")
Back to Journals » International Journal of Nanomedicine » Volume 19
T Lymphocyte-Macrophage Hybrid Membrane-Coated Biomimetic Nanoparticles Alleviate Myocarditis via Suppressing Pyroptosis by Targeting Gene Silencing
Authors Xiong Y, Zhang Z, Liu S, Shen L, Zheng L, Ding L, Liu L, Wu L, Hu Z, Li L , Hu Z, Zhang Z, Zhou L, Xu M, Yao Y
Received 4 September 2024
Accepted for publication 22 November 2024
Published 29 November 2024 Volume 2024:19 Pages 12817—12833
DOI https://doi.org/10.2147/IJN.S487598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Yulong Xiong,1,2,* Zhenhao Zhang,1,2,* Shangyu Liu,1,2 Lishui Shen,1,2 Lihui Zheng,1,2 Ligang Ding,1,2 Limin Liu,1 Lingmin Wu,1 Zhicheng Hu,1 Le Li,1 Zhao Hu,1 Zhuxin Zhang,1 Likun Zhou,1 Mengtong Xu,1 Yan Yao1,2
1Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 2State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Yao, Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, 167A Beilishi Road, Xi Cheng District, Beijing, 100037, People’s Republic of China, Email [email protected]
Introduction: Nanomedicine coated with cell membranes has attracted increasing attention for its enhanced targeting capability and biocompatibility. Based on previous research, we identified interferon regulatory factor 1 (IRF1)-mediated macrophage pyroptosis as a potential therapeutic target for myocarditis. Herein, we fabricated an innovative immune cell membrane-coated zeolitic imidazolate framework-8 (ZIF-8) nano-delivery platform and explored its effects on myocarditis.
Methods: ZIF-8 nanoparticles loaded with siRNA targeting IRF1 (siIRF1) were coated with a T lymphocyte-macrophage hybrid membrane (siIRF1@ZIF@HM NPs) via sonication and extrusion. The morphological and biological characteristics of the nanoparticles were evaluated using transmission electron microscopy (TEM) and dynamic light scattering (DLS). Cellular cytotoxicity was assessed by a cell counting kit-8 assay. Cellular uptake and endo-lysosomal escape in M1-differentiated macrophages were visualized via fluorescence microscopy. The targeting specificity and anti-myocarditis effects were evaluated in an experimental autoimmune myocarditis (EAM) mouse model. The anti-pyroptosis effects were assessed by Western blot analysis both in vivo and in vitro.
Results: Transcriptional sequencing identified T lymphocytes and macrophages as suitable membrane sources. The ZIF-8 nanoparticles exhibited high siRNA loading capacity and pH responsiveness, enabling an efficient release of siIRF1 from endo-lysosomes to the cytoplasm in macrophages. The hybrid membrane coating enabled specific targeting of M1 macrophages both in vivo and in vitro. Furthermore, delivery of siIRF1 effectively suppressed IRF1 expression and inhibited pyroptosis in IFN-γ-stimulated macrophages. Intravenous injection of siIRF1@ZIF@HM NPs significantly alleviated myocarditis progression without evident side effects.
Conclusion: The siIRF1 nanotherapeutic approach shows potential for attenuating myocardial inflammation and mitigating myocarditis progression. Our study highlights the promise of this customized biomimetic nano-delivery system for treating inflammatory diseases.
Keywords: hybrid membrane, myocarditis, macrophage, pyroptosis, small interfering RNA, immunotherapy
Graphical Abstract:

Introduction
Myocarditis, characterized by acute inflammatory responses, chronic fibrosis, and cardiac dysfunction, is a pathogenic precursor to ventricular arrhythmias, heart failure, and dilated cardiomyopathy (DCM).1–3 Despite its increasing prevalence, current therapeutic efficacy remains unsatisfactory.4–7 This can be attributed to the elusive underlying mechanisms, which result in the lack of effective therapeutic targets to halt the inflammatory cascade in myocarditis. Additionally, systemic drug administration often yields suboptimal drug concentrations at the pathological sites in organ-specific diseases, reducing therapeutic potential and precipitating adverse effects.8 Consequently, there is an imperative demand for refined strategies that ensure precise targeting and localized intervention.
The strategic deployment of small interfering RNA (siRNA) for gene silencing marks a new era in therapeutic interventions for various diseases.9 However, the crux of siRNA therapy largely depends on identifying an effective target, as this determines the therapeutic efficacy. Pyroptosis is a highly inflammatory form of programmed cell death characterized by cell lysis and release of pro-inflammatory cytokines.10 Our previous investigations have highlighted the crucial role of pyroptosis in myocarditis development, which occurs primarily in pro-inflammatory macrophages (M1 macrophages).11 This process may be driven by interferon-γ (IFN-γ)-mediated upregulation of interferon regulatory factor 1 (IRF1), known for its role in modulating inflammation and cell death.12 Therefore, directly targeting IRF1 in M1 macrophages with siRNA (siIRF1) represents a promising therapeutic avenue for myocarditis treatment by inhibiting pyroptosis and interrupting the inflammatory cascade.
Although IRF1 has been identified as a potential therapeutic target for myocarditis, the practical application of siIRF1 faces significant challenges that hinder its in vivo delivery and efficacy. In the intricate in vivo environment, siIRF1 molecules encounter numerous obstacles, including degradation, phagocytosis, and unintended immune responses.13 Additionally, physiological barriers presented by the vascular endothelium, cellular membrane, and endo-lysosome require innovative delivery mechanisms to ensure that siRNA reaches the cytosol of target cells (M1 macrophages).14 Consequently, developing stable, effective nanocarriers capable of targeting M1 macrophages within the myocardium and promoting endo-lysosomal escape of siIRF1 into the cytosol remains a critical yet challenging goal in myocarditis treatment.
To address these challenges, innovative functionalized nano-delivery systems have emerged, with particular interest in biomimetic strategies for directing therapeutic agents to pathological sites, including atherosclerotic plaques,15 inflammation,16 and tumors.17 Cell membrane-based nanoplatforms derived from erythrocytes, platelets, immune cells, and cancer cells have been generated for diverse applications based on their intrinsic properties.18–21 In the context of myocarditis, while macrophages predominantly infiltrate the myocardium, our transcriptomic analysis revealed marked upregulation of chemokine (C-X-C motif) ligand 9/10/11 (CXCL9/10/11), which are pivotal in T cell recruitment via interacting with chemokine receptor CXCR3.22 Thus, we have engineered a novel biomimetic approach to precisely deliver siIRF1 to the myocardial inflammatory loci, specifically targeting the M1 macrophages. Zeolitic imidazolate framework-8 (ZIF-8) was designed as the core of the delivery system (denoted as siIRF1@ZIF) to achieve its substantial loading efficiency, superior biocompatibility, and pH-induced biodegradability aimed for endo-lysosomal escape.23,24 To enable targeting capability, we employed a hybrid membrane (HM) derived from EL4 T lymphocytes25 and RAW 264.7 macrophages26 to cloak the siIRF1@ZIF nanoparticles, fabricating biomimetic nanocarriers with a core-shell structure (referred to as siIRF1@ZIF@HM). This hybrid membrane coating on our nanoparticles facilitates cellular uptake by pro-inflammatory M1 macrophages and promotes preferential accumulation in myocarditis. Therapeutic investigations revealed that siIRF1 effectively attenuates IRF1 expression and consequently suppresses IFN-γ-mediated macrophage pyroptosis (Figure 1). In summary, this innovative nanotherapeutic strategy shows strong potential to mitigate myocarditis progression, offering a promising option for future drug development.
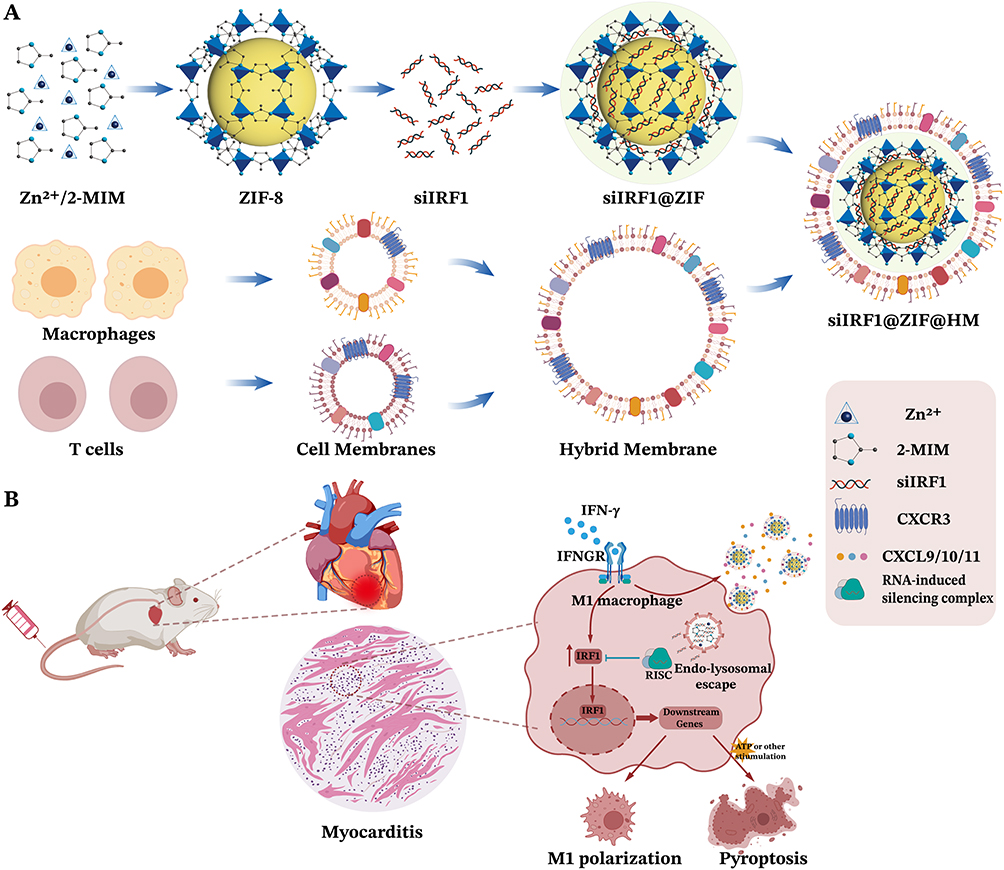 |
Figure 1 Schematic illustration of myocarditis treatment with T lymphocyte-macrophage hybrid membrane-coated siIRF1-loaded ZIF-8 nanoparticles (siIRF1@ZIF@HM NPs). (A) Preparation of siIRF1@ZIF@HM. ZIF-8 nanoparticles were first synthesized with Zn2+ and 2-methylimidazole (2-MIM) and loaded with siIRF1 (siIRF1@ZIF). The hybrid membrane (HM) derived from T cells and macrophages was then coated on the siIRF1@ZIF NPs. (B) Sketch of targeted therapy of myocarditis with siIRF1@ZIF@HM NPs by silencing IFN-γ-induced IRF1 upregulation and subsequent pyroptosis in macrophages. |
Materials and Methods
Materials
Zn(NO3)2·6H2O (99%), 2-methylimidazole (C4H6N2, 2-MIM, 99%), Complete Freund’s Adjuvant (CFA), Phorbol-12-myristate-13-acetate (PMA), and lipopolysaccharides (LPS) were purchased from Sigma-Aldrich (Shanghai, China). Cy5-labeled siRNA for IRF1 silencing (termed as siIRF1 in the study) were synthesized by GenePharma (Shanghai, China). Protease and phosphatase inhibitor cocktail, and adenosine triphosphate (ATP) were purchased from MedChemExpress (MCE, Monmouth Junction, NJ, USA). The interferon γ (IFN-γ) and M-CSF were purchased from PeproTech (USA). The DMEM medium, RPMI 1640 medium, fetal bovine serum (FBS), penicillin-streptomycin, TRIzol reagent, and bicinchoninic acid (BCA) assay were bought from Thermo Fisher Scientific (USA).
Isolation of Cell Membrane
Macrophage and T lymphocyte cell membranes were isolated with a modified protocol using Membrane Protein Extraction Kit.27 The mouse macrophage cell line, RAW264.7, and the mouse T lymphocyte cell line, EL4, were cultured and utilized as the sources of the cell membrane for hybrid membrane production. Cells were firstly suspended in an extraction buffer containing protease and phosphatase inhibitor cocktails and then subjected to an ice bath for 15 minutes. Homogenization on ice ensued until cell disruption reached 90%. After an initial centrifugation at 2,500 g for 10 min at 4 °C, the supernatant underwent a further centrifugation at 14,000 g for 30 minutes at 4 °C to procure cell membrane fragments. Membrane protein concentrations were quantified using a BCA assay. Equal protein quantities of T lymphocyte and macrophage membranes were sonicated on ice, followed by sequential extrusion through polycarbonate membranes with pore sizes of 1 μm, 400 nm, and 200 nm using a mini extruder (Avanti Polar Lipids) to synthesize the hybrid membrane (HM) vesicles.
Preparation of siIRF1@ZIF and siIRF1@ZIF@HM Nanoparticles
The ZIF-8 nanoparticles were synthesized following the literature with some modifications.28 Briefly, solution A was prepared by mixing 2-MIM (325 mg) in 10 mL of methanol and stirred for 30 min at room temperature. Solution B was prepared by dispersing Zn(NO3)2 (147 mg) in 5 mL of methanol and sonicated for 5 min. Subsequently, solution A was added dropwise to solution B and incubated at 37 °C with stirring for 30 min. The ZIF-8 nanoparticles were collected by centrifugation at 8,000 rpm for 5 min and washed with ultrapure water to remove the methanol. To fabricate the siIRF1@ZIF-8 nanoparticles, 25 nmol (330 μg) of Cy5-labeled siIRF1 was added to ZIF-8 (4.5 mg) nanoparticle solution and stirred overnight. Then, the nanoparticles were collected by centrifugation and washed three times with ultrapure water to obtain the siIRF1@ZIF nanoparticles. For membrane coating, the collected HM vesicles and siIRF1@ZIF nanoparticles were mixed and sonicated until the potential remained stable.29 The siIRF1@ZIF@HM nanoparticles were obtained by centrifuging the resulting products to remove the excess membrane fragments.
Measurement of siIRF1 Encapsulation Efficiency
The calibration curve of Cy5 fluorescence intensity versus the concentration of Cy5-labeled siIRF1 was obtained to determine the encapsulated siIRF1 in ZIF-8 nanoparticles (Figure S2C). The weight of total siIRF1 (WT), the weight of unencapsulated siIRF1 (WF), and the weight of nanomaterial (Wnano) were measured. The encapsulation efficiency and drug loading capacity of siIRF1 could be calculated according to the following formulae.
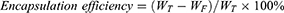
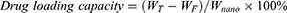
In vitro siIRF1 Release Study
To investigate the pH-responsive release profile of siIRF1 from ZIF-8 nanoparticles, a time-course experiment was performed at various pH levels (7.4, 6.0, 5.5). A total of 1 mL of the siIRF1@ZIF nanoparticles (with 264 μg siIRF1, 20 μM) was enclosed in a dialysis bag, which was then immersed in 20 mL of release medium buffered at the desired pH levels. At predetermined time points (0, 1, 2, 3, 4, 8, 12, 24, 36, and 48 h), 1 mL aliquots of the medium were sampled and immediately replenished with an equal volume of fresh PBS adjusted to the same pH level. The amount of siIRF1 released at each time point was quantified by the spectroscopic absorbance.
Isolation of Bone Marrow-Derived Macrophages (BMDMs)
Mouse BMDMs were isolated from wild-type Balb/c mice as described previously with modifications.30 In general, femurs and tibias were excised from euthanized mice, and bone marrow cells were flushed out using precooled PBS. Bone marrow cells were filtered through a 40-μm cell strainer, collected, and washed twice before cultured in complete RPMI 1640 medium in the presence of 40 ng/mL M-CSF. BMDMs were used on day 7 for subsequent experiments.
Cell Culture and Treatments
Cells were maintained at 37 °C, 5% CO2 in a humidified tissue culture incubator. The mouse macrophage cell lines, RAW264.7 and J774A.1, and the mouse T lymphocyte cell line, EL4, were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China). The cells were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin. The EL4 T lymphocytes were stimulated with 50 ng/mL PMA for 48 h to increase the expression of CXCR3 before membrane isolation. To induce M1 macrophage differentiation, RAW264.7 macrophages and BMDMs were treated with 100 ng/mL LPS and 50 ng/mL IFN-γ for 48 h. To induce macrophage pyroptosis, J774A.1 macrophages and BMDMs were incubated with 100 ng/mL IFN-γ for 48 h and subsequently stimulated with 5 mM ATP for another 1 h. Cell lysates and supernatants were collected respectively for further analysis.
Cell Viability Assay
The cell counting kit-8 (CCK-8 kit, MCE) was used to determine the effect of ZIF-8 on macrophage viability. RAW264.7 cells were seeded into 96-well plates and then treated with various concentrations of ZIF-8 for 12, 24, and 36 hours. Subsequently, 10 µL of CCK-8 reagent was added to each well and incubated for 4 h. The absorbance of each well was measured at 450 nm using a Multiskan Spectrum (Thermo Fisher Scientific) microplate reader.
Evaluation of Cellular Uptake
The cellular uptake behavior of siIRF1@ZIF NPs and siIRF1@ZIF@HM NPs was investigated by confocal laser scanning microscopy. RAW 264.7 macrophages and BMDMs were seeded on glass coverslips in 12-well plates. Subsequently, half of the wells were subjected to 100 ng/mL LPS and 50 ng/mL IFN-γ for 48 h to induce M1 macrophage differentiation. After being washed with PBS, M0 and M1 macrophages were incubated with 40 μg/mL siIRF1@ZIF NPs or siIRF1@ZIF@HM NPs at 37 °C for 4 h. The macrophages were then washed with ice-cold PBS and fixed in 4% paraformaldehyde solution for 20 min. Subsequently, the cells were permeabilized with 0.1% Triton X-100 and blocked with normal blocking goat serum solution (ZLI-9056, ZSGB-BIO, Beijing, China). After blocking, the cells were incubated with CD68 or iNOS primary antibodies overnight at 4 °C, followed by PBS washing and incubation with secondary antibodies at room temperature for 1 h. The cell nuclei were counterstained with DAPI and examined using a fluorescence microscope. The mean fluorescence intensity was quantified using Image J software version 1.5.1.
SDS-PAGE Assay
The protein profile of the hybrid membrane on siIRF1@ZIF@HM was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The extracted membranes and siIRF1@ZIF@HM were lysed with RIPA lysis buffer containing protease inhibitor cocktail on ice for 30 min followed by centrifugation at 13,000 rpm at 4 °C for 5 min. The supernatants were collected, and the protein concentrations were measured by BCA assay. An equivalent amount of protein per sample was mixed with SDS-PAGE loading buffer and heated at 95 °C for 10 min. Samples were equally loaded into the wells of 10% SDS-PAGE gel for electrophoresis analysis. Subsequently, proteins were stained with Coomassie blue fast staining solution (Beyotime) and washed with deionized water three times for 10 min.
Western Blot Analysis
Heart tissues and collected cells were lysed in RIPA lysis/extraction buffer (Beyotime) with protease/phosphatase inhibitor cocktails. Heart tissues were then homogenized by tissue homogenizer. The supernatants were obtained by centrifugation at 12,000 rpm/min for 20 min at 4 °C. Protein concentration was determined via BCA assay. An equivalent amount of protein of each sample was fractionated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore). After blocking in 5% non-fat milk for 2 h, the membranes were washed with TBS-T (TBS with 0.1% Tween 20, pH 8.0) and incubated with the following primary antibodies at 4 °C overnight, including Caspase-1 (1:1000), GSDMD (1:1000), IL-1β (1:1000), CXCL9 (1:1000), IRF1 (1:1000), ITGA4 (1:1000), ITGB1 (1:1000), TCR-β (1:1000), and GAPDH (1:10,000). The membranes were then incubated with the corresponding secondary antibodies Goat-anti-Rabbit IgG (HRP) or Goat-anti-mouse IgG (HRP) (Proteintech) for 1 h at room temperature. After washing 3 times with TBS-T, the immunoreactive bands were visualized using an enhanced chemiluminescence (ECL) kit (Millipore). The gray values of the bands were quantified with ImageJ software 1.5.1.
Reverse Transcription and Real-Time Quantitative (q) PCR
Total RNA was extracted from heart tissues and cultured cells using TRIzol reagent (Thermo Fisher Scientific). Reverse transcription was performed with the GoScriptTM Reverse Transcription System (A3500, Promega, USA). Realtime qPCR was carried out using GoTaq® qPCR and RT-qPCR Systems (A6002, Promega, USA). The expression levels of the target genes were normalized to the housekeeping gene GAPDH using the 2−ΔΔCT method. The sequences of qPCR primers are listed in Table S1.
In vivo Pharmacokinetics and Targeting Study
Animal experiments were approved by the Animal Ethics Committee of Fuwai Hospital, and the welfare of the laboratory animals was ensured in accordance with the Accreditation Criteria for the Quality and Competence of Laboratory Animal Institutions for Care and Using (CNAS-CL06). Six-week-old male Balb/c mice (~20 g weight) were purchased from Vital River Laboratories (Beijing, China). The mice were maintained in a specific pathogen-free (SPF) environment and provided with free access to food and water. To assess the in vivo pharmacokinetics, the same volume of PBS (n=3), siIRF1@ZIF NPs (n=3, 10 mg/kg, 0.73 mg/kg siIRF1), and siIRF1@ZIF@HM NPs (n=3, 10 mg/kg, 0.73 mg/kg siIRF1) were injected into Balb/c mice through the tail vein. At predefined time points, 100 μL of blood samples were collected. The fluorescence intensities were measured using a Multiskan Spectrum (Thermo Fisher Scientific) microplate reader. EAM was induced via subcutaneous injection with 250ug αMHC (Ac-RSLKLMATLFSTYASADR-OH) peptide emulsified with CFA containing 1mg/mL heat-killed Mycobacterium tuberculosis H37Ra (Sigma Aldrich, 1:1, v/v) on days 0 and 7. To study the targeting capability toward myocarditis, the EAM mice were randomly divided into 3 groups on day 21: PBS (n=3), siIRF1@ZIF NPs (n=3, 10 mg/kg), and siIRF1@ZIF@HM NPs (n=3, 10 mg/kg). At predefined intervals post-intravenous injection, mice were euthanized, and major organs, including the heart, liver, spleen, lungs, and kidneys, were harvested for imaging and fluorescence quantification with an in vivo imaging system (InVivo Smart-LF, VISQUE).
In vivo Therapeutic Efficacy Study
Six-week-old Balb/c mice were randomly divided into 5 groups: Control, EAM + PBS (termed as EAM group), EAM + free siIRF1 (0.73 mg/kg), EAM + siIRF1@ZIF NPs (10 mg/kg), EAM + siIRF1@ZIF@HM NPs (10 mg/kg). Mice were treated via tail vein injection on day 7 and every three days thereafter (5 times in total). The body weight of mice was recorded every three days. Mice were euthanized at the acute inflammatory phase (day 21) and the organs were harvested and embedded in OCT. Samples were sliced into 8 μm thick cryosections and stained with hematoxylin-eosin (HE) to evaluate the severity of inflammation. The myocardial inflammation score was determined by HE microscopic approximation of the percent area of myocardium infiltrated with mononuclear cells from five sections per heart based on the scoring system in Table S2. In addition, the heart sections were stained with CD68 and iNOS antibodies for immunofluorescence analysis. Further, the myocardial injury, pyroptosis intensity, and inflammatory cytokines were also measured to evaluate the therapeutic efficacy.
Statistical Analysis
The statistical analysis was conducted using GraphPad Prism 9 software, and all results were presented as mean ± standard error of the mean (s.e.m). The Student’s unpaired t-test was employed to compare two independent groups, while separate t-tests were conducted for experiments comparing multiple time points. One-way ANOVA followed by Tukey’s post-hoc test was applied for comparisons involving three or more groups. All tests were two-tailed, with a significance level set at P < 0.05.
Results and Discussion
Preparation and Characterization of the siIRF1-Loaded Nanoparticles
To simulate the potential mechanisms driving the migration of immune cells to the myocardium, transcriptional sequencing analysis of heart tissues was conducted. KEGG pathway analysis revealed significant enrichment in the cell adhesion molecules and the chemokine signaling pathway (Figure S1A). The heatmap of the identified differential chemokines predominantly highlighted the upregulation of CXCL9/10/11 (Figure S1B), indicating a potential mechanism for immune cell infiltration in myocarditis. Notably, CXCL9/10/11 interact with the chemokine receptor CXCR3, which is primarily expressed on T lymphocytes and macrophages.31–33 Moreover, a previous study demonstrated that macrophages constituted the primary immune cell population in myocarditis, as shown by single-cell sequencing.34 Consequently, to maximize targeting capability, a hybrid membrane (HM) derived from RAW 264.7 macrophages and activated EL4 T lymphocytes was used to fabricate biomimetic nanocarriers for the targeted therapy of myocarditis. Confocal fluorescence imaging was performed to observe the fusion of macrophage and T cell membranes stained with Dio and Dil, respectively (Figure S1C). The fused hybrid membrane exhibited significant colocalization of fluorescence signals, with stronger green fluorescence observed as the proportion of macrophage membrane increased. To ensure maximal expression of CXCR3, PMA was utilized to activate the EL4 T lymphocytes before membrane isolation35 (Figure S1D).
The siIRF1@ZIF@HM nanoparticles were synthesized through a multi-step procedure, modified from previous studies.21,36 Initially, the ZIF-8 nanoparticles (ZIF-8 NPs) were precipitated via the dropwise addition of a zinc ion solution to a vigorously stirred 2-methylimidazole (2-MIM) solution. The siIRF1 labeled with cyanine 5 NHS ester fluorescent dye (Cy5) was then mixed with the prepared ZIF-8 NPs in an aqueous medium and stirred overnight to produce the siIRF1@ZIF nanoparticles (siIRF1@ZIF NPs). Subsequently, these siIRF1@ZIF NPs were coated with hybrid membrane vesicles (Figure 2A) for biomimetic modification. Transmission electron microscopy (TEM) showed a rhombic shape of siIRF1@ZIF NPs with an average size of 113.8 ± 1.5 nm (Figure 2B), while an additional dim layer (17.71 ± 3.1 nm) was observed around the siIRF1@ZIF@HM NPs after HM coating (Figure 2C). Element mapping confirmed the uniform distribution of C, N, O, Zn, and P elements within the nanoparticles (Figure 2D). Dynamic light scattering (DLS) analysis measured the hydrodynamic sizes of ZIF-8, siIRF1@ZIF, and siIRF1@ZIF@HM NPs as approximately 110.7 ± 2.8 nm, 120.3 ± 1.4 nm, and 132.9 ± 1.8 nm, respectively, consistent with the TEM results (Figure 2E). Zeta potential analysis showed a decrease from 9.53 ± 0.88 mV for ZIF-8 NPs to −20.93 ± 0.91 mV after siIRF1 encapsulation, with a further change to −12.57 ± 1.18 mV after HM modification (Figure 2F). Meanwhile, UV-vis absorbance spectra presented a siIRF1-specific absorption peak at approximately 650 nm due to Cy5 conjugation, which was not observed in pure ZIF-8 (Figure 2G). These results confirmed the successful encapsulation of siIRF1, as well as the HM coating on the surface of siIRF1@ZIF NPs. Moreover, the stability tests in both PBS and 30% FBS buffers showed that the nanoparticles maintained consistent hydrodynamic diameters and morphologies without significant aggregation or degradation (Figure S2A and B).
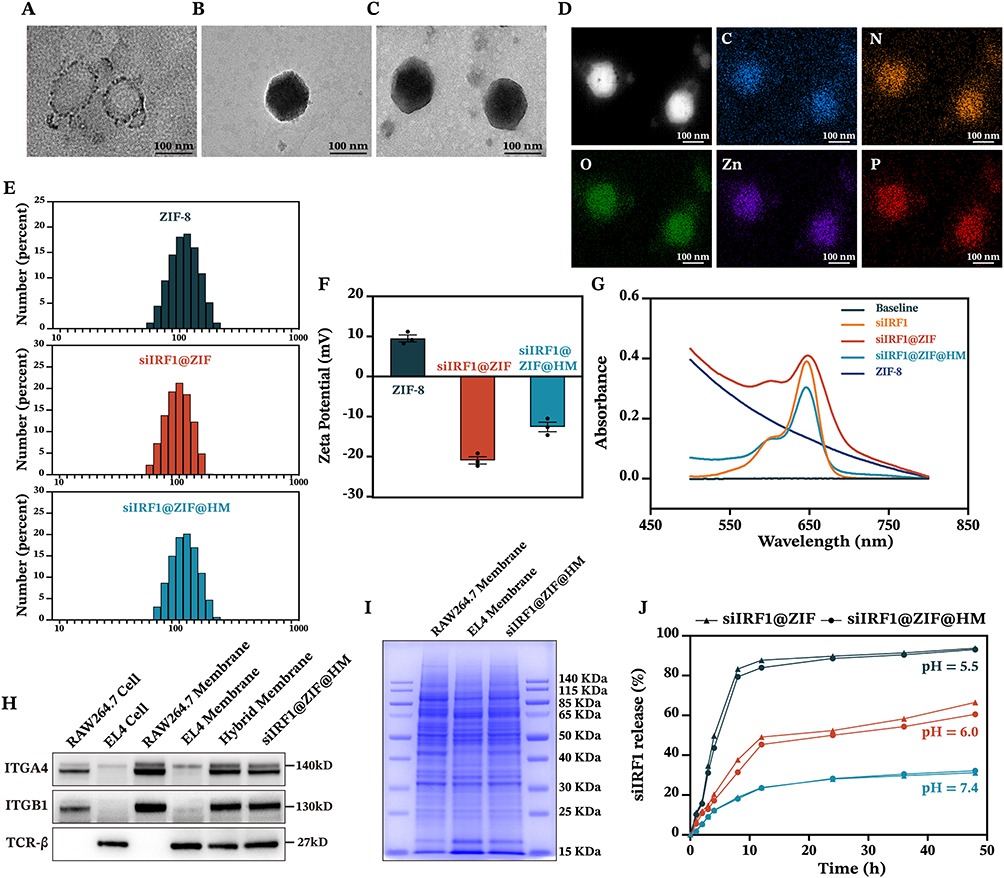 |
Figure 2 Characterization of siIRF1-loaded nanoparticles. (A–C) TEM images of hybrid membrane vesicles, siIRF1@ZIF NPs, and siIRF1@ZIF@HM NPs. (D) Element mapping of siIRF1@ZIF@HM NPs. (E and F) Hydrodynamic diameter and ζ potential of ZIF-8 NPs, siIRF1@ZIF NPs, and siIRF1@ZIF@HM NPs. (G) UV-vis absorbance spectra of pure siIRF1, ZIF-8 NPs, siIRF1@ZIF NPs, and siIRF1@ZIF@HM NPs. (H) Western blot analysis of typical markers of macrophages and T cells. (I) SDS-PAGE analysis of protein profiles in macrophage membrane, T cell membrane, and siIRF1@ZIF@HM NPs. (J) Release curves of siIRF1 from siIRF1@ZIF NPs and siIRF1@ZIF@HM NPs. Scale bars: 100 nm. |
To confirm the functionalization of siIRF1@ZIF with HM, Western blot analysis revealed that typical markers of macrophages (ITGA4 and ITGB1)37 and T lymphocytes (TCR-β38) were successfully transferred from cell membranes to the nanoparticles (Figure 2H). Furthermore, SDS-PAGE analysis indicated that siIRF1@ZIF@HM NPs exhibited protein profiles of both T cells and macrophages (Figure 2I). The Zn2+ ions form acid noncovalent bonds with 2-MIM, leading to pH-responsive biodegradation of the ZIF-8 nanoparticles.39 As illustrated in Figure 2J, the siIRF1@ZIF NPs and siIRF1@ZIF@HM NPs demonstrated a pH-dependent release of siIRF1 in the acidic milieu (pH 5.5 and 6.0), while physiological condition (pH 7.4) resulted in a minimal release. The loading efficiency of siIRF1 was calculated to be 99.8% according to the calibration curve shown in Figure S2C, with a drug loading capacity (DLC) of 7.3%.
Cellular Uptake and Endo-Lysosomal Escape of siIRF1@ZIF@HM Nanoparticles
We have identified M1 macrophages as the target cell type due to their pathological and pyroptotic properties in myocarditis. Consequently, we sought to explore the targeting capability of siIRF1@ZIF@HM NPs towards M1 macrophages in vitro. ZIF-8 has been reported to promote the generation of reactive oxygen species (ROS), which may pose a potential risk to healthy cells.24 Therefore, nanoparticle cytotoxicity on mouse macrophages was first evaluated using the CCK-8 assay. Cell viability remained stable at concentrations up to 40 μg/mL (Figure S3A–D), indicating their good biocompatibility. After inducing M1 macrophage polarization, M0 (unpolarized) and M1 macrophages were incubated with 40 μg/mL siIRF1@ZIF NPs or siIRF1@ZIF@HM NPs for 4 h. The Cy5 fluorescent dye enabled monitoring of cellular uptake, while immunofluorescence staining of CD68 or iNOS confirmed macrophage polarization status. As depicted in Figure 3A–C, M0 macrophages treated with either type of nanoparticles displayed minimal mean fluorescence intensity (MFI), with negligible differences between groups. In contrast, M1 macrophages exposed to siIRF1@ZIF NPs exhibited a relatively higher MFI compared to M0 macrophages, implying an enhanced endocytic activity in M1 macrophages. However, M1 macrophages treated with siIRF1@ZIF@HM NPs showed a significantly elevated MFI, in stark contrast to other groups, suggesting a preferential uptake of siIRF1@ZIF@HM NPs by M1 macrophages. These findings, consistent in both RAW 264.7 macrophages and bone marrow-derived macrophages (BMDMs), demonstrated the specific targeting capability of siIRF1@ZIF@HM NPs towards M1 macrophages, attributed to the selective uptake rather than a generalized increase in endocytosis.
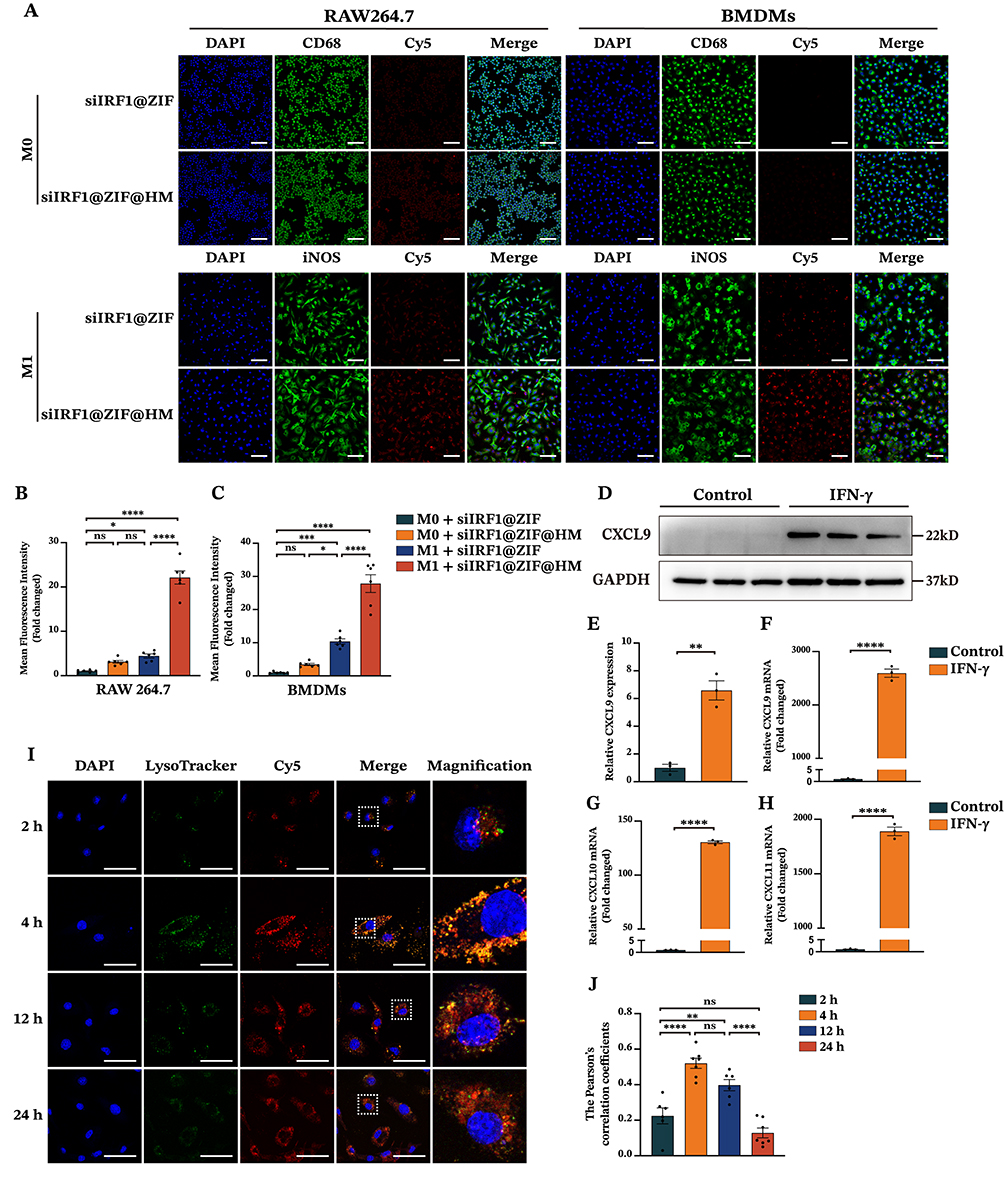 |
Figure 3 Cellular uptake and endo-lysosomal escape of siIRF1-loaded nanoparticles in macrophages. (A) Confocal fluorescence images of macrophages for cellular uptake detection. RAW 264.7 macrophages and BMDMs were pretreated with or without LPS (100 ng/mL) and IFN-γ (50 ng/mL) for 48 h, followed by incubation with 40 μg/mL of siIRF1@ZIF NPs or siIRF1@ZIF@HM NPs for 4 h. CD68 and iNOS were visualized by immunofluorescence staining, and siIRF1 was tracked with Cy5 fluorescence labeling. (B and C) Quantifications of mean fluorescence intensity. (D and E) Western blot analysis of CXCL9 expression in RAW 264.7 macrophages treated with IFN-γ (50 μg/mL). (F–H) The mRNA expressions of CXCL9, CXCL10, and CXCL11 in IFN-γ (50 μg/mL) stimulated RAW 264.7 macrophages. (I and J) Endo-lysosomal escape of siIRF1@ZIF@HM NPs in M1 macrophages. M1-polarized RAW 264.7 macrophages were incubated with siIRF1@ZIF@HM NPs for 2 h, 4 h, 12 h, and 24 h, followed by staining with 50 nM Lyso-Tracker Green for 10 min and subsequently with 1× Hoechst 33342 for 10 min. Data were presented as mean ± SEM (n ≥ 3). Scale bar: 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviation: ns, not significant. |
M1 macrophages are commonly induced by IFN-γ stimulation, which has also been reported to promote the expressions of CXCL9/10/11 in various cell types, including macrophages.22,40 Using Western blot and qPCR, we verified a substantial increase in these chemokine levels in macrophages upon IFN-γ stimulation (Figure 3D–H). To further investigate the mechanisms underlying the specific targeting, the CXCL9-CXCR3 axis was inhibited. Pre-blockade of CXCR3 on the hybrid membrane or CXCL9 of BMDMs reduced the uptake of siIRF1@ZIF@HM NPs by M1-polarized macrophages (Figure S3E and F). These results suggest that the enhanced targeting capability of the HM coating on nanoparticles may be attributed to the interaction between the CXCL9/10/11 chemokines and the CXCR3 receptor.
Subsequently, we proceeded to investigate the release of siIRF1 from endo-lysosomal compartments, a critical step for effective gene silencing by siRNA within the cytoplasm.41 To visualize the dynamic escape of siIRF1, M1 macrophages were incubated with 40 μg/mL siIRF1@ZIF@HM NPs for various time periods (2 h, 4 h, 12 h, and 24 h). Confocal microscopy was utilized to capture colocalization images of siIRF1 and lysosomes, labeled with the Cy5 and Lyso-Tracker Green, respectively (experimental procedures in the supporting information). As shown in Figure 3I and J, the signals of Cy5 and Lyso-Tracker were separated after 2 h incubation, representing the initial stage of endocytosis. After 4 h incubation, a significant overlay of the Cy5 and Lyso-Tracker signals was observed, indicating the entry of siIRF1 into lysosomes. The Cy5 signal became separate from the Lyso-Tracker at 12 h, which was more evident at 24 h, reflecting the release of siIRF1 into the cytosol. Mean Pearson’s correlation coefficients of Cy5 and Lyso-Tracker signals were 0.22, 0.52, 0.39, and 0.13 at 2 h, 4 h, 12 h, and 24 h, respectively. Overall, these data indicated that the siIRF1 was successfully discharged into the cytosol as a result of the pH-responsive dissociation of the MOF scaffold, thereby facilitating the endo-lysosomal disruption.23
Therapeutic Effects of siIRF1-Loaded Nanoparticles in vitro
Our previous research has demonstrated that M1 macrophage pyroptosis is a pivotal factor in the pathogenesis of myocarditis.11 IFN-γ, a key inducer of M1 differentiation, is implicated in the lytic programmed cell death, including pyroptosis and PANoptosis, potentially through the IFN-γ-induced upregulation of IRF1.42–44 To validate the anti-pyroptotic effects of siIRF1-loaded nanoparticles in vitro, we first investigated their cytotoxicity and gene silencing efficiency in mouse J774A.1 macrophages (Figure S4A–D, experimental procedures in the supporting information). Subsequently, mRNA levels of IRF1 and its downstream genes, including Caspase-1, ZBP1, and GBP1, were determined by qPCR to assess the gene silencing capability of the siIRF1@ZIF NPs and siIRF1@ZIF@HM NPs. As a result, treatment with siIRF1@ZIF@HM NPs significantly reduced the mRNA expression of IRF1 and its downstream genes, whereas siIRF1@ZIF NPs showed less pronounced gene silencing ability due to lower uptake (Figure 4A–4D). These findings indicated a high gene silencing efficiency of the siIRF1 in this study.
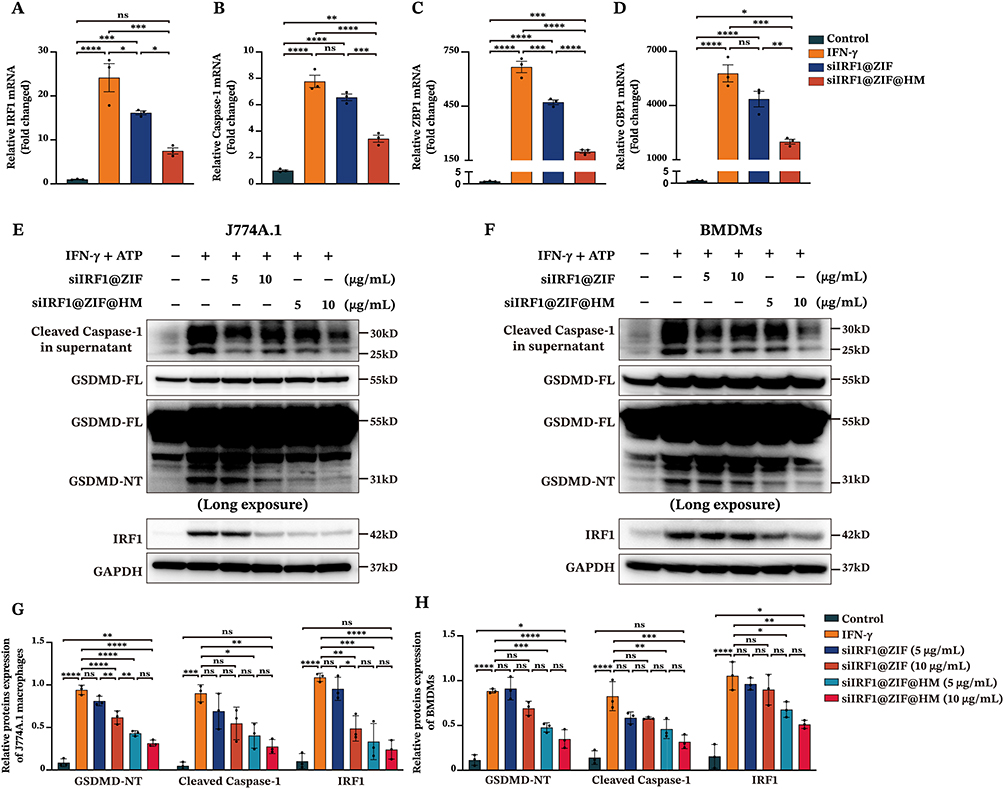 |
Figure 4 In vitro therapeutic effects of siIRF1-loaded nanoparticles on macrophage pyroptosis. (A–D) The mRNA levels of IRF1, Caspase-1, ZBP1, and GBP1 in J774A.1 macrophages determined by qPCR. The J774A.1 macrophages were stimulated with 50 ng/mL IFN-γ to induce IRF1 upregulation, simultaneously incubated with 10 μg/mL siIRF1@ZIF NPs or 10 μg/mL siIRF1@ZIF@HM NPs. (E and F) The expression levels of pyroptosis-related proteins in J774A.1 macrophages and BMDMs (100 ng/mL IFN-γ for 48 h followed by 5 mM ATP for 1 h) measured by Western blotting. Cells were co-incubated with siIRF1@ZIF NPs or siIRF1@ZIF@HM NPs (5 μg/mL or 10 μg/mL). (G and H) Quantifications of protein levels in the supernatants and cell lysates. Data were presented as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviation: ns, not significant. |
Subsequently, the anti-pyroptotic effects of the nanoparticles were determined in both J774A.1 macrophages and BMDMs subjected to pyroptosis induction via IFN-γ incubation followed by ATP stimulation. As shown in Figure 4E–H, the siIRF1@ZIF@HM NPs significantly inhibited the expression of IRF1 expression upon IFN-γ stimulation, thereby suppressing the pyroptosis in both macrophages, as evidenced by the decreased levels of the N-terminal of GSDMD (GSDMD-NT) and Cleaved Caspase-1 (the purification procedures detailed in the supporting information). Notably, both low and high concentrations (5 μg/mL and 10 μg/mL) displayed high therapeutic efficiency with no obvious differences between the two groups. In contrast, the siIRF1@ZIF NPs exhibited relatively lower anti-pyroptotic capacity, achieving noticeable effects only at a high concentration (10 μg/mL) in J774A.1 macrophages, with diminished effects in BMDMs. These findings demonstrated that targeting IRF1 in M1 macrophages with siIRF1@ZIF@HM NPs is an effective approach to inhibit IFN-γ-mediated macrophage pyroptosis in vitro.
In vivo Pharmacokinetics and Targeting Profiles of siIRF1-Loaded Nanoparticles
Our in vitro findings confirmed that siIRF1@ZIF@HM NPs effectively target pro-inflammatory M1 macrophages and attenuate IFN-γ-induced pyroptosis. To investigate these effects in vivo, we assessed the circulation time of siIRF1@ZIF NPs and siIRF1@ZIF@HM NPs after intravenous injection. Serial blood samples were collected at designated intervals, and the pharmacokinetic evaluation was performed by measuring fluorescence intensity (Figure 5A). Notably, the clearance rates of siIRF1@ZIF@HM NPs showed no significant difference compared to those of siIRF1@ZIF NPs. Subsequently, an experimental autoimmune myocarditis (EAM) mouse model was used to evaluate the targeting efficacy of the nanoparticles toward inflamed myocardium and their overall biodistribution. Consistent with the results of transcriptional sequencing, mRNA levels of chemokines CXCL9, CXCL10, and CXCL11 were markedly upregulated in EAM cardiac tissues (Figure 5B). Following intravenous administration of PBS, siIRF1@ZIF NPs, and siIRF1@ZIF@HM NPs to EAM mice, major organs (heart, liver, spleen, lung, and kidney) were harvested at 4 h, 12 h, and 24 h post-injection for analysis. Ex vivo imaging revealed pronounced cardiac fluorescence intensity in the siIRF1@ZIF@HM NP-treated group, evident at both gross and sectional levels at all time points (Figure 5C–E). Quantitative biodistribution assessments of the nanoparticles in major organs are presented in Figure S5A–C. Intriguingly, both nanoparticles predominantly accumulated in the kidneys rather than the liver. Specifically, kidneys from mice receiving siIRF1@ZIF NPs showed a peak in fluorescence intensity at 4 h, which diminished over time. In contrast, the siIRF1@ZIF@HM NPs group exhibited a relatively stable renal fluorescence, implying initial preferential cardiac retention followed by renal elimination. Immunofluorescence staining was performed to evaluate nanoparticles targeting to M1 macrophages. Fluorescence imaging of cardiac cryosections revealed that siIRF1@ZIF@HM NPs accumulated within M1 macrophages marked by CD68 and iNOS expression (the immunofluorescence procedures detailed in the supporting information). In comparison, the siIRF1@ZIF NPs group displayed a less intense and non-specific signal (Figure 5F). Furthermore, the fluorescence of Cy5-labeled siIRF1 was also observed in other organs with confocal microscopy (Figure S5D), which coincided with the ex vivo imaging results. These data indicated that the HM modification substantially enhances the targeting precision of siIRF1@ZIF@HM NPs to the inflammatory myocardium, particularly favoring M1 macrophages in vivo.
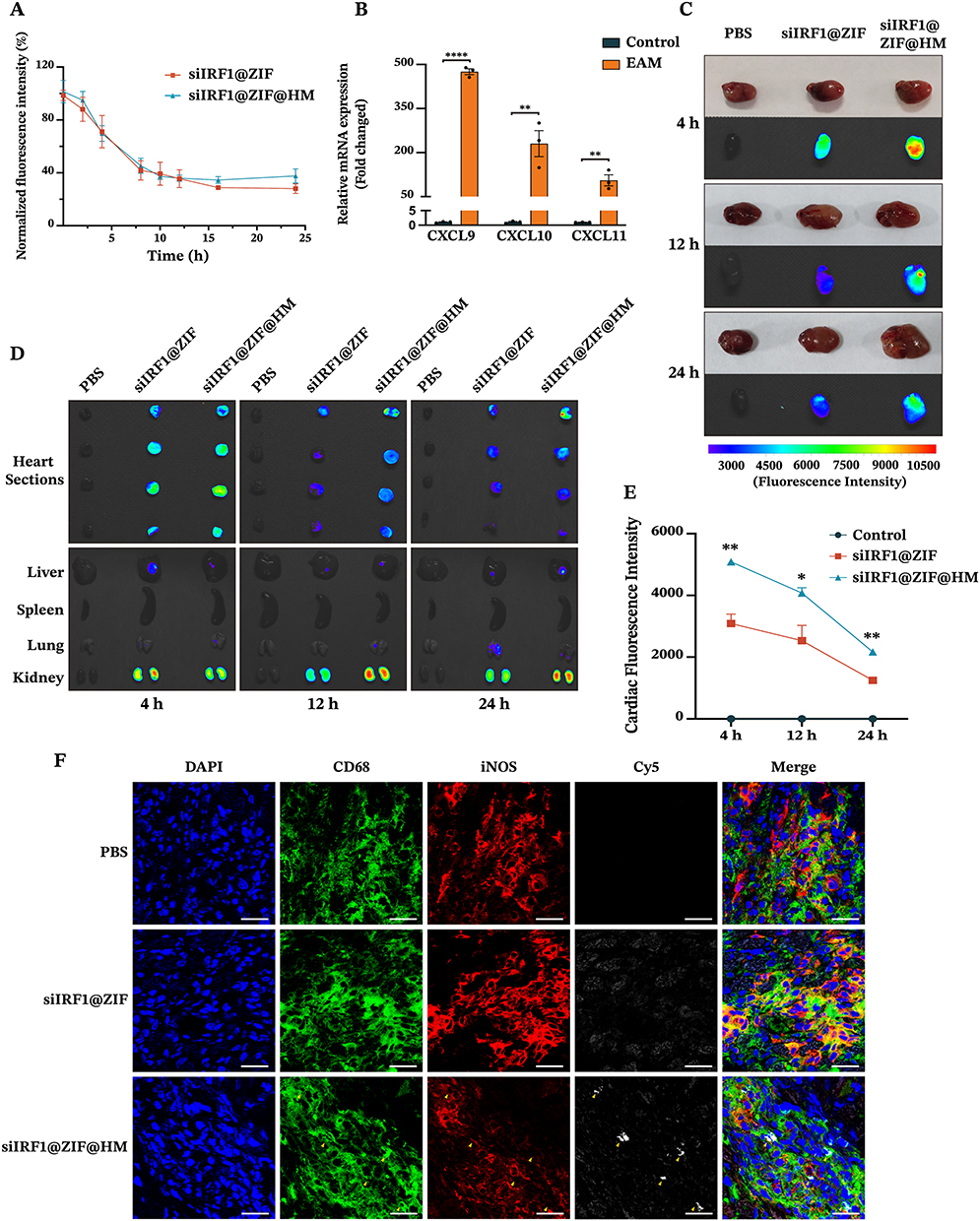 |
Figure 5 In vivo pharmacokinetics and targeting capability of siIRF1@ZIF@HM NPs in EAM mice after intravenous injection. (A) In vivo circulation of siIRF1@ZIF NPs (10 mg/kg) and siIRF1@ZIF@HM NPs (10 mg/kg) in Balb/c mice. Blood samples were collected at various time points after intravenous injection and the fluorescence intensities were quantified. (B) The mRNA levels of CXCL9, CXCL10, and CXCL11 in EAM heart tissues. (C–E) Ex vivo fluorescence images and quantitative analysis of Cy5 fluorescence signal in the heart and other organs. On day 21 post-EAM induction, mice were intravenously injected with PBS (Negative Control), siIRF1@ZIF NPs (10 mg/kg), and siIRF1@ZIF@HM NPs (10 mg/kg). At 4 h, 12 h, and 24 h after administration, mice were euthanized, and major organs were harvested for ex vivo imaging. (F) Immunofluorescence staining analysis of colocalization of Cy5-labeled siIRF1 with CD68+ iNOS+ macrophages in cardiac cryosections. Data were presented as mean ± SEM (n = 3). Scale bar: 25 μm. *P < 0.05, **P < 0.01, ****P < 0.0001. Abbreviation: ns, not significant. |
Therapeutic Efficacy and Biosafety of siIRF1 Nanotherapies in EAM Mice
The therapeutic effects of the nanoparticles against pyroptosis and myocarditis were subsequently evaluated. EAM mice were randomly allocated into five groups. On day 7 post-EAM induction, each group received intravenous injections of either PBS, free siIRF1, siIRF1@ZIF NPs, or siIRF1@ZIF@HM NPs, at a dosage of 0.73 mg/kg siIRF1 per administration for five times (Figure S6). On day 21, corresponding to the acute phase of EAM, major organs were harvested for analysis. Compared with the control group, hematoxylin and eosin (HE) staining of myocardial sections revealed extensive inflammation in the EAM group, characterized by leukocyte infiltration exceeding 50% of the cardiac section (Figure 6A–C). The groups treated with siIRF1 and siIRF1@ZIF NPs exhibited a reduction in inflammation scores and ratios, although the differences were not statistically significant. In contrast, the administration of siIRF1@ZIF@HM NPs substantially reduced cardiac inflammation. Additionally, EAM induction notably increased the serum levels of CK-MB, which were effectively suppressed by siIRF1@ZIF@HM NPs treatment, thereby alleviating myocardial injury (Figure 6D). Furthermore, the EAM group showed significant upregulation in the transcriptional levels of IRF1 and pro-inflammatory cytokines IL-1β, IL-6, and TNF-α, primarily produced by M1 polarized macrophages.45 Consistent with the histological findings, the siIRF1@ZIF NPs group showed a downward trend in the expression of IRF1 and these cytokines, while siIRF1@ZIF@HM NPs significantly decreased their mRNA levels (Figure 6E–H). These findings demonstrated that siIRF1@ZIF@HM NPs effectively silenced cardiac IRF1 expression, thereby mitigating the inflammatory responses in myocarditis.
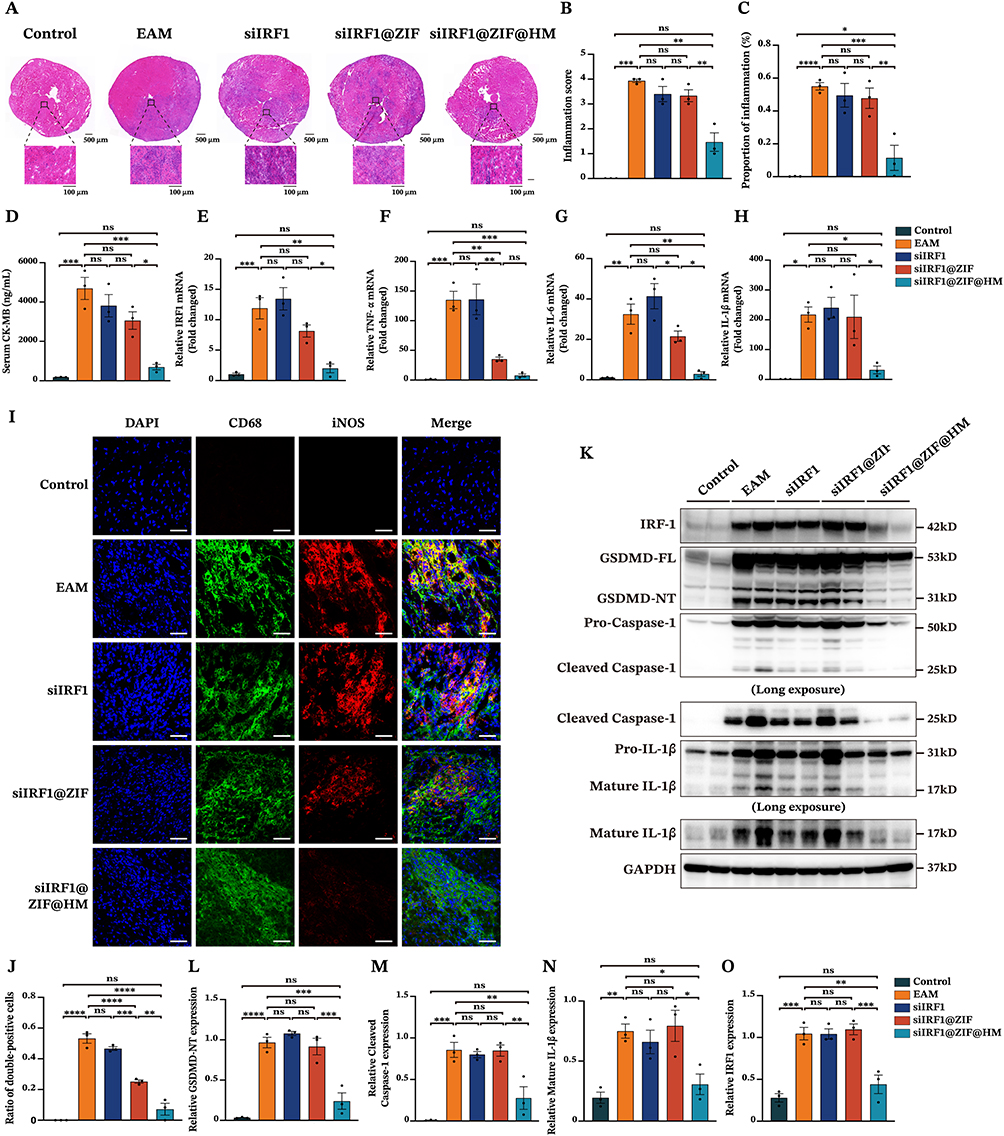 |
Figure 6 Therapeutic efficacy of siIRF1@ZIF@HM NPs in EAM mice. (A) Representative images of hematoxylin and eosin staining of myocardial sections from different groups of mice (Control, EAM + PBS, EAM + free siIRF1, EAM + siIRF1@ZIF NPs, and EAM + siIRF1@ZIF@HM NPs). (B and C) Quantitative analysis of histoscore and proportion of inflammation in heart sections. (D) Serum levels of CK-MB. (E–H) The cardiac mRNA expressions of IRF1 and pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) analyzed by qPCR. (I and J) Representative coimmunostaining of CD68 (green) and iNOS (red). (K–O) The cardiac expressions of pyroptosis-related proteins in various groups determined by Western blotting. Data were presented as mean ± SEM (n = 3). Scale bar: 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviation: ns, not significant. |
To elucidate the therapeutic mechanisms, we performed immunofluorescence staining and Western blot analysis. The cardiac cryosections from each group were stained with CD68 and iNOS antibodies to assess macrophage polarization within myocarditis. As depicted in Figure 6I and J, the EAM and siIRF1 groups showed substantial colocalization of CD68 and iNOS at the inflammatory loci. In contrast, macrophages from the siIRF1@ZIF NPs group exhibited decreased double-positive staining, while the iNOS signal was notably diminished in the siIRF1@ZIF@HM NPs group, indicating inhibited macrophage polarization. In addition, Western blot analysis revealed significant activation of pyroptosis-associated proteins in the EAM group, indicated by cleavage of GSDMD-FL, Pro-Caspase-1, and Pro-IL-1β (Figure 6K–O). The administration of siIRF1 and siIRF1@ZIF NPs had minimal effects on pyroptosis. However, siIRF1@ZIF@HM NPs treatment suppressed the cleavage of the GSDMD-FL, Pro-Caspase-1, and Pro-IL-1β, thereby impeding the pyroptotic cascade. Notably, IRF1 expression levels correlated with pyroptosis intensity, suggesting the underlying mechanisms of pyroptosis in myocarditis. Collectively, these findings demonstrated that siIRF1@ZIF@HM NPs downregulated IRF1 expression, suppressed macrophage polarization and pyroptosis, and consequently alleviated myocarditis.
Excessive Zn2+ exhibit potent toxicity and its rapid degradation may cause great damages to healthy cells. To evaluate the safety profiles of the nano-formulations, the body weights of mice in each group were monitored throughout the therapy. As illustrated in Figure S7A, no significant fluctuations in body weight were observed. Furthermore, biochemical analysis of the blood samples indicated stable serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and creatinine (CREA) relative to the control group (Figure S7B–D), suggesting unimpaired hepatic and renal function. Concurrently, serum levels of total cholesterol (TC), triglyceride (TG), and creatine kinase (CK) were within normal ranges and comparable to those of the control group (Figure S7E–G), indicating metabolic stability. Histological assessment of the major organs, including the liver, spleen, lung, and kidney, also revealed no significant tissue abnormalities with HE staining (Figure S7H). Notably, there is still limited data and experimental evidence on the in vivo toxicity of ZIF-8. Given its potential risks, achieving precise control over ZIF-8 biodegradation and drug release is essential to minimize possible harm.
Conclusion
In the current study, we successfully engineered a biomimetic nanocomplex tailored for the directed delivery of nucleic acids to myocarditis sites and pinpointed IRF1 as a viable target for myocarditis treatment. Based on transcriptional sequencing and our previous research on myocarditis, the biomimetic nanoparticles were constructed with a siIRF1-loaded ZIF-8 nanoparticle core and encased within a T lymphocyte-macrophage hybrid membrane as the outer shell. The MOF nanoparticles not only possessed a high loading capacity of nucleic acid payloads but also facilitated the endo-lysosomal release of siIRF1 into the cytosol through pH-responsive dissociation. In vitro experiments demonstrated the specific targeting of siIRF1@ZIF@HM NPs to M1 macrophages, culminating in the efficient silencing of IRF1 and the consequent suppression of pyroptosis in IFN-γ-stimulated macrophages. In vivo results confirmed the precise accumulation of siIRF1@ZIF@HM NPs in myocardial tissue, specifically within M1 macrophages at the inflammatory sites. The siIRF1@ZIF@HM NPs effectively alleviated the progression of myocarditis, primarily by inhibiting IFN-γ-mediated macrophage pyroptosis. In summary, this nanotherapeutic strategy shows promising potential as a precise intervention for myocarditis, with prospects for broader applications to a range of inflammatory disorders in the future.
Abbreviations
ATP, adenosine triphosphate; BMDMs, bone marrow-derived macrophages; CCK8, cell counting kit 8; CFA, complete Freund’s adjuvant; CXCL9, chemokine (C-X-C motif) ligand 9; CXCL10, chemokine (C-X-C motif) ligand 10; CXCL11, chemokine (C-X-C motif) ligand 11; CXCR3, C-X-C Motif Chemokine Receptor 3; DCM, dilated cardiomyopathy; DLS, dynamic light scattering; EAM, experimental autoimmune myocarditis; FBS, fetal bovine serum; GSDMD-FL, full length of gasdermin D; GSDMD-NT, N-terminal of gasdermin D; HE, hematoxylin and eosin; HM, hybrid membrane; IFN-γ, interferon-γ; IRF1, interferon regulatory factor 1; IRF1 siRNA, siIRF1; KEGG, Kyoto encyclopedia of genes and genomes; LPS, lipopolysaccharides; PMA, Phorbol-12-myristate-13-acetate; RISC, RNA-induced silencing complex; siRNA, small interfering RNA; ZIF-8, zeolitic imidazolate framework-8; 2-MIM, 2-methylimidazole.
Funding
This research was supported by grants from the National Natural Science Foundation of China (No.81970285, No.81570309 and No. 82000323).
Disclosure
The authors declare that they have no competing interests in this work. This paper has been uploaded to ResearchSquare as a preprint: https://doi.org/10.21203/rs.3.rs-4206914/v1
References
1. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18(12):733–744. doi:10.1038/s41577-018-0065-8
2. Ammirati E, Frigerio M, Adler ED, et al. Management of acute myocarditis and chronic inflammatory cardiomyopathy. Circ Heart Fail. 2020;13(11):e007405. doi:10.1161/CIRCHEARTFAILURE.120.007405
3. Kindermann I, Barth C, Mahfoud F, et al. Update on myocarditis. J. Am. Coll. Cardiol. 2012;59(9):779–792. doi:10.1016/j.jacc.2011.09.074
4. Basso C. Myocarditis. N. Engl. J. Med. 2022;387(16):1488–1500. doi:10.1056/NEJMra2114478
5. Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation. 2001;104(1):39–45. doi:10.1161/01.CIR.104.1.39
6. Frustaci A, Russo MA, Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur. Heart J. 2009;30(16):1995–2002. doi:10.1093/eurheartj/ehp249
7. Chimenti C, Russo MA, Frustaci A. Immunosuppressive therapy in virus-negative inflammatory cardiomyopathy: 20-year follow-up of the TIMIC trial. Eur. Heart J. 2022;43(36):3463–3473. doi:10.1093/eurheartj/ehac348
8. Vargason AM, Anselmo AC, Mitragotri S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021;5(9):951–967. doi:10.1038/s41551-021-00698-w
9. Ranasinghe P, Addison ML, Dear JW, et al. Small interfering RNA: discovery, pharmacology and clinical development—an introductory review. Br. J. Pharmacol. 2023;180(21):2697–2720.
10. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 2009;7(2):99–109. doi:10.1038/nrmicro2070
11. Xiong Y, Zhang Z, Liu S, et al. Lupeol alleviates autoimmune myocarditis by suppressing macrophage pyroptosis and polarization via PPARα/LACC1/NF-κB signaling pathway. Phytomedicine. 2024;123:155193. doi:10.1016/j.phymed.2023.155193
12. Feng H, Zhang Y-B, Gui J-F, et al. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLOS Pathog. 2021;17(1):e1009220. doi:10.1371/journal.ppat.1009220
13. Setten RL, Rossi JJ, Han S. The current state and future directions of RNAi-Based therapeutics. Nat. Rev. Drug Discov. 2019;18(6):421–446. doi:10.1038/s41573-019-0017-4
14. Sahoo S, Kariya T, Ishikawa K. Targeted delivery of therapeutic agents to the heart. Nat. Rev. Cardiol. 2021;18(6):389–399. doi:10.1038/s41569-020-00499-9
15. Liu Y, He M, Yuan Y, et al. Neutrophil-membrane-coated biomineralized metal–organic framework nanoparticles for atherosclerosis treatment by targeting gene silencing. ACS Nano. 2023;17(8):7721–7732. doi:10.1021/acsnano.3c00288
16. Zhang Q, Dehaini D, Zhang Y, et al. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 2018;13(12):1182–1190. doi:10.1038/s41565-018-0254-4
17. Fang RH, Hu C-MJ, Luk BT, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–2188. doi:10.1021/nl500618u
18. Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. U. S. A. 2011;108(27):10980–10985. doi:10.1073/pnas.1106634108
19. Zhuang J, Gong H, Zhou J, et al. Targeted gene silencing in vivo by platelet membrane–coated metal-organic framework nanoparticles. Sci. Adv. 2020;6(13):eaaz6108. doi:10.1126/sciadv.aaz6108
20. Zhu L, Yu X, Cao T, et al. Immune cell membrane-based biomimetic nanomedicine for treating cancer metastasis. Acta Pharm. Sin. B. 2023;13(6):2464–2482. doi:10.1016/j.apsb.2023.03.004
21. Li Z, Cai H, Li Z, et al. A tumor cell membrane-coated self-amplified nanosystem as a nanovaccine to boost the therapeutic effect of Anti-PD-L1 antibody. Bioact. Mater. 2023;21:299–312. doi:10.1016/j.bioactmat.2022.08.028
22. Tokunaga R, Zhang W, Naseem M, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat. Rev. 2018;63:40–47. doi:10.1016/j.ctrv.2017.11.007
23. Zhuang J, Kuo C-H, Chou L-Y, Liu D-Y, Weerapana E, Tsung C-K. Optimized metal–organic-framework nanospheres for drug delivery: evaluation of small-molecule encapsulation. ACS Nano. 2014;8(3):2812–2819. doi:10.1021/nn406590q
24. Wang D, Wu Q, Ren X, Niu M, Ren J, Meng X. Tunable Zeolitic Imidazolate Framework‐8 Nanoparticles for Biomedical Applications. Small Methods. 2023;8:2301270.
25. Kang M, Hong J, Jung M, et al. T‐cell‐mimicking nanoparticles for cancer immunotherapy. Adv. Mater. 2020;32(39):2003368. doi:10.1002/adma.202003368
26. Gao C, Huang Q, Liu C, et al. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat. Commun. 2020;11:2622. doi:10.1038/s41467-020-16439-7
27. Li S-Y, Cheng H, Xie B-R, et al. Cancer cell membrane camouflaged cascade bioreactor for cancer targeted starvation and photodynamic therapy. ACS Nano. 2017;11(7):7006–7018. doi:10.1021/acsnano.7b02533
28. Cravillon J, Münzer S, Lohmeier S-J, et al. Rapid room-temperature synthesis and characterization of nanocrystals of a prototypical zeolitic imidazolate framework. Chem. Mater. 2009;21(8):1410–1412. doi:10.1021/cm900166h
29. Yang F, Cabe MH, Ogle SD, Sanchez V, Langert KA. Optimization of critical parameters for coating of polymeric nanoparticles with plasma membrane vesicles by sonication. Sci. Rep. 2021;11(1):23996. doi:10.1038/s41598-021-03422-5
30. Toda G, Yamauchi T, Kadowaki T, Ueki K. Preparation and culture of bone marrow-derived macrophages from mice for functional analysis. STAR Protoc. 2021;2(1):100246. doi:10.1016/j.xpro.2020.100246
31. Muehlinghaus G, Cigliano L, Huehn S, et al. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood. 2005;105(10):3965–3971. doi:10.1182/blood-2004-08-2992
32. Satarkar D, Patra C. Evolution, expression and functional analysis of CXCR3 in neuronal and cardiovascular diseases: a narrative review. Front. Cell Dev. Biol. 2022;10:882017. doi:10.3389/fcell.2022.882017
33. Hasegawa T, Venkata Suresh V, Yahata Y, et al. Inhibition of the CXCL9-CXCR3 axis suppresses the progression of experimental apical periodontitis by blocking macrophage migration and activation. Sci. Rep. 2021;11(1):2613. doi:10.1038/s41598-021-82167-7
34. Hua X, Hu G, Hu Q, et al. Single-cell RNA sequencing to dissect the immunological network of autoimmune myocarditis. Circulation. 2020;142(4):384–400. doi:10.1161/CIRCULATIONAHA.119.043545
35. Wang Q, Ren Y, Mu J, et al. Grapefruit-derived nanovectors use an activated leukocyte trafficking pathway to deliver therapeutic agents to inflammatory tumor sites. Cancer Res. 2015;75(12):2520–2529. doi:10.1158/0008-5472.CAN-14-3095
36. Chen Q, Xu M, Zheng W, et al. Se/Ru-decorated porous metal–organic framework nanoparticles for the delivery of pooled siRNAs to reversing multidrug resistance in taxol-resistant breast cancer cells. ACS Appl. Mater. Interfaces. 2017;9(8):6712–6724. doi:10.1021/acsami.6b12792
37. Cao H, Dan Z, He X, et al. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano. 2016;10(8):7738–7748. doi:10.1021/acsnano.6b03148
38. Dutta A, Zhao B, Love PE. New insights into TCR β-selection. Trends Immunol. 2021;42(8):735–750. doi:10.1016/j.it.2021.06.005
39. Mete D, Yemeztaşlıca E, Şanlı-mohamed G. Sorafenib loaded ZIF-8 metal-organic frameworks as a multifunctional nano-carrier offers effective hepatocellular carcinoma therapy. J. Drug Deliv. Sci. Technol. 2023;82:104362. doi:10.1016/j.jddst.2023.104362
40. Bronger H, Singer J, Windmüller C, et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br. J. Cancer. 2016;115(5):553–563. doi:10.1038/bjc.2016.172
41. Dowdy SF. Overcoming Cellular Barriers for RNA Therapeutics. Nat. Biotechnol. 2017;35(3):222–229. doi:10.1038/nbt.3802
42. Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184(1):149–168.e17. doi:10.1016/j.cell.2020.11.025
43. Karki R, Sharma BR, Lee E, et al. Interferon regulatory factor 1 regulates panoptosis to prevent colorectal cancer. JCI Insight. 2020;5:12. doi:10.1172/jci.insight.136720
44. Guo M, Yan R, Ji Q, et al. IFN regulatory factor-1 induced macrophage pyroptosis by modulating m6A modification of Circ_0029589 in patients with acute coronary syndrome. Int. Immunopharmacol. 2020;86:106800. doi:10.1016/j.intimp.2020.106800
45. Murray PJ. Macrophage polarization. Annu. Rev. Physiol. 2017;79(1):541–566. doi:10.1146/annurev-physiol-022516-034339
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.