the control NPs (less than 15%). The DIG-decorating NPs become internalized along with the anti-CD33 bsAb and accumulate in the endosomal compartments as observed by confocal microscopy.
BsAbs offer precise targeting capabilities by simultaneously binding to two different antigens. This design enhances specificity, reducing the off-target immune responses. This can be advantageous in minimizing collateral damage to healthy tissues. Also, bsAbs are constructed from humanized Ab fragments, which display low immunogenicity in patients.90 The orientation of Abs when combined with NPs is a critical aspect that can significantly influence the performance and efficacy of the resulting conjugates. The anti-mPEG segment of the bsAbs is designed to specifically bind to the methoxy ends of mPEG molecules on the surface of PEGylated NPs. This results in a uniform orientation of the anti-cancer scFv portion of the bsAbs, facilitating precise targeting of PEGylated NPs to cancer cells. The one-step method involves simply mixing the bsAbs with PEGylated NPs, demonstrating a straightforward approach to confer tumor-targeting capability to PEGylated NPs. Under the one-step formulation strategy, how bsAbs stably remain on the NPs becomes a major issue. Cheng et al23 evaluated the stability of mPEG × HER2 on PLD. The results showed that αHER2/PLD remained HER2-bound after storage at 4°C for 7 days or incubation in 10% human serum for 72 h at 37°C. The results showed that the one-step formulation of bsAbs was retained on the NPs, and the HER2-targeting ability of targeted NPs might not be influenced. On the other hand, Howard et al reported that bsAb-functionalized PEG particles showed similar accumulation (p > 0.05) in EGFR-expressing tumours in vivo compared to their non-targeted counterparts in mouse xenografts. Furthermore, when the concentration of bsAbs exceeded 170 bsAbs per particle, the targeted PEG particles exhibited notably increased accumulation in the spleen compared to non-targeted particles or those with a lower bsAb density. This suggests that elevating the functionalization of bsAbs beyond a specific threshold compromises the stealth characteristic of PEGs, resulting in unintended off-target accumulation in vivo.
Pharmacokinetics of Non-Covalent Antibody-Conjugated PEGylated Nanoparticles
The pharmacokinetics of non-covalent antibody-conjugated PEGylated nanoparticles are crucial because they influence the biodistribution, stability, and therapeutic efficacy. The high-affinity interaction between biotin and streptavidin (affinity: Kd ~10−15 M) can be compromised in vivo due to competitive displacement by endogenous biotin. This displacement can significantly alter the biodistribution of nanoparticles, leading to increased uptake by the liver, kidney and spleen, primarily due to rapid clearance by the reticuloendothelial system (RES).91,92 Studies have demonstrated that such a mechanism can result in a substantial proportion of nanoparticles being sequestered in these organs, thereby reducing their availability for target engagement. Although biotin-streptavidin interactions are initially stable, endogenous biotin can displace streptavidin, resulting in the dissociation of the complex and a decreased half-life of the nanoparticles.93,94 The dissociated components are rapidly cleared by the liver and kidneys, further shortening the circulation time of these nanoparticles.
Nanoparticles conjugated to the anti-Fc domain exhibit a stable interaction with antibodies under physiological conditions. However, the dynamic environment of the bloodstream can gradually lead to the dissociation antibody from the Fc-binding domain.78 Dissociated antibodies or nanoparticles, particularly those small enough, may be filtered by the kidneys, leading to renal clearance.95 This process reduces the half-life of the nanoparticles and may impact their therapeutic effectiveness.
Bispecific antibodies are engineered to enhance targeting specificity by binding both to the nanoparticle and the target antigen. However, the non-covalent nature of this interaction introduces the risk of dissociation in vivo, which can reduce targeting efficiency and increase the rate of clearance. This, in turn, affects the biodistribution of the nanoparticles and potentially diminishes therapeutic efficacy. Research highlights that the balance between stability and dissociation is critical for maintaining the desired biodistribution and clearance profiles. Although these nanoparticles are designed to remain in circulation long enough to effectively target specific antigens, the non-covalent binding of bispecific antibodies to nanoparticles may lead to a reduced half-life if dissociation occurs, resulting in faster clearance from the bloodstream.
Comparison of One-Step Formulations for Targeted Liposomal Drug Delivery
In the case of one-step formulation, the direct attachment of engagers to liposomes may expose the entire construct to the immune system, potentially leading to an immune response against the liposomal drug delivery system.96 The immunogenicity profiles of three one-step formulation targeted NP strategies differ based on their molecular components and origins. Biotin-based and streptavidin-tagged targeted NPs generally have higher immunogenicity due to biotin-streptavidin, which are foreign substances that the immune system may react to. Targeted NPs based on Fc-binding domains have the potential to trigger immunological responses, particularly when sourced from non-human sources like protein A/G and peptides. Therefore, it is preferable to use human-derived or modified domains to reduce immunogenicity. BsAb targeted NPs offer enhanced targeting through dual specificity and may not induce immune responses, particularly when derived from humanized or full Abs are preferred to reduce immunogenicity. Overall, while bsAbs may offer lower immunogenicity, careful consideration and optimization are required for all strategies to ensure the development of safe and effective targeted drug delivery systems.
The biotin/streptavidin or bsAbs are directly attached to the liposome surface, potentially leading to varied orientations and spatial heterogeneity. This direct coupling may result in a mix of orientations, affecting the accessibility of binding domains and potentially influencing targeting efficiency. The orientation of targeting ligands or engagers on liposomes can significantly influence the binding affinity and targeting efficiency of NPs. In biotin-based and streptavidin-tagged targeted NPs, the orientation of biotin or streptavidin molecules is generally heterogeneous, as Streptavidin’s tetrameric structure provides numerous binding sites, which can limit the application of the NPs. In Fc-binding domain-based targeted NPs, the orientation of Fc-binding domains may vary depending on the coupling strategy, potentially affecting the binding affinity and stability of the NPs. For bsAb-targeted NPs, the anti-mPEG component of bsAb binds to the methoxy ends of the mPEG molecules on the surface of PEGylated NPs. This enables the anti-cancer scFv portion of the bsAbs to be homogeneously oriented and allows for the targeted delivery of PEGylated NPs to cancer cells. Overall, the precise orientation of engagers coupled to liposomes is essential for optimizing the binding affinity, stability, and targeting efficiency of targeted NPs across all strategies, highlighting the importance of careful design and characterization in NP development.
The modification complexity of Abs and NPs utilizing a one-step formulation differs significantly from the two-step targeted technique. In the one-step formulation, both the Ab and NPs are modified simultaneously, which simplifies the process. However, a one-step formulation might make it more difficult to fine-tune particular changes that increase complexity. In biotin-based and streptavidin-tagged targeted NPs, the modification process is generally straightforward, involving the attachment of biotin or streptavidin molecules to liposomes followed by the binding of their respective partners, which require more complex engineering to achieve optimal targeting efficiency. For Fc-binding domain-based targeted NPs, the coupling process may require more intricate engineering to ensure proper orientation and stability of the Fc-binding domains on liposomes, potentially increasing the complexity of the modification process. In bsAb-targeted NPs, the modification may offer simpler modification processes. BsAbs can simplify and expedite the conversion of non-targeted PEGylated NPs into targeted NPs without any chemical modification compared to biotin-based or Fc-binding domain-based strategies (Table 2).
 |
Table 2 Comparison of One-Step Formulation for Targeted Liposomal Drugs Delivery |
Two-Step Targeting Strategy for NP Active Targeting
Pre-Targeting Bispecific Ab + PEGylated NPs
Pre-targeting bsAbs can be accumulated on the surface of target cells to capture the subsequently administered NPs. These pre-targeting strategies can solve problems such as conformational changes, compromise of the stealth feature and hindrance of tumor uptake of NPs97,98 (Figure 5). Su et al generated a humanized bsAb (PEG engager) by merging a humanized anti-mPEG Fab with a human anti-EGFR scFv which can induce endocytosis of PEGylated NPs into EGFR+ triple-negative breast cancers (TNBCs). This strategy could remarkably enhance the antitumor efficacy of PEGylated therapeutic agents in vitro and in vivo.99 However, loss of the Fc fragment led to loss-of-function of ADCC and CDC of Abs that might decrease the therapeutic efficacy. A bsAb made by fusion of an anti-mPEG scFv to the C-terminus of an anti-HER2 Ab named HER2 ×mPEG BsAb (IgG-scFv format) was introduced by Chen.100 Results indicated that bsAbs enhance accumulation and retention by 2.2-fold in the HER2high MCF7/HER2 tumor compared to the HERlow MCF7/neo1 tumors. Previously, we also proved that the IgG-scFv format of bsAbs can solve the non-specific targeting cytotoxicity and reduced EPR effect seen in liquid tumors.101 By using clinical anti-CD20 mAb (Ofatumumab)102 fused with humanized anti-mPEG scFv, the bsAb enhanced internalization of NPs and internalized up to 56% into CD20-expressing Raji cells compared to the control groups which show no detectable signal of internalization within 24 h. Huckaby et al generated a “Fab-IgG” bsAb against ICAM-1, which is constitutively expressed on the luminal surface of the gastrointestinal (GI) epithelium and PEG on the surface of “mucus-penetrating” particles (MPP). The results indicated that the uptake of both non-targeted and actively targeted NPs was substantially decreased by half or more (p < 0.0001) when compared to pre-targeted NPs.103 Parker et al used OrthoMab,104 a bsAb platform, to construct two bispecific pretargeting molecules (tandem Fab and Fab-IgG1) that bind both breast cancer cell-overexpressed HER2 receptors and PEG on PEGylated liposomal doxorubicin (PLD) and polystyrene beads.105 In mice treated with a pretargeting dosage of bsAb, the ratio of doxorubicin concentration in the tumor to serum increased by ~3-fold compared to mice treated with PLD alone or bsAb (Table 3).
 |
Table 3 Two-Step Targeting Strategy for Nanoparticles Active Targeting |
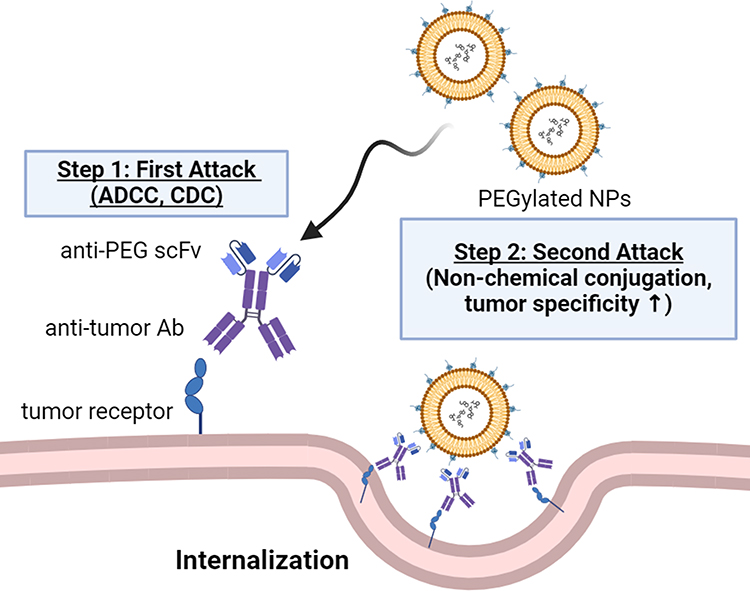 |
Figure 5 Schematic representation of the two-step strategy for bsAb (PEG × tumor marker) combined with PEGylated NPs. The bsAbs accumulate on the tumor cell surface to activate the first attack of Ab-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Then, bsAbs can capture PEGylated NPs to trigger a second attack by increasing internalization and cytotoxicity. Without chemical modification, the two-step strategy maintains naïve NPs with tumor specificity and enhances drug accumulation in tumor cells. |
Pre-targeting bsAbs first bind to the tumor antigen and then capture the NPs without affecting conformational changes to enhance the accumulation and internalization in the tumor. By using fully human or humanized Abs to construct the bsAbs the immune response of the bsAbs can be minimized during treatment. Also, the pre-targeting strategy does not affect the physicochemical properties of NPs, including size, shape, surface charge, and composition, which play crucial roles in determining their immunogenicity. BsAbs utilize 15–2b or 6.3 scFv to specifically attach to the methyl group or backbone of PEG.108,109 These scFv alter NP interactions and properties by specifically targeting locations on PEG molecules. This adaptability enhances the stability, distribution, and effectiveness in targeting for medical treatment and diagnostic purposes, thus advancing nanomedicine. By engineering the variable domains of the bsAbs, researchers can tailor the binding specificity to different tumor antigens or epitopes, allowing for precise targeting of cancer cells. A significant hurdle arises when considering higher doses of pre-targeting bsAbs, as this approach might potentially reduce the binding and accumulation of NPs in tumor cells. The challenge stems from the spatial and temporal heterogeneity in tumor receptor expression.110 Hence, developing a “personalized” pre-targeting strategy could be optimized and allow an ideal concentration of bsAbs against the heterogeneous nature of cancer cells, especially in cases of relapse or highly metastatic tumors.106 It is known that attaching targeting ligands directly to NPs may compromise the stealth characteristics of the NPs, leading to faster clearance and diminished uptake in tumors.97 The two-step targeting strategy of bsAbs is not expected to impact the pharmacokinetics (PK) and stability of NPs during treatment. However, Yang et al indicated that extra circulating bsAbs binding to NPs afterward can enhance NP clearance before NPs extravasate into tumors.106,107 To optimize both tumor distribution and retention while addressing systemic clearance, diverse strategies have been employed, such as modulation of treatment timing,53,54 size,111 valency,112 and composition113–115 of pre-targeting bsAbs. The chemical conjugation of Abs onto NPs may impact the translation of drugs to the clinic including the amount of time required, the ligand aggregation phenomenon, the bioactivity of targeting ligand reduction, and cost.116 Also, Su et al mentioned that variations between different batches of targeted NPs can impede the process of applying them to clinical use and making them available for commercial purposes.99
Pre-Targeting Biotinylated Ab + PEGylated NPs
Pre-targeting with biotinylated Abs can capture streptavidin based on PEGylated NPs to improve tumor targeting and drug accumulation at the tumor area. In the biotin-streptavidin ligation technique, the tumor-targeting proteins such as Abs and derivatives conjugate either biotin or streptavidin, and then attach the NPs to the complementary biotin or streptavidin component (Figure 6). Lehtinen et al employed EGFR Abs conjugated with biotin, which were affixed onto the exterior of PEGylated NPs with streptavidin. They explored the process of cellular uptake influenced by receptors and assessed the effectiveness of EGFR-targeted liposomes in causing cell death in human ovarian adenocarcinoma cells.117 In summary, the direct targeting of liposomes to EGFR proved to be both specific and effective in ovarian cancer cells when tested in vitro. Hang et al used biotinylated anti-CEA Abs to bind cancer cells and collect streptavidin-based NPs for molecular ultrasound imaging and chemotherapy in ovarian cancer treatment. Red fluorescence from the confocal laser scanning microscope showed that DiI-labeled NPs were more attracted to SKOV3 cells than the drug-loaded direct-targeting group and the control group. Yang et al also examined a pre-targeting approach to enhance the delivery of NPs to various target cells. These cells were pre-treated with diverse bispecific streptavidin-scFv fusion proteins (eg, CD20, tumor-associated glycoprotein) and then the absorption of biotinylated NPs was assessed.118 Pre-targeting of Raji cells with streptavidin-αCD20 scFv resulted in a ~15-fold greater uptake of fully biotinylated PS-PEG-biotin NPs compared to PEGylated NPs without biotin functionalization (p < 0.01) (Table 3).
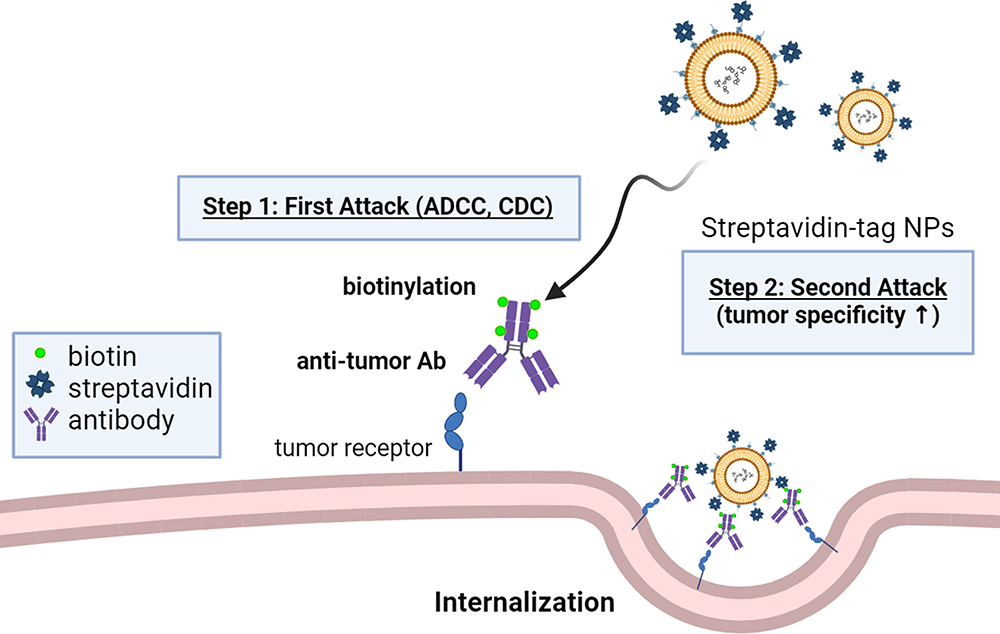 |
Figure 6 Schematic representation of the two-step strategy for biotinylated Abs combined with streptavidin-tagged NPs. The N-hydroxysuccinimide biotin cross-links to a primary amine of Ab to form biotinylated Abs. The biotinylated Abs accumulate on the tumor cell surface to activate the first attack of ADCC and CDC. Then, the biotin on the Abs can interact with the streptavidin which is modified on the NPs to trigger a second attack by increasing internalization and cytotoxicity. This two-step strategy offers a versatile approach for the development of streptavidin-tagged NPs, allowing for enhanced specificity and efficacy of drug delivery applications. |
Product-related impurities in bsAb or fusion proteins, such as aggregation and fragmentation are critical considerations in the development and manufacturing of biotinylated Abs. Aggregation refers to the clumping or clustering of protein molecules. In biotinylated Abs, it can occur when individual protein units form aggregates, compromising the product’s quality, safety, and efficacy resulting in loss of targeting function and immune response.119–122 Process-related impurities like imidazole can arise during the manufacturing and purification processes. Improper washing and elution steps during chromatography can lead to the carryover of imidazole into the final product. Also, the presence of imidazole above permissible levels may have safety implications. Those impurities are essential for ensuring the safety, efficacy, and regulatory approval of biotinylated Abs in therapeutic applications. Continuous monitoring, optimization of manufacturing processes, and adherence to regulatory standards are key elements in mitigating these challenges. The immunogenicity of biotinylated Abs is an important consideration in drug development, as an immune response can impact the safety and efficacy of the therapeutic. The risk of immunogenicity is influenced by several factors, including the degree of humanization,123 choice of expression systems,124,125 and post-translational modifications.126 Monitoring immunogenicity during clinical trials through robust assays for anti-drug Abs is standard practice, allowing for early detection and evaluation of potential immune reactions. Addressing immunogenicity challenges in biotinylated Abs is crucial to ensure their safety, efficacy, and successful clinical translation. The stability of biotinylated Abs on NPs is crucial for their effective therapeutic application. The interaction between biotinylated Abs and NPs can impact the stability of both entities, influencing their pharmacokinetics and overall therapeutic performance.127 Proper stabilization is crucial to prevent protein unfolding, or degradation during storage and administration.128,129 Factors such as surface modifications of NPs, choice of coupling techniques, and the physicochemical properties of the fusion protein play essential roles in maintaining stability.130 Advanced analytical techniques, such as size-exclusion chromatography and dynamic light scattering, rigorously assess the stability of biotinylated Abs on NPs. Addressing stability challenges ensures the preservation of the therapeutic integrity, bioactivity, and safety of these complexes, promoting their successful application in targeted drug delivery and precision medicine.
Comparison of Two-Step Targeting Strategies for Targeted Liposomal Drug Delivery
On the other hand, pre-targeting bsAbs or fusion proteins capture liposomes involving a two-step process, where bsAbs or fusion proteins first bind selectively to target cells before the introduction of liposomes. This strategy may mitigate immunogenicity concerns, as the liposomes are introduced after the initial targeting step, potentially reducing the exposure of the entire construct to the immune system. The immunogenicity of bsAbs and biotinylated antibodies is influenced by their molecular characteristics and origins. Biotinylated antibodies are typically derived from natural Abs and undergo biotinylation, which involves the attachment of biotin molecules to specific sites on the Ab structure. The biotinylation process is generally well-tolerated, and biotin itself is a naturally occurring molecule in biological systems, leading to low immunogenicity. Humanized or fully human bsAbs are developed to minimize immunogenicity by reducing the potential for immune recognition of non-human sequences. However, due to the foreign protein component, streptavidin-tagged NPs, as opposed to PEGylated NPs, could cause immunogenicity by repeated administration over time.
The modification strategies for PEGylated NPs in comparison with streptavidin-tagged NPs differ in their complexity and the nature of the surface modifications. Clinical use of PEGylated NPs has been established for an extended period. In contrast, Streptavidin-tagged NPs require further incorporation of streptavidin through chemical conjugation increasing the complexity and potential for off-target effects. The additional chemical conjugation steps can complicate the nanoparticle synthesis process, potentially leading to reduced reproducibility and increased variability in nanoparticle properties, which can affect their stability, biocompatibility, and targeting efficiency. The choice of nanoparticle modification strategy should be carefully considered based on the desired properties, application requirements, and potential immunological considerations to optimize the performance and safety of the nanoparticles in therapeutic and diagnostic applications (Table 4).
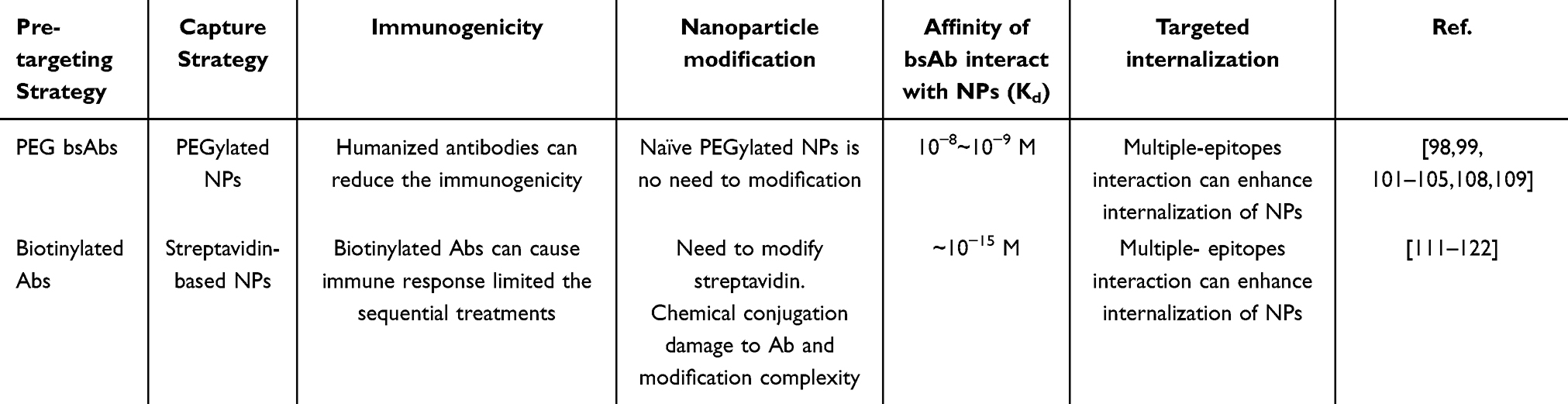 |
Table 4 Comparison of Two-Step Targeting Strategies for Targeted Liposomal Drugs Delivery |
Comparison of the One-Step Formulation and Two-Step Targeting Strategies
A comparison between one-step formulation and two-step targeting is pivotal in understanding their distinct advantages. Both approaches contribute to advancing targeted drug delivery, offering tailored solutions for diverse therapeutic applications. In the one-step formulation, the Ab and NPs are simultaneously formulated, potentially leading to faster production times and simpler procedures. However, this method may result in lower stability due to non-specific interactions between the Ab and NPs during formulation. Increasing the affinity of Abs might ensure a stable and specific binding, preventing premature dissociation of bsAbs from liposomes in physiological conditions. This stability is essential for maintaining the integrity of the targeted liposomes during circulation in the bloodstream, preventing nonspecific interactions, and reducing the risk of premature payload release.131 Ultimately, the choice between a one-step or a two-step strategy depends on the specific requirements of the application and the desired balance between efficiency and stability.
The comparison between the one-step formulation and the two-step targeting strategy of Ab and NPs also extends to treatment timing. In the one-step formulation, immediate administration can be conducted upon completion. This streamlined approach may be advantageous for situations requiring rapid treatment initiation. However, the two-step targeting strategy involves separate optimization of Ab and NP modifications before conjugation, which may result in a longer preparation time before treatment administration. In the two-step targeting strategy, it is crucial to minimize the circulating levels of pre-targeting bsAbs during the administration of NPs. Elevated concentrations of bsAbs can result in the entrapment of NPs within the bloodstream, impeding their efficient extravasation to the intended targeted tissues. This emphasizes the need for a precise balance in the timing and concentration of bsAbs to support, rather than hinder, the effective delivery of NPs to the intended target sites.105,132 Maintaining the persistence of bsAbs on the tumor cell surface at the time of NP administration is crucial, preventing premature internalization that could hinder subsequent NP binding. Achieving an optimal time interval between bsAb and NP administration, balancing systemic clearance and tumor surface retention, is vital for effective pre-targeting drug delivery.133 Understanding the kinetics of bsAb clearance and their behavior at the tumor site is imperative for developing an efficient pre-targeting drug delivery system. Therefore, the choice between one-step and two-step strategies depends on the urgency of treatment initiation and the desired level of control over the conjugation process (Table 5).
 |
Table 5 Comparison of Two Strategies for Targeted Liposomal Drugs Delivery |
Conclusion and Future Prospects
The comparison of the one-step formulation technique and the two-step targeting strategy highlights the intricate factors involved in creating efficient, targeted drug delivery systems. Combining biotin/streptavidin, Fc-binding domain, or bsAbs directly onto liposomes in a one-step process simplifies manufacturing but could present issues with immunogenicity and inconsistent Ab orientations, which might affect targeting effectiveness. Pre-targeting bsAb or fusion proteins that trap liposomes offer a two-step method that may decrease immunogenicity and provide more consistent Ab orientations, potentially improving targeting accuracy. The selection of a strategy depends on therapeutic objectives, taking into account aspects including immunogenicity, targeted specificity, and the complexity of the drug delivery system.
PEGylated products remain widely accessible in clinical practice. The continuous advancement of nanomedicine and biotechnology holds promise for enhancing these techniques and ultimately improving the precise delivery of therapeutic agents for various biological applications. Anti-PEG bsAbs give PEGylated NPs targeted capabilities, improving their clinical potential. The incorporation of anti-PEG bispecific antibodies offers a transformative approach to overcome this limitation by conferring PEGylated NPs with targeted capabilities. Anti-PEG bispecific antibodies enable the specific and dual targeting of PEGylated NPs to both PEG and a specific cell surface marker, enhancing the precision, efficiency, and selectivity of nanoparticle delivery. This targeted approach can significantly improve therapeutic efficacy, reduce systemic toxicity, and enhance patient compliance by minimizing off-target effects and the required drug dosage. Furthermore, the active targeting facilitated by anti-PEG bispecific antibodies can broaden the clinical applications of PEGylated NPs, particularly in the treatment of heterogeneous or resistant cell populations and in immunotherapy. Overall, the one-step formulation of anti-PEG bsAb and PEGylated NP not only accelerates the development of targeted nanomedicine but also enhances its clinical feasibility, scalability, and translational potential, paving the way for the advancement of personalized and precision medicine approaches in various therapeutic areas.
In current clinical trials, bispecific antibody-targeted lipopolysaccharides (LPS)-packaged micelles are a leading strategy for targeted cancer therapy, as shown by the Phase I trial of EGFR-targeted, paclitaxel-loaded minicells.134 While these micelles are advancing in clinical studies, PEGylated nanoparticles, despite extensive preclinical research, have yet to enter clinical trials, underscoring the challenges of bringing PEGylated formulations to clinical use. Currently, non-covalent conjugation methods, particularly in the context of PEGylated nanoparticles, are still primarily in the preclinical or early clinical phases, and thus not extensively represented in the available clinical trial databases or published studies. Additionally, much of the existing literature and ongoing trials tend to focus on more established covalent conjugation techniques, further contributing to the scarcity of clinical data on non-covalent methods. We recognize the importance of this area and understand that more detailed clinical reporting and further research are essential to fully elucidate the clinical translation potential of these nanoplatforms.
Abbreviations
Ab, antibody; ADCC, Ab-dependent cellular cytotoxicity; AUC, area under the curve; bsAb, bispecific antibody; CDC, complement-dependent cytotoxicity; DIG, digoxigenin; EDC, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide; EPR, enhanced permeability and retention; FcBP, Fc-binding peptide; GI, gastrointestinal; HER2, Human epidermal growth factor receptor 2; LGA-PEI, lactic-co-glycolic acid-polyethylenimine; MR, magnetic resonance; NP, nanoparticles; PEG, polyethylene glycol; PILP, PEGylated immuno-lipopolyplexes; PK, pharmacokinetic; PLD, PEGylated liposomal doxorubicin; QD, quantum dot; RES, reticuloendothelial system; SAL, streptavidin-tagged liposome; scFv, single chain Fv; TfR, transferrin receptor; TNBC, triple-negative breast cancer.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study is also supported by grants from the Ministry of Science and Technology, Taipei, Taiwan (MOST 110-2320-B-037 −010 -MY3, MOST 111-2124-M-037-001, MOST 111-2314-B-037 −051 -MY3, NSTC 112-2124-M-037-001 - and NSTC 112-2320-B-037-011-MY3.); the Ministry of Economic Affairs (112-EC-17-A-17-S6-021); the KMU-KMUH Co-Project of Key Research (KMU-DK(B)112001, KMU-DK(B)112001-1 and KMUH-DK(B)112001-2); NTHU-KMU Joint Research Project (KT112P002).
Disclosure
The authors report no competing interests in this work.
References
1. Chen BM, Cheng TL, Roffler SR. Polyethylene Glycol Immunogenicity: theoretical, Clinical, and Practical Aspects of Anti-Polyethylene Glycol Antibodies. ACS Nano. 2021;15(9):14022–14048. doi:10.1021/acsnano.1c05922
2. Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Contrele. 2010;145(3):178–181. doi:10.1016/j.jconrel.2010.03.016
3. Estelrich J, Sanchez-Martin MJ, Busquets MA. Nanoparticles in magnetic resonance imaging: from simple to dual contrast agents. Int J Nanomed. 2015;10:1727–1741. doi:10.2147/IJN.S76501
4. Perry JL, Reuter KG, Kai MP, et al. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett. 2012;12(10):5304–5310. doi:10.1021/nl302638g
5. Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. BBA. 2009;1788(10):2259–2266. doi:10.1016/j.bbamem.2009.06.022
6. Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi:10.1016/j.ijpharm.2005.10.010
7. Gao H, Liu J, Yang C, et al. The impact of PEGylation patterns on the in vivo biodistribution of mixed shell micelles. Int J Nanomed. 2013;8:4229–4246. doi:10.2147/IJN.S51566
8. Ahsan F, Rivas IP, Khan MA, Torres Suarez AI. Targeting to macrophages: role of physicochemical properties of particulate carriers--liposomes and microspheres--on the phagocytosis by macrophages. J Contrele. 2002;79(1–3):29–40. doi:10.1016/S0168-3659(01)00549-1
9. Barenholz Y. Doxil(R)--The first FDA-approved nano-drug: lessons learned. J Contrele. 2012;160(2):117–134. doi:10.1016/j.jconrel.2012.03.020
10. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–436. doi:10.2165/00003088-200342050-00002
11. Wu D, Si M, Xue HY, Wong HL. Nanomedicine applications in the treatment of breast cancer: current state of the art. Int J Nanomed. 2017;12:5879–5892. doi:10.2147/IJN.S123437
12. Xu L, Wang W, Sheng YC, Zheng QS. Pharmacokinetics and its relation to toxicity of pegylated-liposomal doxorubicin in Chinese patients with breast tumours. J Clin Pharm Ther. 2010;35(5):593–601. doi:10.1111/j.1365-2710.2009.01128.x
13. Frampton JE. Liposomal Irinotecan: a Review in Metastatic Pancreatic Adenocarcinoma. Drugs. 2020;80(10):1007–1018. doi:10.1007/s40265-020-01336-6
14. Lamb YN, Scott LJ. Liposomal Irinotecan: a Review in Metastatic Pancreatic Adenocarcinoma. Drugs. 2017;77(7):785–792. doi:10.1007/s40265-017-0741-1
15. Adiwijaya BS, Kim J, Lang I, et al. Population Pharmacokinetics of Liposomal Irinotecan in Patients With Cancer. Clin Pharmacol Ther. 2017;102(6):997–1005. doi:10.1002/cpt.720
16. Brendel K, Bekaii-Saab T, Boland PM, et al. Population pharmacokinetics of liposomal irinotecan in patients with cancer and exposure-safety analyses in patients with metastatic pancreatic cancer. CPT Pharmacometrics Syst Pharmacol. 2021;10(12):1550–1563. doi:10.1002/psp4.12725
17. Biancacci I, De Lorenzi F, Theek B, et al. Monitoring EPR Effect Dynamics during Nanotaxane Treatment with Theranostic Polymeric Micelles. Adv Sci. 2022;9(10):e2103745. doi:10.1002/advs.202103745
18. Shim MK, Yang S, Park J, et al. Preclinical development of carrier-free prodrug nanoparticles for enhanced antitumor therapeutic potential with less toxicity. J Nanobiotechnology. 2022;20(1):436. doi:10.1186/s12951-022-01644-x
19. Fu Y, Jang MS, Liu C, Li Y, Lee JH, Yang HY. Oxygen-Generating Organic/Inorganic Self-Assembled Nanocolloids for Tumor-Activated Dual-Model Imaging-Guided Photodynamic Therapy. ACS Appl Mater Interfaces. 2023;15(30):36013–36024. doi:10.1021/acsami.3c07008
20. Verhoef JJ, Anchordoquy TJ. Questioning the Use of PEGylation for Drug Delivery. Drug Deliv Transl Res. 2013;3(6):499–503. doi:10.1007/s13346-013-0176-5
21. Sapra P, Allen TM. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res. 2002;62(24):7190–7194.
22. Chuang KH, Wang HE, Chen FM, et al. Endocytosis of PEGylated agents enhances cancer imaging and anticancer efficacy. Mol Cancer Ther. 2010;9(6):1903–1912. doi:10.1158/1535-7163.MCT-09-0899
23. Cheng YA, Chen IJ, Su YC, et al. Enhanced drug internalization and therapeutic efficacy of PEGylated nanoparticles by one-step formulation with anti-mPEG bispecific antibody in intrinsic drug-resistant breast cancer. Biomater Sci. 2019;7(8):3404–3417. doi:10.1039/C9BM00323A
24. Munster P, Krop IE, LoRusso P, et al. Safety and pharmacokinetics of MM-302, a HER2-targeted antibody-liposomal doxorubicin conjugate, in patients with advanced HER2-positive breast cancer: a Phase 1 dose-escalation study. Br J Cancer. 2018;119(9):1086–1093. doi:10.1038/s41416-018-0235-2
25. Chen H-J, Cheng Y-A, Chen Y-T, et al. Targeting and internalizing PEGylated nanodrugs to enhance the therapeutic efficacy of hematologic malignancies by anti-PEG bispecific antibody (mPEG × CD20). Cancer Nanotechnol. 2023;14(1):78. doi:10.1186/s12645-023-00230-6
26. Shim G, Kim D, Lee S, Chang RS, Byun J, Oh YK. Staphylococcus aureus-mimetic control of antibody orientation on nanoparticles. Nanomedicine. 2019;16:267–277. doi:10.1016/j.nano.2018.09.007
27. Wang Y, Yang M, Qian J, et al. Sequentially self-assembled polysaccharide-based nanocomplexes for combined chemotherapy and photodynamic therapy of breast cancer. Carbohydr Polym. 2019;203:203–213. doi:10.1016/j.carbpol.2018.09.035
28. Yang Z, Li H, Zhang W, et al. CD163 Monoclonal Antibody Modified Polymer Prodrug Nanoparticles for Targeting Tumor-Associated Macrophages (TAMs) to Enhance Anti-Tumor Effects. Pharmaceutics. 2023;15:4. doi:10.3390/pharmaceutics15041241
29. Hallam TJ, Wold E, Wahl A, Smider VV. Antibody conjugates with unnatural amino acids. Mol Pharmaceut. 2015;12(6):1848–1862. doi:10.1021/acs.molpharmaceut.5b00082
30. Richards DA, Maruani A, Chudasama V. Antibody fragments as nanoparticle targeting ligands: a step in the right direction. Chem Sci. 2017;8(1):63–77. doi:10.1039/C6SC02403C
31. Gao Y, Kyratzis I. Covalent immobilization of proteins on carbon nanotubes using the cross-linker 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide--a critical assessment. Bioconjug Chem. 2008;19(10):1945–1950. doi:10.1021/bc800051c
32. Devaraj NK, Finn MG. Introduction: click Chemistry. Chem Rev. 2021;121(12):6697–6698. doi:10.1021/acs.chemrev.1c00469
33. Guesdon JL, Ternynck T, Avrameas S. The use of avidin-biotin interaction in immunoenzymatic techniques. J Histochem Cytochem. 1979;27(8):1131–1139. doi:10.1177/27.8.90074
34. Gitlin G, Bayer EA, Wilchek M. Studies on the biotin-binding site of avidin. Tryptophan residues involved in the active site. Biochem J. 1988;250(1):291–294. doi:10.1042/bj2500291
35. Wilchek M, Bayer EA. Introduction to avidin-biotin technology. Methods Enzymol. 1990;184:5–13.
36. Hu Y, Li K, Wang L, Yin S, Zhang Z, Zhang Y. Pegylated immuno-lipopolyplexes: a novel non-viral gene delivery system for liver cancer therapy. J Contrele. 2010;144(1):75–81. doi:10.1016/j.jconrel.2010.02.005
37. Salvati E, Re F, Sesana S, et al. Liposomes functionalized to overcome the blood-brain barrier and to target amyloid-beta peptide: the chemical design affects the permeability across an in vitro model. Int J Nanomed. 2013;8:1749–1758. doi:10.2147/IJN.S42783
38. Papadia K, Markoutsa E, Antimisiaris SG. A simplified method to attach antibodies on liposomes by biotin-streptavidin affinity for rapid and economical screening of targeted liposomes. J Biomed Nanotechnol. 2014;10(5):871–876. doi:10.1166/jbn.2014.1792
39. Chen MH, Soda Y, Izawa K, et al. A versatile drug delivery system using streptavidin-tagged pegylated liposomes and biotinylated biomaterials. Int J Pharm. 2013;454(1):478–485. doi:10.1016/j.ijpharm.2013.06.031
40. Lee HY, Jung HS, Fujikawa K, et al. New antibody immobilization method via functional liposome layer for specific protein assays. Biosens Bioelectron. 2005;21(5):833–838. doi:10.1016/j.bios.2005.01.020
41. Redelmeier TE, Guillet J-G, Ballyt MB. High-affinity targeting of biotin-labeled liposomes to streptavidin-conjugated ligands. Drug Delivery. 1995;2(2):98–109. doi:10.3109/10717549509031357
42. Conrad M, Proll G, Builes-Munden E, Dietzel A, Wagner S, Gauglitz G. Tools to compare antibody gold nanoparticle conjugates for a small molecule immunoassay. Mikrochim Acta. 2023;190(2):62. doi:10.1007/s00604-023-05637-x
43. Bruckner M, Simon J, Landfester K, Mailander V. The conjugation strategy affects antibody orientation and targeting properties of nanocarriers. Nanoscale. 2021;13(21):9816–9824. doi:10.1039/D0NR08191D
44. Janu L, Stanisavljevic M, Krizkova S, et al. Electrophoretic study of peptide-mediated quantum dot-human immunoglobulin bioconjugation. Electrophoresis. 2013;34(18):2725–2732. doi:10.1002/elps.201300088
45. Jin T, Tiwari DK, Tanaka S, Inouye Y, Yoshizawa K, Watanabe TM. Antibody-protein A conjugated quantum dots for multiplexed imaging of surface receptors in living cells. Mol Biosyst. 2010;6(11):2325–2331. doi:10.1039/c0mb00056f
46. Rodriguez-Serrano F, Mut-Salud N, Cruz-Bustos T, et al. Functionalized immunostimulating complexes with protein A via lipid vinyl sulfones to deliver cancer drugs to trastuzumab-resistant HER2-overexpressing breast cancer cells. Int J Nanomed. 2016;11:4777–4785. doi:10.2147/IJN.S112560
47. Watanabe TM, Higuchi H. Stepwise movements in vesicle transport of HER2 by motor proteins in living cells. Biophys J. 2007;92(11):4109–4120. doi:10.1529/biophysj.106.094649
48. Thorpe SJ, Turner C, Heath A, et al. Clonal analysis of a human antimouse antibody (HAMA) response. Scand J Immunol. 2003;57(1):85–92. doi:10.1046/j.1365-3083.2003.01189.x
49. Jiang CT, Chen KG, Liu A, et al. Immunomodulating nano-adaptors potentiate antibody-based cancer immunotherapy. Nat Commun. 2021;12(1):1359. doi:10.1038/s41467-021-21497-6
50. Kedmi R, Veiga N, Ramishetti S, et al. A modular platform for targeted RNAi therapeutics. Nat Nanotechnol. 2018;13(3):214–219. doi:10.1038/s41565-017-0043-5
51. Kampel L, Goldsmith M, Ramishetti S, et al. Therapeutic inhibitory RNA in head and neck cancer via functional targeted lipid nanoparticles. J Contrele. 2021;337:378–389. doi:10.1016/j.jconrel.2021.07.034
52. Ma J, Mo Y, Tang M, et al. Bispecific Antibodies: from Research to Clinical Application. Front Immunol. 2021;12:626616. doi:10.3389/fimmu.2021.626616
53. Beishenaliev A, Loke YL, Goh SJ, et al. Bispecific antibodies for targeted delivery of anti-cancer therapeutic agents: a review. J Contrele. 2023;359:268–286. doi:10.1016/j.jconrel.2023.05.032
54. Kao CH, Wang JY, Chuang KH, et al. One-step mixing with humanized anti-mPEG bispecific antibody enhances tumor accumulation and therapeutic efficacy of mPEGylated nanoparticles. Biomaterials. 2014;35(37):9930–9940. doi:10.1016/j.biomaterials.2014.08.032
55. Cheng YA, Wu TH, Wang YM, et al. Humanized bispecific antibody (mPEG x HER2) rapidly confers PEGylated nanoparticles tumor specificity for multimodality imaging in breast cancer. J Nanobiotechnology. 2020;18(1):118. doi:10.1186/s12951-020-00680-9
56. Lin WW, Cheng YA, Li CC, et al. Enhancement of tumor tropism of mPEGylated nanoparticles by anti-mPEG bispecific antibody for ovarian cancer therapy. Sci Rep. 2021;11(1):7598. doi:10.1038/s41598-021-87271-2
57. Cheng WJ, Lin SY, Chuang KH, et al. Combined Docetaxel/Pictilisib-Loaded mPEGylated Nanocarriers with Dual HER2 Targeting Antibodies for Synergistic Chemotherapy of Breast Cancer. Int J Nanomed. 2022;17:5353–5374. doi:10.2147/IJN.S388066
58. Cui J, Ju Y, Houston ZH, et al. Modulating Targeting of Poly(ethylene glycol) Particles to Tumor Cells Using Bispecific Antibodies. Adv Healthc Mater. 2019;8(9):e1801607. doi:10.1002/adhm.201801607
59. Jain A, Cheng K. The principles and applications of avidin-based nanoparticles in drug delivery and diagnosis. J Contrele. 2017;245:27–40. doi:10.1016/j.jconrel.2016.11.016
60. Leonetti M, Thai R, Cotton J, et al. Increasing immunogenicity of antigens fused to Ig-binding proteins by cell surface targeting. J Iimmunol. 1998;160(8):3820–3827. doi:10.4049/jimmunol.160.8.3820
61. Xiao Z, McQuarrie SA, Suresh MR, Mercer JR, Gupta S, Miller GG. A three-step strategy for targeting drug carriers to human ovarian carcinoma cells in vitro. J Biotechnol. 2002;94(2):171–184. doi:10.1016/S0168-1656(01)00424-2
62. Ren C-L, Carvajal D, Shull KR, Szleifer I. Streptavidin−Biotin Binding in the Presence of a Polymer Spacer. A Theoretical Description. Langmuir. 2009;25(20):12283–12292. doi:10.1021/la901735d
63. Xu D, Heck AJ, Kuan SL, Weil T, Wegner SV. Precise tetrafunctional streptavidin bioconjugates towards multifaceted drug delivery systems. Chem Commun. 2020;56(68):9858–9861. doi:10.1039/D0CC04054A
64. Chidley C, Mosiewicz K, Johnsson K. A designed protein for the specific and covalent heteroconjugation of biomolecules. Bioconjug Chem. 2008;19(9):1753–1756. doi:10.1021/bc800268j
65. Lee NK, Wang CJ, Lim J, et al. Impact of the conjugation of antibodies to the surfaces of polymer nanoparticles on the immune cell targeting abilities. Nano Converg. 2021;8(1):24. doi:10.1186/s40580-021-00274-7
66. Dimasi N, Fleming R, Zhong H, et al. Efficient Preparation of Site-Specific Antibody-Drug Conjugates Using Cysteine Insertion. Mol Pharmaceut. 2017;14(5):1501–1516. doi:10.1021/acs.molpharmaceut.6b00995
67. Hama S, Sakai M, Itakura S, Majima E, Kogure K. Rapid modification of antibodies on the surface of liposomes composed of high-affinity protein A-conjugated phospholipid for selective drug delivery. Biochem Biophys Rep. 2021;27:101067. doi:10.1016/j.bbrep.2021.101067
68. Mori S, Abe A, Ishikawa N, Rafique A, Ito Y. A novel site-specific chemical conjugation of IgG antibodies by affinity peptide for immunoassays. J Biochem. 2021;169(1):35–42. doi:10.1093/jb/mvaa084
69. Yu C, Tang J, Loredo A, et al. Proximity-Induced Site-Specific Antibody Conjugation. Bioconjug Chem. 2018;29(11):3522–3526. doi:10.1021/acs.bioconjchem.8b00680
70. Gil-Garcia M, Ventura S. Multifunctional antibody-conjugated coiled-coil protein nanoparticles for selective cell targeting. Acta Biomater. 2021;131:472–482. doi:10.1016/j.actbio.2021.06.040
71. Cheng WW, Allen TM. The use of single chain Fv as targeting agents for immunoliposomes: an update on immunoliposomal drugs for cancer treatment. Expert Opin Drug Deliv. 2010;7(4):461–478. doi:10.1517/17425240903579963
72. Lu JM, Liang Z, Liu D, Zhan B, Yao Q, Chen C. Two Antibody-Guided Lactic-co-Glycolic Acid-Polyethylenimine (LGA-PEI) Nanoparticle Delivery Systems for Therapeutic Nucleic Acids. Pharmaceuticals. 2021;14:9. doi:10.3390/ph14090841
73. Bjorck L, Kronvall G. Purification and some properties of streptococcal protein G, a novel IgG-binding reagent. J Iimmunol. 1984;133(2):969–974. doi:10.4049/jimmunol.133.2.969
74. Stone GC, Sjobring U, Bjorck L, Sjoquist J, Barber CV, Nardella FA. The Fc binding site for streptococcal protein G is in the C gamma 2-C gamma 3 interface region of IgG and is related to the sites that bind staphylococcal protein A and human rheumatoid factors. J Iimmunol. 1989;143(2):565–570. doi:10.4049/jimmunol.143.2.565
75. Uhlen M, Guss B, Nilsson B, Gatenbeck S, Philipson L, Lindberg M. Complete sequence of the staphylococcal gene encoding protein A. A gene evolved through multiple duplications. J Biol Chem. 1984;259(3):1695–1702. doi:10.1016/S0021-9258(17)43463-6
76. Yang L, Biswas ME, Chen P. Study of binding between protein A and immunoglobulin G using a surface tension probe. Biophys J. 2003;84(1):509–522. doi:10.1016/S0006-3495(03)74870-X
77. Lund LN, Christensen T, Toone E, Houen G, Staby A, St hilaire PM. Exploring variation in binding of Protein A and Protein G to immunoglobulin type G by isothermal titration calorimetry. J Mol Recognit. 2011;24(6):945–952. doi:10.1002/jmr.1140
78. Lin PC, Chen SH, Wang KY, et al. Fabrication of oriented antibody-conjugated magnetic nanoprobes and their immunoaffinity application. Anal Chem. 2009;81(21):8774–8782. doi:10.1021/ac9012122
79. Yang H, Gurgel PV, Williams DK Jr, et al. Binding site on human immunoglobulin G for the affinity ligand HWRGWV. J Mol Recognit. 2010;23(3):271–282. doi:10.1002/jmr.967
80. Menegatti S, Bobay BG, Ward KL, et al. Design of protease-resistant peptide ligands for the purification of antibodies from human plasma. J Chromatogr A. 2016;1445:93–104. doi:10.1016/j.chroma.2016.03.087
81. Reese HR, Xiao X, Shanahan CC, et al. Novel peptide ligands for antibody purification provide superior clearance of host cell protein impurities. J Chromatogr A. 2020;1625:461237. doi:10.1016/j.chroma.2020.461237
82. Zhao WW, Shi QH, Sun Y. FYWHCLDE-based affinity chromatography of IgG: effect of ligand density and purifications of human IgG and monoclonal antibody. J Chromatogr A. 2014;1355:107–114. doi:10.1016/j.chroma.2014.05.083
83. Kruljec N, Molek P, Hodnik V, Anderluh G, Bratkovic T. Development and Characterization of Peptide Ligands of Immunoglobulin G Fc Region. Bioconjug Chem. 2018;29(8):2763–2775. doi:10.1021/acs.bioconjchem.8b00395
84. Moles E, Howard CB, Huda P, et al. Delivery of PEGylated liposomal doxorubicin by bispecific antibodies improves treatment in models of high-risk childhood leukemia. Sci Transl Med. 2023;15(696):eabm1262. doi:10.1126/scitranslmed.abm1262
85. Song D, Cui J, Ju Y, et al. Cellular Targeting of Bispecific Antibody-Functionalized Poly(ethylene glycol) Capsules: do Shape and Size Matter? ACS Appl Mater Interfaces. 2019;11(32):28720–28731. doi:10.1021/acsami.9b10304
86. Chuang K-H, Kao C-H, Roffler SR, et al. Development of an Anti-Methoxy Poly(ethylene glycol) (α-mPEG) Cell-Based Capture System to Measure mPEG and mPEGylated Molecules. Macromolecules. 2014;47(19):6880–6888. doi:10.1021/ma501156r
87. O’Connell JP, Campbell RL, Fleming BM, Mercolino TJ, Johnson MD, McLaurin DA. A highly sensitive immunoassay system involving antibody-coated tubes and liposome-entrapped dye. Clin Chem. 1985;31(9):1424–1426. doi:10.1093/clinchem/31.9.1424
88. Thorey IS, Grote M, Mayer K, Brinkmann U. Hapten-Binding Bispecific Antibodies for the Targeted Delivery of SiRNA and SiRNA-Containing Nanoparticles. Methods Mol Biol. 2016;1364:219–234.
89. Schneider B, Grote M, John M, et al. Targeted siRNA Delivery and mRNA Knockdown Mediated by Bispecific Digoxigenin-binding Antibodies. Mol Ther Nucleic Acids. 2012;1(9):e46. doi:10.1038/mtna.2012.39
90. Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36(1):3–10. doi:10.1016/j.ymeth.2005.01.001
91. D’Agata R, Palladino P, Spoto G. Streptavidin-coated gold nanoparticles: critical role of oligonucleotides on stability and fractal aggregation. Beilstein J Nanotechnol. 2017;8:1–11. doi:10.3762/bjnano.8.1
92. Chirra HD, Sexton T, Biswal D, Hersh LB, Hilt JZ. Catalase-coupled gold nanoparticles: comparison between the carbodiimide and biotin-streptavidin methods. Acta Biomater. 2011;7(7):2865–2872. doi:10.1016/j.actbio.2011.01.003
93. Lee JH, Chapman DV, Saltzman WM. Nanoparticle Targeting with Antibodies in the Central Nervous System. BME Front. 2023;4:0012. doi:10.34133/bmef.0012
94. Perrault SD, Chan WC, In vivo assembly of nanoparticle components to improve targeted cancer imaging.
95. Wang J, Masehi-Lano JJ, Chung EJ. Peptide and antibody ligands for renal targeting: nanomedicine strategies for kidney disease. Biomater Sci. 2017;5(8):1450–1459. doi:10.1039/C7BM00271H
96. Wang AZ, Gu F, Zhang L, et al. Biofunctionalized targeted nanoparticles for therapeutic applications. Expert Opin Biol Ther. 2008;8(8):1063–1070. doi:10.1517/14712598.8.8.1063
97. Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338(6109):903–910. doi:10.1126/science.1226338
98. Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. doi:10.1038/nnano.2011.166
99. Su YC, Burnouf PA, Chuang KH, Chen BM, Cheng TL, Roffler SR. Conditional internalization of PEGylated nanomedicines by PEG engagers for triple negative breast cancer therapy. Nat Commun. 2017;8:15507. doi:10.1038/ncomms15507
100. Chen IJ, Cheng YA, Ho KW, et al. Bispecific antibody (HER2 x mPEG) enhances anti-cancer effects by precise targeting and accumulation of mPEGylated liposomes. Acta Biomater. 2020;111:386–397. doi:10.1016/j.actbio.2020.04.029
101. Ho KW, Chen IU, Cheng YA, et al. Double attack strategy for leukemia using a pre-targeting bispecific antibody (CD20 Ab-mPEG scFv) and actively attracting PEGylated liposomal doxorubicin to enhance anti-tumor activity. J Nanobiotechnology. 2021;19(1):16. doi:10.1186/s12951-020-00752-w
102. Gravanis I, Ersboll J, Skovlund E, Abadie E, Marty M, Pignatti F. The European Medicines Agency review of ofatumumab (Arzerra(R)) for the treatment of chronic lymphocytic leukemia in patients refractory to fludarabine and alemtuzumab: summary of the scientific assessment of the European medicines agency committee for medicinal products for human use. Oncologist. 2010;15(12):1335–1343. doi:10.1634/theoncologist.2010-0255
103. Huckaby JT, Parker CL, Jacobs TM, et al. Engineering Polymer-Binding Bispecific Antibodies for Enhanced Pretargeted Delivery of Nanoparticles to Mucus-Covered Epithelium. Angew Chem Int Ed Engl. 2019;58(17):5604–5608. doi:10.1002/anie.201814665
104. Lewis SM, Wu X, Pustilnik A, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nature Biotechnol. 2014;32(2):191–198. doi:10.1038/nbt.2797
105. Parker CL, McSweeney MD, Lucas AT, et al. Pretargeted delivery of PEG-coated drug carriers to breast tumors using multivalent, bispecific antibody against polyethylene glycol and HER2. Nanomedicine. 2019;21:102076. doi:10.1016/j.nano.2019.102076
106. Yang Q, Parker CL, McCallen JD, Lai SK. Addressing challenges of heterogeneous tumor treatment through bispecific protein-mediated pretargeted drug delivery. J Contrele. 2015;220(Pt B):715–726. doi:10.1016/j.jconrel.2015.09.040
107. Longman SA, Cullis PR, Choi L, de Jong G, Bally MB. A two-step targeting approach for delivery of doxorubicin-loaded liposomes to tumour cells in vivo. Cancer Chemother Pharmacol. 1995;36(2):91–101. doi:10.1007/BF00689191
108. McCallen J, Prybylski J, Yang Q, Lai SK. Cross-Reactivity of Select PEG-Binding Antibodies to Other Polymers Containing a C-C-O Backbone. ACS Biomater Sci Eng. 2017;3(8):1605–1615. doi:10.1021/acsbiomaterials.7b00147
109. Chen BM, Su YC, Chang CJ, et al. Measurement of Pre-Existing IgG and IgM Antibodies against Polyethylene Glycol in Healthy Individuals. Anal Chem. 2016;88(21):10661–10666. doi:10.1021/acs.analchem.6b03109
110. Janku F. Tumor heterogeneity in the clinic: is it a real problem? Ther Adv Med Oncol. 2014;6(2):43–51. doi:10.1177/1758834013517414
111. Goldenberg DM, Rossi EA, Sharkey RM, McBride WJ, Chang CH. Multifunctional antibodies by the Dock-and-Lock method for improved cancer imaging and therapy by pretargeting. J Nucl Med. 2008;49(1):158–163. doi:10.2967/jnumed.107.046185
112. Sharkey RM, Karacay H, Litwin S, et al. Improved therapeutic results by pretargeted radioimmunotherapy of non-Hodgkin’s lymphoma with a new recombinant, trivalent, anti-CD20, bispecific antibody. Cancer Res. 2008;68(13):5282–5290. doi:10.1158/0008-5472.CAN-08-0037
113. Forero A, Weiden PL, Vose JM, et al. Phase 1 trial of a novel anti-CD20 fusion protein in pretargeted radioimmunotherapy for B-cell non-Hodgkin lymphoma. Blood. 2004;104(1):227–236. doi:10.1182/blood-2003-09-3284
114. Lin Y, Pagel JM, Axworthy D, Pantelias A, Hedin N, Press OW. A genetically engineered anti-CD45 single-chain antibody-streptavidin fusion protein for pretargeted radioimmunotherapy of hematologic malignancies. Cancer Res. 2006;66(7):3884–3892. doi:10.1158/0008-5472.CAN-05-3443
115. Steiner M, Gutbrodt K, Krall N, Neri D. Tumor-targeting antibody-anticalin fusion proteins for in vivo pretargeting applications. Bioconjug Chem. 2013;24(2):234–241. doi:10.1021/bc300567a
116. Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. doi:10.1039/c2cs15344k
117. Lehtinen J, Raki M, Bergstrom KA, et al. Pre-targeting and direct immunotargeting of liposomal drug carriers to ovarian carcinoma. PLoS One. 2012;7(7):e41410. doi:10.1371/journal.pone.0041410
118. Yang Q, Parker CL, Lin Y, Press OW, Park SI, Lai SK. Pretargeting with bispecific fusion proteins facilitates delivery of nanoparticles to tumor cells with distinct surface antigens. J Contrele. 2017;255:73–80. doi:10.1016/j.jconrel.2017.03.388
119. Yan Y, Xing T, Wang S, Daly TJ, Li N. Coupling Mixed-Mode Size Exclusion Chromatography with Native Mass Spectrometry for Sensitive Detection and Quantitation of Homodimer Impurities in Bispecific IgG. Anal Chem. 2019;91(17):11417–11424. doi:10.1021/acs.analchem.9b02793
120. Tustian AD, Endicott C, Adams B, Mattila J, Bak H. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs. 2016;8(4):828–838. doi:10.1080/19420862.2016.1160192
121. Lee HY, Contreras E, Register AC, et al. Development of a bioassay to detect T-cell-activating impurities for T-cell-dependent bispecific antibodies. Sci Rep. 2019;9(1):3900. doi:10.1038/s41598-019-40689-1
122. Pham NB, Meng WS. Protein aggregation and immunogenicity of biotherapeutics. Int J Pharm. 2020;585:119523. doi:10.1016/j.ijpharm.2020.119523
123. Lu RM, Hwang YC, Liu IJ, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1. doi:10.1186/s12929-019-0592-z
124. Wang N, Shang J, Jiang S, Du L. Subunit Vaccines Against Emerging Pathogenic Human Coronaviruses. Front Microbiol. 2020;11:298. doi:10.3389/fmicb.2020.00298
125. Du L, Zhao G, Chan CC, et al. Recombinant receptor-binding domain of SARS-CoV spike protein expressed in mammalian, insect and E. coli cells elicits potent neutralizing antibody and protective immunity. Virology. 2009;393(1):144–150. doi:10.1016/j.virol.2009.07.018
126. Alpizar A, Marino F, Ramos-Fernandez A, et al. A Molecular Basis for the Presentation of Phosphorylated Peptides by HLA-B Antigens. Mol Cell Proteomics. 2017;16(2):181–193. doi:10.1074/mcp.M116.063800
127. Nguyen TTK, Pham KY, Yook S. Engineered therapeutic proteins for sustained-release drug delivery systems. Acta Biomater. 2023;171:131–154. doi:10.1016/j.actbio.2023.09.018
128. Rahban M, Ahmad F, Piatyszek MA, Haertle T, Saso L, Saboury AA. Stabilization challenges and aggregation in protein-based therapeutics in the pharmaceutical industry. RSC Adv. 2023;13(51):35947–35963. doi:10.1039/D3RA06476J
129. Choi JS, Jang WS, Park JS. Comparison of adsorption and conjugation of Herceptin on poly(lactic-co-glycolic acid) nanoparticles - Effect on cell internalization in breast cancer cells. Mater Sci Eng C Mater Biol Appl. 2018;92:496–507. doi:10.1016/j.msec.2018.06.059
130. Chehelgerdi M, Chehelgerdi M, Allela OQB, et al. Progressing nanotechnology to improve targeted cancer treatment: overcoming hurdles in its clinical implementation. Mol Cancer. 2023;22(1):169. doi:10.1186/s12943-023-01865-0
131. Takakura Y, Takahashi Y. Strategies for persistent retention of macromolecules and nanoparticles in the blood circulation. J Contrele. 2022;350:486–493. doi:10.1016/j.jconrel.2022.05.063
132. van de Watering FC, Rijpkema M, Robillard M, Oyen WJ, Boerman OC. Pretargeted imaging and radioimmunotherapy of cancer using antibodies and bioorthogonal chemistry. Front Med Lausanne. 2014;1:44. doi:10.3389/fmed.2014.00044
133. Liu G, Dou S, Pretorius PH, et al. Tumor pretargeting in mice using MORF conjugated CC49 antibody and radiolabeled complimentary cMORF effector. Q J Nucl Med Mol Imaging. 2010;54(3):333–340.
134. Solomon BJ, Desai J, Rosenthal M, et al. A First-Time-In-Human Phase I Clinical Trial of Bispecific Antibody-Targeted, Paclitaxel-Packaged Bacterial Minicells. PLoS One. 2015;10(12):e0144559. doi:10.1371/journal.pone.0144559
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.