")
Back to Journals » International Journal of Nanomedicine » Volume 19
Targeted Delivery and ROS-Responsive Release of Lutein Nanoassemblies Inhibit Myocardial Ischemia-Reperfusion Injury by Improving Mitochondrial Function
Authors Shi P, Sha Y, Wang X, Yang T, Wu J, Zhou J, Liu K, Guan X, Wang S , Liu Y, Gao J, Sun H, Ban T, Cao Y
Received 11 September 2024
Accepted for publication 5 November 2024
Published 19 November 2024 Volume 2024:19 Pages 11973—11996
DOI https://doi.org/10.2147/IJN.S488532
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Kamakhya Misra
Pilong Shi,1,* Yuetong Sha,1,* Xinran Wang,1,* Tao Yang,1 Jiawei Wu,1 Jiajun Zhou,1 Kai Liu,1 Xue Guan,2 Song Wang,1 Yongsheng Liu,1 Jingquan Gao,3 Hongli Sun,1 Tao Ban,1 Yonggang Cao1
1Department of Pharmacology, Harbin Medical University, Heilongjiang, 163319, People’s Republic of China; 2Morphological Experiment Center, Harbin Medical University, Heilongjiang, 163319, People’s Republic of China; 3Department of Nursing, School of Medicine, Lishui University, Lishui, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yonggang Cao; Hongli Sun, Email [email protected]; [email protected]
Purpose: Myocardial ischemia-reperfusion injury (MI/RI) is associated with increased oxidative damage and mitochondrial dysfunction, resulting in an elevated risk of mortality. MI/RI may be alleviated by protecting cardiomyocytes from oxidative stress. Lutein, which belongs to a class of carotenoids, has proven to be effective in cardiovascular disease treatment due to its remarkable antioxidant properties, but its application is limited due to its poor stability and low bioavailability in vivo.
Methods: In this study, a delivery system was developed based on distearoyl phosphatidyl ethanolamine (DSPE)-thiol-ketone (TK)-PEG2K (polyethylene glycol 2000) (abbreviated as DTP) and PCM-SH (CWLSEAGPVVTVRALRGTGSW) to deliver lutein (abbreviated as lutein@DTPP) to damaged myocardium. First, lutein, lutein@DTP, or lutein@DTPP were injected through the tail vein once a day for 3 days and then MI/RI model rats were established by exposing rats to ischemia for 45 min and reperfusion for 6 h. We employed a range of experimental techniques including qRT-PCR, Western blotting, transmission electron microscopy, immunohistochemistry, immunofluorescence, flow cytometry, immunoprecipitation, molecular docking, and molecular dynamics simulations.
Results: Lutein@DTPP exhibited good myocardial targeting and ROS-responsive release. Our data suggested that lutein@DTPP effectively suppresses ferroptosis in cardiomyocytes. Mechanistically, we observed an upregulation of mouse double minute-2 (MDM2) in the hearts of MI/RI models and cardiomyocytes exposed to hypoxia/reoxygenation (H/R) conditions. In addition, NADH-ubiquinone oxidoreductase 75 kDa Fe-S protein 1 (NDUFS1) translocation from the cytosol to the mitochondria was inhibited by MDM2 upregulation. Notably, no significant variation in the total NDUFS1 expression was observed in H/R-exposed cardiomyocytes following treatment with siMDM2. Further study indicated that lutein facilitates the translocation of NDUFS1 from the cytosol to mitochondria by directly binding and sequestering MDM2, thereby improving mitochondrial function and inhibiting ferroptosis.
Conclusion: Lutein@DTPP promoted the mitochondrial translocation of NDUFS1 to restore mitochondrial function and inhibited the ferroptosis of cardiomyocytes by directly binding and sequestering MDM2.
Keywords: myocardial ischemia-reperfusion injury, lutein, ROS, MDM2, NDUFS1, ferroptosis
Graphical Abstract:

Introduction
Myocardial ischemia and its subsequent ischemia-reperfusion injury are significant contributors to myocardial injury and key pathophysiological processes in several cardiovascular diseases (CVDs), leading to increased healthcare costs and mortality rates.1 Currently, therapeutic strategies for managing myocardial ischemia-reperfusion injury (MI/RI) primarily focus on protecting cardiomyocytes by removing reactive oxygen species (ROS), controlling inflammation, and limiting apoptosis.2 Due to the substantial energy demands associated with contractile activity, the heart relies heavily on the preservation of normal mitochondrial respiration function. Emerging evidence indicates that mitochondrial dysfunction plays a crucial role in cardiomyocyte death and cardiac failure during energy-stress conditions, such as MI/RI.3,4 Enhanced mitochondrial function ultimately enhances cell survival by aiding cardiomyocytes in coping with cellular ischemia stressors.5 Thus, innovative cardioprotective strategies may be provided by novel therapeutics effective in mitigating mitochondrial dysfunction in MI/RI.6,7
As the cell’s power plant, mitochondria are responsible for generating adenosine triphosphate (ATP), the energy needed to support cellular life processes.8 Under stress conditions, mitochondria regulate various biological activities, including signal transduction and metabolism.9 The contraction and relaxation of the heart rely on a continuous supply of large amounts of ATP, which is produced by oxidative phosphorylation in the mitochondria of cardiomyocytes. Mitochondrial complex I (MCI), the largest enzyme in the mitochondrial respiratory chain, initiates this process by transferring electrons from NADH to coenzyme Q10 and facilitating proton translocation from the mitochondrial matrix to the intermembrane space.10 In mammals, seven of the fourteen core subunits of the 45-subunit MCI are encoded by nuclear genes.11,12 One of these subunits, NDUFS1, acts as a catalyst in the initial stage of NADH oxidation in the mitochondrial respiratory chain and is essential for maintaining MCI function and stability.13 ATP synthesis is facilitated by an electrochemical gradient generated by the translocation of protons across the inner membrane of the mitochondria. As a by-product of synchronous ATP synthesis, ROS is also produced in some mitochondrial complexes, primarily MCI.14,15 Research has indicated that the absence or mutation of NDUFS1 leads to a reduction in the levels and catalytic performance of MCI.13,16 A previous study demonstrated that NDUFS1 deficiency in myocardial hypertrophy induced by pressure overload worsens the dysfunction of mitochondrial membrane potential.17 Moreover, Qi et al demonstrated that diabetes-related cardiomyopathy is worsened by the deficiency of akap1, which inhibits the translocation of NDUFS1 from the mitochondria, resulting in cardiomyocyte apoptosis and mitochondrial dysfunction.18 A recent study revealed that cardiac-specific overexpression of NDUFS1 alleviates cardiac dysfunction following myocardial infarction by suppressing apoptosis and mitochondrial dysfunction.19 Although the downregulation of NDUFS1 expression is involved in MI/RI, the underlying mechanism remains poorly understood.
Lutein (C40H56O2), an essential carotenoid, is not naturally synthesized in the human body.20 Lutein-rich foods and fruits include avocado, kiwi, spinach, kale, celery, dark green or yellow vegetables, marigolds, eggs, carrots, celery, and broccoli.21 Existing literature has confirmed various pharmacological properties of lutein, including anti-inflammatory, antioxidant, and anti-apoptotic effects.22 Additionally, lutein has been demonstrated to protect the skin from ultraviolet damage and reduce diabetes-related oxidative stress.23 Recent studies have demonstrated that lutein has therapeutic effects on cardiovascular diseases. For example, animal studies have demonstrated that lutein can protect against atherosclerosis.24 Furthermore, Liu et al found that interferon regulatory factor (IRF) is positively regulated by lutein in cardiac hypertrophy, inhibiting ferroptosis of cardiac microvascular endothelial cells.25 Notably, lutein has poor water solubility and stability due to its unsaturated structure. Lutein is prone to degradation and deactivation, and its oral bioavailability is low, which significantly limits its therapeutic efficacy.26 Therefore, delivering lutein directly to diseased areas could greatly enhance its bioavailability and pharmacological effects.
In recent years, targeted drug delivery has become a research hotspot. One study employed a macrophage cell membrane coated with polydopamine to target the heart for treating MI/RI.27 In our preliminary experiment, we successfully developed a delivery strategy using a neutrophil membrane-camouflaged mesoporous silica nanocomplex to inhibit cardiac hypertrophy.28 Moreover, we have designed a PCL-PEG-MAL-PCM-SH-based delivery system for PRT4165 to inhibit MI/RI.29 However, the targeted delivery of drug monomer components to damaged myocardium remains an area requiring further exploration in MI/RI treatment.
In the current study, a delivery system was developed using DSPE-TK-PEG2K and PCM-SH to deliver lutein to damaged myocardium. PCM-SH, a 21-mer peptide isolated through phage display, exhibited a 180-fold higher binding affinity for primary cardiomyocytes compared to controls,29 suggesting high specificity. Under MI/RI conditions, the levels of ROS, particularly the most abundant and stable types, are significantly elevated, indicating the potential of ROS as a physiological stimulus. For ROS-responsive linkage, we used the TK functional group, which is known to be stable against protease-catalyzed degradation and under both acidic and basic conditions. Lutein@DTPP nanoparticles were successfully prepared by surface modification with PCM-SH and incorporating a ROS-sensitive TK functional group, maintaining the intact structure of lutein@DTPP. Through this approach, Lutein@DTPP nanoparticles were developed to target cardiomyocytes, improving mitochondrial function, and inhibiting MI/RI.
This study explores the therapeutic effects of lutein@DTPP in an ischemia reperfusion-induced myocardial injury model and its regulatory mechanisms. Our findings demonstrated that lutein@DTPP significantly inhibits myocardial infarction size and improves cardiac function in MI/RI rats. Mechanistically, MDM2 deficiency was shown to restore mitochondrial respiratory function, reduce ROS production, and suppress ferroptosis in cardiomyocytes exposed to hypoxia/reoxygenation (H/R). Additionally, immunoprecipitation analysis indicated that MDM2 interacts with NDUFS1 in cardiomyocytes. Specifically, lutein@DTPP improved mitochondrial function by facilitating the translocation of NDUFS1 from the cytosol to the mitochondria through competitive binding to MDM2. These findings suggest that targeted regulation of NDUFS1 levels in mitochondria could be a promising therapeutic strategy for MI/RI treatment.
Materials and Methods
Nanoparticle Preparation and Characterization
Soy lecithin, cholesterol, DSPE-TK-PEG2K, DSPE-PEG2K-PCM-SH (Xi’an ruixi Biological Technology Co., Ltd. China), and lutein (HY-N6947, purity 89.49%, MedChemExpress) were dissolved together in 6 mL chloroform and evaporated into a membrane. Next, the nanoparticles were collected using ultrasound and a liposome extruder (Polycarbonate membrane, pore size: 200 nm). The unloaded lutein was removed by filtration through the polycarbonate membrane. The size and morphology of the nanoparticles were examined using a transmission electron microscope (TEM), while a dynamic light scattering (DLS) detector (Zetasizer-ZS90) was used to measure the zeta potential and size distribution of the nanoparticles.
Cell Uptake of Nanoparticles
The uptake of lutein@DTP and lutein@DTPP by cardiomyocytes was evaluated using flow cytometry and fluorescence microscopy. Cardiomyocytes were cultured for 24 hours on 15 mm glass coverslips in plates containing culture media, and then exposed to H/R for fluorescence microscopy analysis. Subsequently, under standard cell culture conditions, the cardiomyocytes were incubated in Dulbecco’s modified eagle medium (DMEM) containing fluorescein isothiocyanate (FITC)-labeled lutein@DTP and lutein@DTPP. Following three washes with ice-cold phosphate-buffered saline (PBS) (BL601A, Biosharp), the cells were fixed with 4% paraformaldehyde (PFA) for 10 min at 37°C. After three additional PBS washes, the sample nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (BL105B, Biosharp) for 10 min. Fluorescence microscopy was then used to capture images of the cells. For flow cytometry analysis, cardiomyocytes were cultured in 24-well plates and treated with FITC-labeled lutein@DTP and lutein@DTPP under the same conditions. After treatment, the cells were detached using 0.05% trypsin, collected by centrifugation at 500 rpm, and resuspended in PBS. A flow cytometer was subsequently used to assess the uptake of lutein@DTP and lutein@DTPP by the cardiomyocytes.
Animal Experiment
Male Wistar rats (200–250 g), 6–8 weeks old, were randomly divided into five groups (n=6 per group): (1) the sham group, which underwent a sham operation and received a vehicle (saline, administered via tail vein injection); (2) the model group, which underwent the operation and received a vehicle (saline, tail vein injection); (3) the model + lutein group; (4) the model + lutein@DTP group; and (5) the model + lutein@DTPP group, with lutein administered at a dose of 20 mg/kg. Anesthesia was induced in the rats through the inhalation of isoflurane gas, after which they were positioned in a supine posture. An animal ventilator was then used to assist their breathing during the surgical procedure. A horizontal incision was made in the 3rd or 4th intercostal space to perform a left thoracotomy. During this procedure, 6–0 sutures were used to ligate the left anterior descending coronary artery (LAD) around a PE-10 tube. The occlusion of the LAD was confirmed by ST-segment elevation on the electrocardiogram (ECG). After 45 min of occlusion, the PE-10 tube was removed, and the rats underwent 6 h of reperfusion. The groups receiving lutein, lutein@DTP, or lutein@DTPP were treated with their respective interventions once daily for 3 days before the operation. The rats were randomly subjected to MI/RI and the corresponding interventions. The efficacy of the interventions was assessed through morphological analysis, transthoracic echocardiography, and biomarker analysis, all conducted in a blinded manner.
Isolation and Culture of Cardiac Microvascular Endothelial Cells (CMECs)
Cardiac microvascular endothelial cells (CMECs) were isolated from Wistar rats (weighing 100–120 g), as previously described. Briefly, the left ventricles of male rats were harvested, and the epicardial coronary arteries and endocardial endothelium were removed. The ventricular tissue was then minced into 1 mm³ pieces. These tissue fragments were dissociated using collagenase II and cultured in DMEM/F12 medium supplemented with 1% vascular endothelial growth factor (VEGF), 1% endothelial cell growth supplement (ECGS), 10% fetal bovine serum (FBS), and 100 mg/mL penicillin/streptomycin.
Isolation and Culture of Primary Cardiomyocyte
Myocardial tissue was isolated from rats of 1–2 days old rats. The heart tissue was digested using 2.5 mg/mL trypsin (Aladdin Co., Ltd., Shanghai, China), followed by the addition of a digestion buffer containing 2 mg/mL DNase I (D7291, Sigma) for perfusion to isolate cardiomyocytes. After centrifuging the tissue suspension to collect the cardiomyocytes, they were cultured in DMEM (C11995500BT, HyClone) supplemented with 20% FBS (11011–8611, EVERY GREEN), 100 μg/mL streptomycin, and 100 μg/mL penicillin.
Hypoxia/Reoxygenation (H/R) Cardiomyocyte Preparation
Cardiomyocytes were subjected to hypoxia for 12 h, followed by reoxygenation for 24 h. The cells were then randomly divided into five groups: (1) Control group; (2) Model group; (3) Model + lutein group; (4) Model + lutein@DTP group; and (5) Model + lutein@DTPP group. The treatment groups received lutein, lutein@DTP, or lutein@DTPP, respectively, administered at a dose of 20 μM following H/R exposure.
Echocardiographic Assessment
Electrocardiography (ECG) was performed using a previously reported approach.30 The rats were anesthetized with isoflurane gas, and a depilation agent was applied to remove the hair from their chests. Following hair removal, an ultrasound gel was applied to the chest area to facilitate scanning with a contact scanning head.
2,3,5-Triphenyltetrazolium Chloride (TTC) and Hematoxylin and Eosin (HE) Staining
After treatment with lutein, lutein@DTP, or lutein@DTPP, the rats were euthanized, and their hearts were harvested. The hearts were then stored at −20°C for 25 min. Subsequently, the heart tissues were sectioned into 5 slices and incubated in 2% TTC (T8877-10G, Sigma) solution at 37°C for 15 minutes, protected from light. The infarct size was determined using ImageJ software. For histopathological analysis, several heart sections (4–5 μm thick) were stained with hematoxylin and eosin (HE) to examine the tissue structure.31
Laser Speckle Contrast Analysis (Lasca) of Infarcted Tissues
A moor FLPI-2 real-time blood flow zoom laser speckle imaging system (Moor Instruments, Ltd., UK) was utilized for LASCA to evaluate the infarcted area in terms of spatial vascular profile and flow velocity.
Immunofluorescence (IF) Staining
A method described in a previous report32 was used for IF staining. Anti-γH2AX (1:300, Cell Signaling Technology) antibodies or anti-Gpx4 (1:50, Santa Cruz Biotechnology) antibodies were added to the samples before overnight incubation at 4°C. Subsequently, the samples were rinsed three times with PBS before being incubated with species-specific secondary antibodies.
Immunohistochemistry (IHC)
A method described in a previous report was used for the IHC analysis.33 Briefly, after the hearts were removed, they were fixed in formalin for 48 hours, followed by paraffin embedding and sectioning. The sections were then incubated overnight at 4°C with anti-γH2AX antibodies or anti-4-HNE antibodies. The following day, diaminobenzidine (DAB) was applied to detect the sections according to the manufacturer’s instructions.
Electron Microscopy
Following a previously reported procedure,34 electron microscopy was performed to observe the cardiomyocytes for mitochondrial alterations during MI/RI.
Determination of ROS, JC-1 Monomer, and MDA Levels
ROS (S0033S, Beyotime), JC-1 monomer (C2003S, Beyotime), and MDA levels were measured as per the directions of the manufacturer.
RNA Extraction and Real-Time Quantitative PCR Analysis (qRT-PCR)
The RNA extraction and RT-PCR processes were performed following a previously reported method.32 The reference gene used was β-actin, and its expression was used to normalize the results. The specific sequences for each pair of primers are listed as follows: ptgs2, the forward primer sequence was CAACCAGCAGTTCCAGTATCAG, and the reverse primer sequence was GAGCAAGTCCGTGTTCAAGG.
Cell Viability Assay
Cardiomyocytes were cultured overnight in a 96-well plate. The cell viability was then measured using the Cell Counting Kit-8 (CCK-8), following the manufacturer’s instructions.
Determination of Mitochondrial ROS
The fluorescent probe MitoSOX™ (Invitrogen) was utilized to estimate the levels of mitochondrial superoxide. Briefly, 1 mL of MitoSOX™ (5 μM) was introduced and subjected to incubation in darkness at 37 °C for 10 min. A fluorescence microscope was utilized to measure the ROS levels.
Mitochondrial Oxygen Consumption Rate (OCR) Assay
An XF24 Seahorse extracellular flux analyzer (Agilent Seahorse Bioscience, Santa Clara, CA, USA) was used to measure the mitochondrial OCR. In brief, cells were seeded in cell culture plates, and a Seahorse XF calibration solution was added to a sensor probe plate, which was hydrated overnight in a 37°C CO2-free incubator. One hour before the measurements, 180 μL of XF assay medium replaced the cell growth medium, and the microplate was placed in a 37°C CO2-free incubator. The A, B, and C ports of the probe plates were loaded with oligomycin, FCCP, and rotenone/antimycin A stock solutions, respectively, all prepared using the XF Cell Mito Stress Test Kit with XF assay medium. Measurements were taken at 37°C, and data were collected using Wave 2.2.0.276 software (Agilent).
Molecular Dynamics Simulation
To further refine the binding patterns of proteins to small molecule complexes, we performed detailed molecular dynamics (MD) simulations of protein-small molecule complexes using the Desmond program. In the simulation, OPLS2005 force fields were used to parameterize the interactions between proteins and small molecules, while TIP3P models were used to simulate water molecules. The protein-small molecule complex was placed in a cubic simulation box, and chloride and sodium ions were added to neutralize the system’s charge. Before running the simulation, the system’s energy was minimized over 50,000 steps using the steepest descent method to establish a stable initial state. This was followed by two equilibration phases: first, an NVT equilibration with 50,000 steps, followed by an NPT equilibration with the same number of steps. During these two equilibrium stages, we restricted the positions of the heavy atoms to help the system gradually adapt to the simulated environment. Once the equilibration phases were complete, we ran an unconstrained 100 ns simulation to observe the free-state dynamics of the protein-small molecule complexes. To ensure detailed tracking of the simulation, energy and coordinate data were recorded every 10 picoseconds. Throughout the process, the temperature was maintained at 300 K, and the pressure was held at 1 bar to replicate physiological conditions.
Western Blotting Analysis
A RIPA lysis buffer was used to lyse myocardial tissues or cultured cardiomyocytes in preparation for Western blotting analysis. A total of 50 μg of protein was separated by SDS-PAGE, after which the extracts were transferred onto a nitrocellulose membrane. The membrane was incubated overnight at 4°C with primary antibodies, including β-actin (1:2000, ZSGB-BIO), MDM2 (1:1000, Huabio), COX4 (1:1000, Cell Signaling Technology), NDUFS1 (1:1000, Proteintech Group). The protein bands were visualized utilizing an Imaging System (LI-COR Biosciences) with the application of enhanced chemiluminescence (ECL) reagents.
Co-Immunoprecipitation Assays (Co-IP)
Co-IP tests were performed as described previously.35 Cell lysates were incubated overnight at 4°C with either control IgG or the target-specific antibodies. Following this, protein G Sepharose was added, and the mixture was incubated for an additional 4 h at 4°C. Afterward, the samples underwent primary and secondary antibody incubations, followed by Western blotting analysis to detect the immunoprecipitated proteins.
siRNA Transfection
GenePharma (Shanghai, China) supplied the small interfering RNA (siRNA) targeting MDM2 and negative control siRNA. The sequences for MDM2 siRNA are as follows: Forward: GCAGCCAAGAAAGUGGCAATT, Reverse: UUGCCACUUUCUUGGCUGCTT; The sequences for the negative control siRNA are: Forward: UUCUCCGAACGUGUCACGUTT, Reverse: ACGUGACACGUUCGGAGAATT.
Mitochondrial Complex I Activity Assay
A Mitochondrial Complex I Activity Assay Kit (Solarbio) was utilized as directed by the manufacturer to measure the MCI activity.
Statistical Analysis
GraphPad Prism 9.0 and SPSS 19.0 statistical software were utilized to analyze the data. All quantitative data were presented as mean ± standard deviation (SD). For comparisons among multiple groups, a one-way analysis of variance (ANOVA) was performed. Statistical significance was defined as p < 0.05.
Results
The Formula for Preparing DSPE-PEG2K and PCM-SH
To prepare myocardial targeting liposomes, DSPE-PEG2K-PCM-SH was synthesized (Figure 1A) by first dissolving 100 mg of DSPE-PEG2K-mal in 3 mL dimethylformamide (DMF). Next, a 1.1 molar equivalent of the polypeptide was added, and the mixture was stirred at room temperature for 12 h. Following this, the reaction solution was transferred to a dialysis bag (molecular weight 2000 Da) for dialysis for 24 h, and the product was collected after freezing and drying.
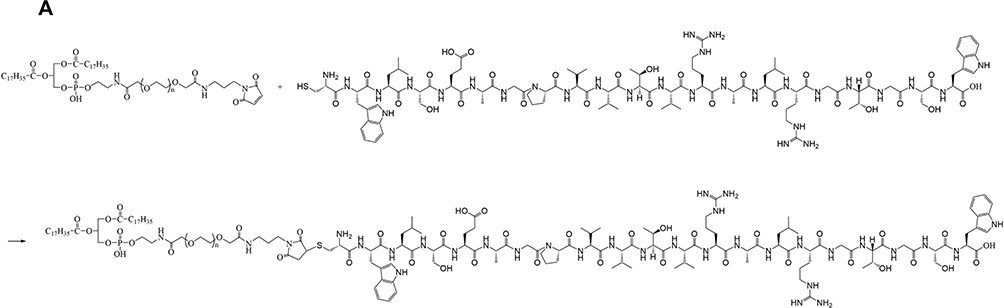 |
Figure 1 The synthesis formula of DSPE-PEG2K-MAL and PCM-SH. (A) The synthetic route of DSPE-PEG2K-MAL and PCM-SH. |
Design, Synthesis, and Characterization of lutein@DTPP
Lutein has poor water solubility and stability, resulting in low bioavailability, which significantly limits its therapeutic potential. To improve lutein’s efficacy, nano complexes that can effectively deliver lutein to injured myocardial tissue may offer additional protection to the heart. Figure 2A illustrates the general approach to synthesizing lutein@DTPP. This involves incorporating lutein during liposome preparation and using extrusion to combine PCM-SH with the liposomes. The zeta potential, shape, and size of the nanoparticles were continuously monitored during formulation. Nuclear magnetic resonance spectroscopy confirmed the successful conjugation of PCM-SH with DSPE-PEG2K (Figure 2B). Transmission electron microscope (TEM) revealed that lutein@DTPP had a uniform, spherical morphology with an average particle size of approximately 135 nm (Figure 2C and D). Fluorescence spectrum analysis demonstrated that both lutein@DTP (without PCM-SH) and lutein@DTPP exhibited strong fluorescence intensity (Figure 2E and F). The zeta potential of lutein@DTP and lutein@DTPP as determined via dynamic laser scattering were 23.22 ± 2.24 mV and 24.79 ± 3.47 mV, respectively (Figure 2G). High-performance liquid chromatography (HPLC) was used to determine lutein loading efficiency and release kinetics. The loading efficiency of lutein was about 7%. The release studies showed that lutein@DTPP gradually released lutein in PBS or 20% serum, achieving 90% release after 24 h. However, in PBS containing 100 µM H2O2-a pathological level of ROS-lutein@DTPP underwent rapid release, with 80% being released within 30 min. In contrast, the non-ROS-responsive control (without the TK functional group) did not exhibit this burst release effect (Figure 2H). Since there is a significant increase in ROS generation during MI/RI,36 our delivery technique leverages ROS-responsive components, making lutein release specific to oxidative conditions and setting it apart from conventional drug delivery approaches.
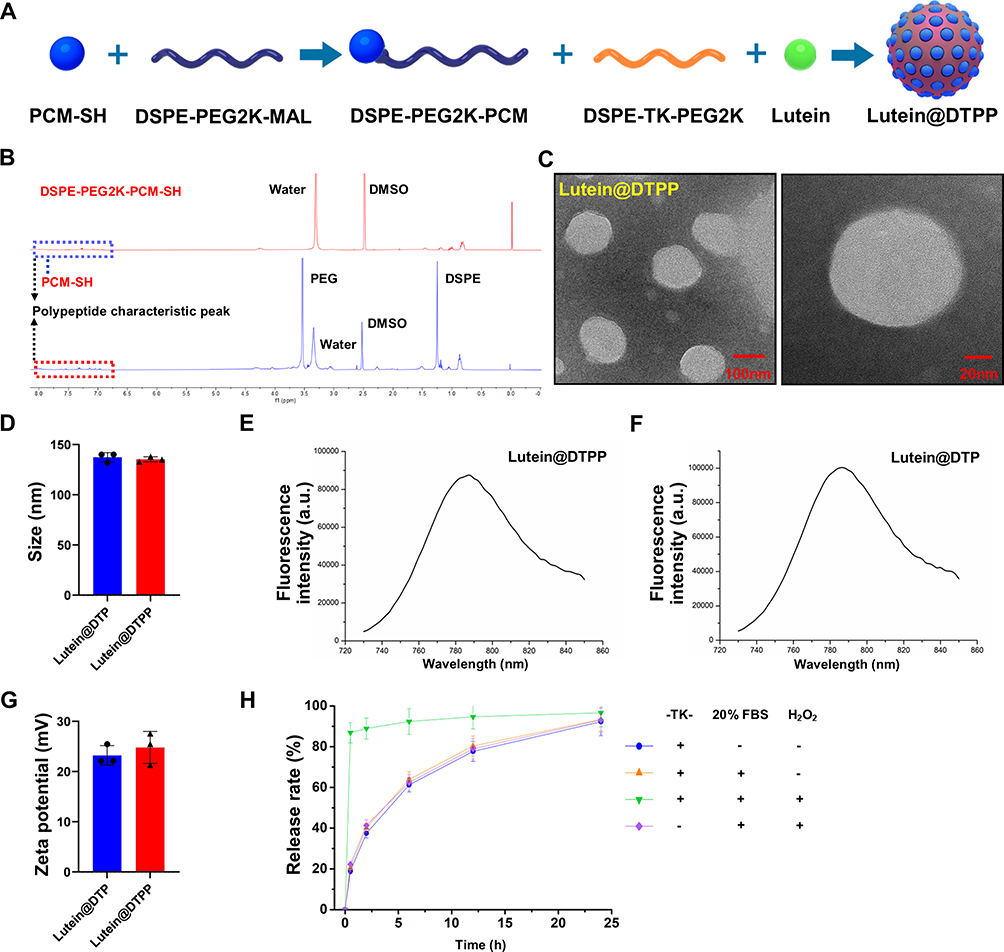 |
Figure 2 Preparation and Characteristics of lutein@DTPP. (A) Schematic illustration of lutein@DTPP preparation. (B) Nuclear magnetic resonance spectroscopy of DSPE-PEG2K-PCM-SH. (C) TEM image of lutein@DTPP. (D) Size of lutein@DTP and lutein@DTPP. (E and F) Fluorescence spectrum analysis of lutein@DTPP and lutein@DTP. (G) Zeta potential of lutein@DTP and lutein@DTPP. (H) Release profiles of lutein from nanoparticles. Data are presented as mean ± SD (n = 3). |
Cellular Uptake and Target Specificity of lutein@DTPP
Rats were injected with lutein@DTP or lutein@DTPP via the tail vein to comprehensively assess their in vivo efficacy. The distribution of lutein@DTP or lutein@DTPP in the hearts of rats exposed to MI/RI was studied using bioimaging techniques. The results demonstrated that PCM-SH enhances the binding of lutein@DTPP to cardiomyocytes, with this interaction being heart-specific; other tissues, such as the lungs, liver, spleen, and kidneys, showed significantly lower distribution of lutein@DTPP (Figure 3A). Additionally, we investigated the potentially toxic effects of lutein@DTPP in normal rats by examining morphological changes in the heart, liver, spleen, lungs, and kidneys through HE staining. The findings indicated that lutein@DTPP did not have any toxic effects on these organs (Figure 3B). For lutein@DTPP nanospheres to be therapeutically effective, cells must internalize them. Homologous targeting specificity is anticipated from the lutein@DTPP nanoplatforms that have been functionalized with PCM-SH. Cardiomyocytes (CMs) and cardiac microvascular endothelial cells (CMECs) were treated with lutein@DTPP, and the results indicated that CMs exhibited stronger green fluorescence compared to CMECs (Figure 3C and D). Moreover, CMs treated with lutein@DTPP “nano-targeted cells” exhibited more intense green fluorescence compared to those treated with lutein@DTP nanospheres at 1, 2, and 4 h (Figure 3E and F), indicating that the PCM-SH-coating promoted the intracellular uptake of the nanocarriers. This phenomenon was further confirmed by flow cytometry quantitative analyses (Figure 3G). Collectively, these results indicate that the presence of PCM-SH significantly facilitates the intracellular uptake of the nanocarriers, thereby enhancing their therapeutic effects.
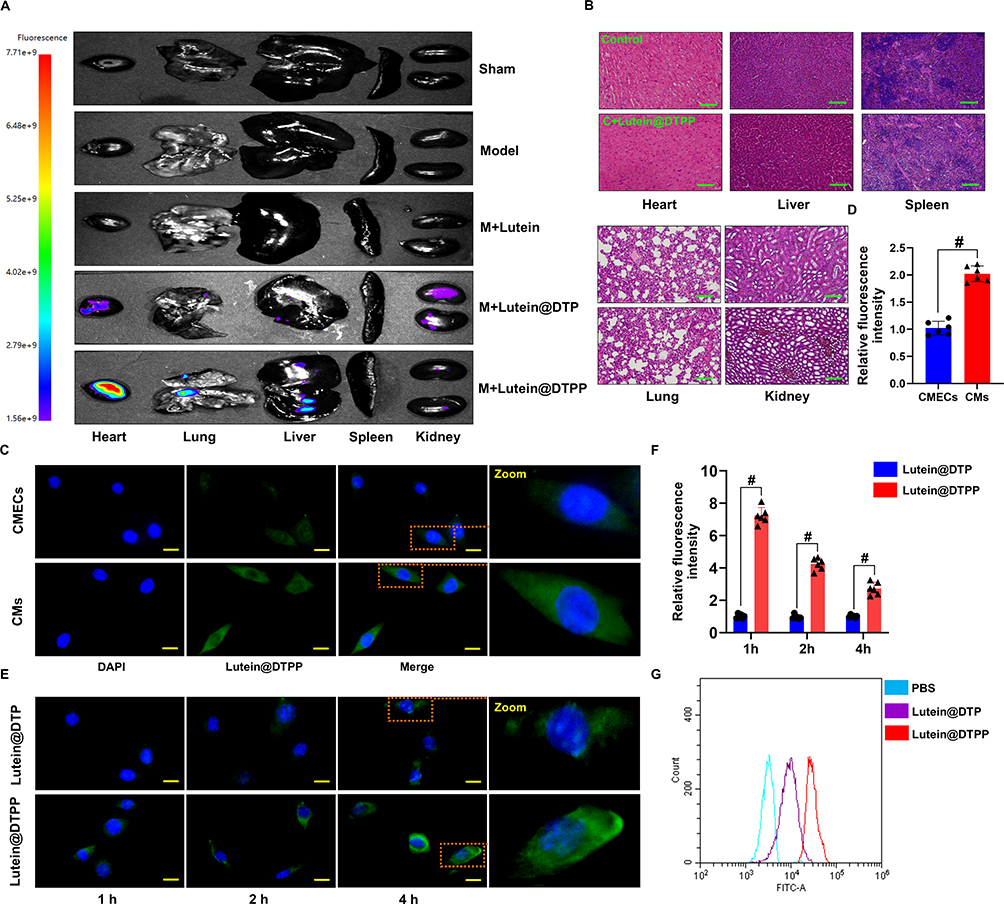 |
Figure 3 Assessment of the targeting ability of lutein@DTPP and cellular uptake. (A) Vivo fluorescence imaging of nanoparticles in the rat heart and other organs following lutein, lutein@DTP, or lutein@DTPP administration. (B) Histology characteristics of major organs of lutein@DTPP treated healthy rat were detected by HE staining. (C and D) Visualization of lutein@DTPP uptake by CMECs and CMs by fluorescence microscopy. Data are presented as mean ± SD (n=6). #P < 0.05 vs CMECs group. (E and F) Intercellular localization of lutein@DTP and lutein@DTPP in CMs at various periods is shown in immunofluorescence imaging. Green-labeled lutein@DTP and lutein@DTPP with DAPI-stained nuclei (blue). (G) Analysis via flow cytometry of the uptake of FITC-labeled nanoparticles by CMs. Data are presented as mean ± SD (n=6). #P < 0.05 vs lutein@DTP group. Model: rats treated with ischemia for 45 min and then reperfusion for 6 h. Scale bars: (B) 50 μm; (C and E) 10 μm. |
Lutein@DTPP Protects the Myocardium Against Ischemia/Reperfusion Injury
The experimental design for the animal studies is illustrated in Figure 4A. Lutein, lutein@DTP, or lutein@DTPP was injected through the tail vein once a day for 3 days, followed by the establishment of MI/RI models through 45 min of ischemia and 6 h of reperfusion. Analysis of cardiac function indicators demonstrated that the lutein@DTPP-20 group (lutein administered at a dose of 20 mg/kg) significantly improved left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF) compared to the model group (Figure 4B–D). Consequently, lutein@DTPP-20 was selected for subsequent experiments. To further compare the therapeutic effects of lutein (20 mg/kg), lutein@DTP, and lutein@DTPP, each treatment was administered to rats with MI/RI in the following experiments. Histological analyses using HE and TTC staining revealed a significant reduction in infarcted areas in the hearts treated with lutein@DTPP (Figure 4E–G). Immunohistochemistry results indicated an increase in γH2AX-positive cells within the myocardial tissue following MI/RI, which were significantly reduced after lutein@DTPP administration (Figure 4H). Furthermore, the restoration of revascularization and blood flow are critical indicators of the therapeutic efficacy on infarcted tissue. LASCA was employed to assess the spatial vascular profiles and flow velocity in the infarcted regions, providing insights into blood flow recovery and revascularization status. Hearts affected by MI/RI exhibited significant blood flow obstruction, as illustrated in Figure 4I and J. In contrast, lutein@DTPP treatment demonstrated a potential to enhance blood flow recovery compared to lutein or lutein@DTP. These findings collectively indicate that lutein@DTPP has a remarkable therapeutic effect on MI/RI in rats.
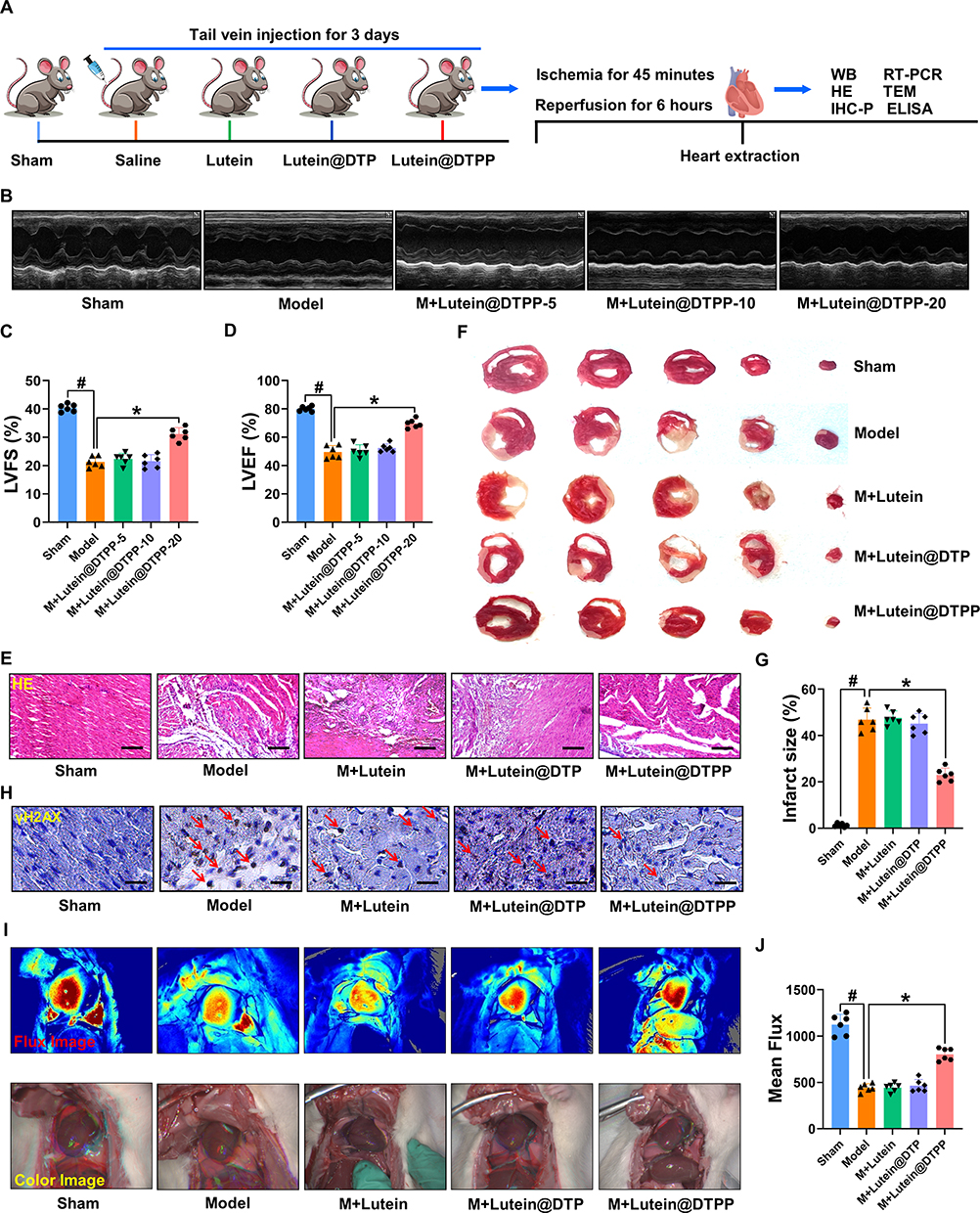 |
Figure 4 Injection of lutein@DTPP decreased the infarct area and improved cardiac function in rats. (A) The time axis scheme that illustrates the experimental design for the animal study. (B) Images of echocardiography that are representative of each group. (C and D) Analysis of LVFS and LVEF. (E) Histopathological images of the cardiac tissue sections of rats stained with HE. (F and G) Detection of infarct areas in cardiac tissue sections with TTC staining. (H) Representative immunohistochemical staining of γH2AX in hearts of rats. γH2AX positive cells were labeled with DAB. DAPI (blue, nuclei); γH2AX positive (brown). (I) The LASCA technique was used to assess the spatial vascular profile and flow velocity in the infarct area of various groups. (J) The mean flux was qualified. Data are presented as mean ± SD (n=6). #P < 0.05 vs Sham group; *P < 0.05 vs Model group. Scale bars: (E) 50 μm; (H) 25 μm. |
Lutein@DTPP Attenuates MI/RI-Induced ROS Production and Ferroptosis
Excessive production ROS is recognized as the primary pathogenic mechanism underlying MI/RI, leading to a series of complex pathological phenomena, including increased infarct size, inflammation, cellular apoptosis, and ferroptosis.37–39 Considering that myocardial protection is closely correlated with the antioxidative properties of lutein, we first examined the ROS levels in rats with MI/RI following treatment with lutein, lutein@DTP, or lutein@DTPP. The results indicated that only lutein@DTPP reduced the ROS levels in the rats (Figure 5A). To further investigate oxidative stress in myocardial tissue, we employed 4-hydroxynonenal (4-HNE) staining and measured malondialdehyde (MDA) content, both of which are indicators of lipid peroxidation. Notably, lutein@DTPP remarkably repressed MI/RI-induced lipid peroxide generation (Figure 5B and C). Next, TEM analysis revealed mitochondrial atrophy and a reduction in mitochondrial cristae morphology, indicating mitochondrial dysfunction (Figure 5D). The qRT-PCR results indicated that lutein@DTPP remarkably inhibited the mRNA expression of ptgs2 (Figure 5E). Collectively, our data suggests that lutein@DTPP decreases ROS levels, thereby inhibiting ferroptosis in cardiomyocytes of rats with MI/RI.
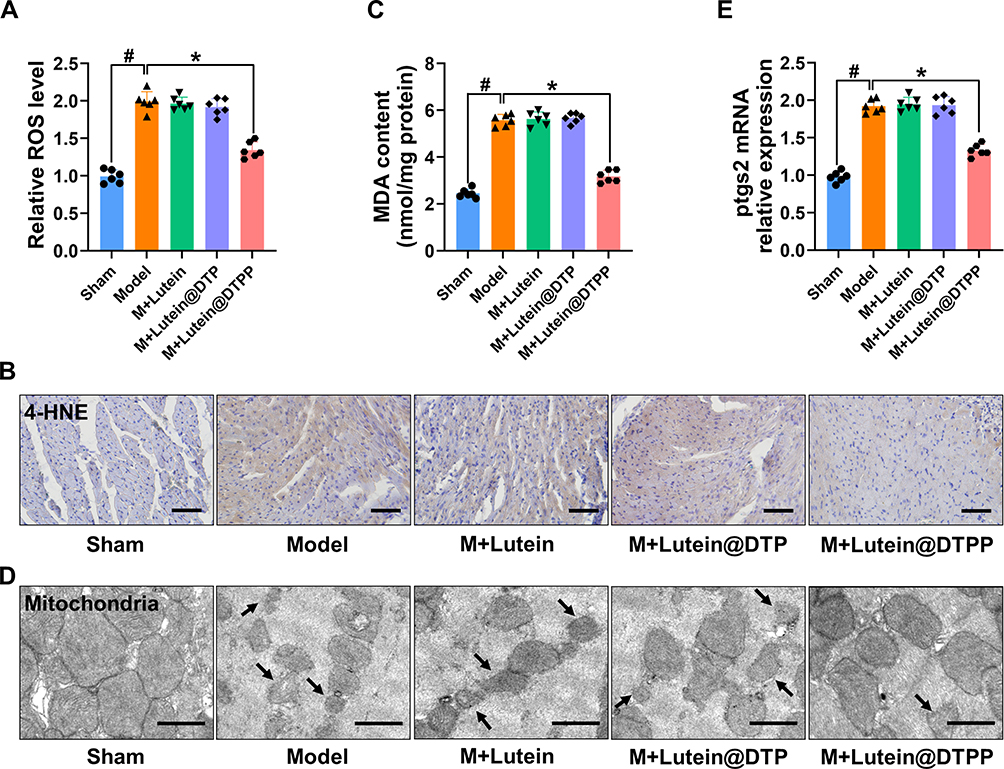 |
Figure 5 Lutein@DTPP decreased ROS to inhibit ferroptosis in MI/RI rats. (A) ROS levels were detected after treatment with lutein, lutein@DTP, or lutein@DTPP in the hearts of MI/RI rats. (B) Representative immunohistochemical staining of 4-HNE. (C) Production of MDA in rats after MI/RI. (D) Representative transmission electron-microscopic images of mitochondria in rats treated with lutein, lutein@DTP, or lutein@DTPP. (E) Quantification of relative ptgs2 mRNA expression in cardiac tissue by qRT-PCR. Data are presented as mean ± SD (n=6). #P < 0.05 vs Sham group; *P < 0.05 vs Model group. Scale bars: (B) 50 μm; (D) 2.0 μm. |
Therapeutic Effects of lutein@DTPP on Cardiomyocytes Exposed to H/R
Lutein has previously been demonstrated to possess antioxidant properties and to inhibit the production of ROS. Nevertheless, the underlying molecular biological mechanism through which lutein@DTPP exerts its antioxidative activity has not yet been elucidated. As a result, this study investigated the potential molecular mechanism underlying the antioxidant activity of lutein@DTPP in MI/RI using cardiomyocytes (Figure 6A). The cardiomyocytes were treated with lutein, lutein@DTP, or lutein@DTPP. First, the optimum lutein@DTPP concentration in cardiomyocytes exposed to H/R was found to be 20 μM, as evidenced by the results of the CCK-8 assay (Figure 6B and C). Immunofluorescence analysis for γH2AX revealed that neither lutein nor lutein@DTP effectively reduced H/R-induced cardiomyocyte damage; however, lutein@DTPP significantly mitigated such injury (Figure 6D). Changes in the levels of ROS in the mitochondria usually accompany mitochondrial dysfunction.40 Consequently, we first identified intracellular and mitochondrial ROS production in cardiomyocytes. Notably, lutein@DTPP was found to markedly decrease ROS production in cardiomyocytes exposed to H/R (Figure 6E and F). Furthermore, while no significant changes were observed in the levels of ptgs2 and malondialdehyde (MDA) in the lutein and lutein@DTP treatment groups compared to the model group, a significant reduction in both MDA and ptgs2 levels was evident in the lutein@DTPP group (Figure 6G and H). These findings indicate that lutein@DTPP markedly inhibits ferroptosis in cardiomyocytes exposed to H/R.
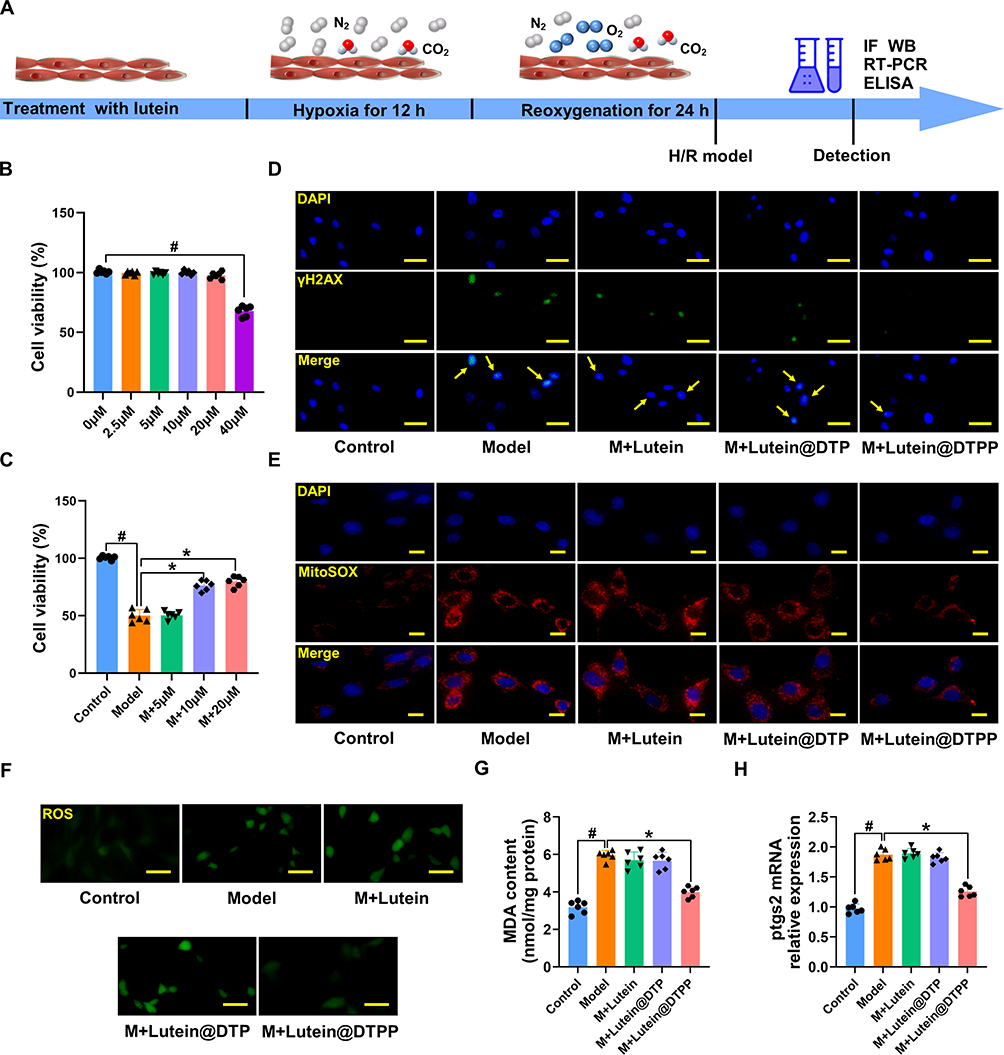 |
Figure 6 Lutein@DTPP inhibited ferroptosis in cardiomyocytes exposed to H/R. (A) The time axis scheme for the H/R-induced cell research is shown. Data are depicted as mean ± SD (n=6). (B) CCK-8 results of normal cardiomyocytes after different concentrations of lutein treatment. #P < 0.05 vs 0 μM group. (C) CCK-8 results of H/R-injured cardiomyocytes treated with lutein, lutein@DTP, or lutein@DTPP. (D) Fluorescence images of γH2AX in H/R-injured cardiomyocytes. (E and F) Representative fluorescence images of mitochondrial and intracellular ROS production. (G) MDA levels in cardiomyocytes. (H) Ptgs2 mRNA expression in cardiomyocytes upon H/R insult. Data are presented as mean ± SD (n=6). #P < 0.05 vs Control group; *P < 0.05 vs Model group. Scale bars: (D and F) 25 μm; (E) 10 μm. Model: cardiomyocytes treated with hypoxia 12 h and then reoxygenation 24 h. |
Lutein@DTPP Improves Mitochondrial Dysfunction in Cardiomyocytes Exposed to H/R
Subsequently, we assessed mitochondrial membrane potential and respiratory capacity to understand how lutein affects mitochondrial dysfunction. The JC-1 monomer was used to evaluate changes in mitochondrial membrane potential, and results showed a significant increase in the number of cardiomyocytes with impaired potential after H/R compared to the control group. However, this rise in JC-1 monomer levels was significantly reduced when lutein@DTPP was administered, indicating improved mitochondrial membrane integrity (Figure 7A). Furthermore, lutein@DTPP remarkably promoted mitochondrial respiration rate, including spare respiration, ATP synthesis, maximal respiration, and basal respiration, in cardiomyocytes exposed to H/R (Figure 7B and C). Overall, our analysis demonstrates that lutein@DTPP improves mitochondrial dysfunction and reduces myocardial injury.
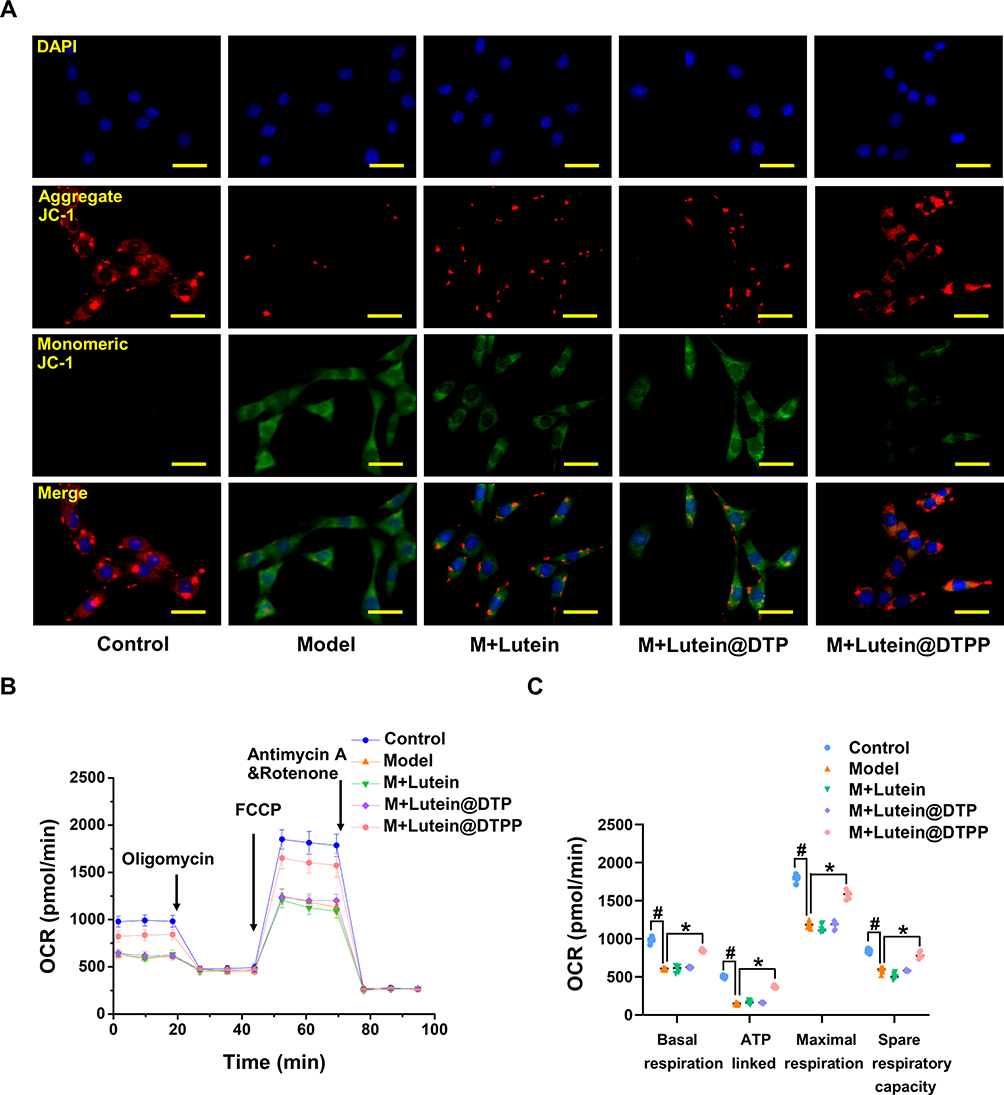 |
Figure 7 Lutein@DTPP improved mitochondrial function of H/R-induced cardiomyocytes. (A) Illustrative images showing the fluorescence of JC-1 in the H/R cardiomyocytes. (B and C) Measure of OCR and respective quantitative analysis in cardiomyocytes. Data are presented as mean ± SD (n=6). #P < 0.05 vs Control group; *P < 0.05 vs Model group. Scale bars: 25 μm. Model: cardiomyocytes treated with hypoxia 12 h and then reoxygenation 24 h. |
MDM2 Knockdown Enhances NDUFS1 Translocation into Mitochondria to Modulate Complex I Activity
Existing research indicates that NDUFS1, which serves as the initial component of the mitochondrial electron transport chain (ETC), functions as a dual-purpose “engine” of ATP and ROS synthesis and is critically involved in cell apoptosis, oxidative stress, and metabolic reprogramming in various pathological conditions.19 In addition, NDUFS1 directly binds and is sequestered by MDM2, impeding its localization inside mitochondria and eventually leading to the instability of complex I and supercomplex as well as the inefficiencies of oxidative phosphorylation. However, it is unclear whether NDFUS1 and MDM2 interactions are implicated in the development of MI/RI. Hence, Western blotting was used to assess the NDUFS1 and MDM2 protein levels. Notably, the NDFUS1 protein level was significantly decreased, whereas that of MDM2 was remarkably increased in the hearts of rats with MI/RI and cardiomyocytes exposed to H/R (Figure 8A–D). A series of additional experiments was performed to further validate the relationship between MDM2 and NDUFS1 in MI/RI. We began by conducting immunoprecipitation (IP) experiments. Unsurprisingly, we found evidence of a clear interplay between MDM2 and NDUFS1 (Figure 8E). The cytosol and mitochondria were isolated from cardiomyocytes exposed to H/R because NDUFS1 is synthesized in the cytosol and subsequently transported to the mitochondria. We discovered that NDUFS1 translocation into mitochondria was greatly enhanced by MDM2 deficiency (Figure 8F–H). There was a significant difference between the control and cardiomyocytes exposed to H/R in terms of the concentration of JC-1 monomer, with the latter showing higher levels. In addition, Knockdown of MDM2 significantly reduced JC-1 monomer levels compared to H/R-induced cardiomyocytes (Figure 8I). NDUFS1 is a key component in the regulation of MCI activity, functioning as a core member of the complex. MCI activity was upregulated in cardiomyocytes exposed to H/R in response to MDM2 deficiency, as anticipated (Figure 8J). Consistent with the aforementioned findings, MDM2 deficiency restored mitochondrial respiratory function, including basal respiration, ATP production, and maximum respiration (Figure 8K and L). Moreover, after knocking down MDM2, ROS levels in cells and mitochondria decreased significantly (Figure 8M and N). Collectively, our results indicate that MDM2 blocks the translocation of NDUFS1 into mitochondria and aggravates mitochondrial dysfunction.
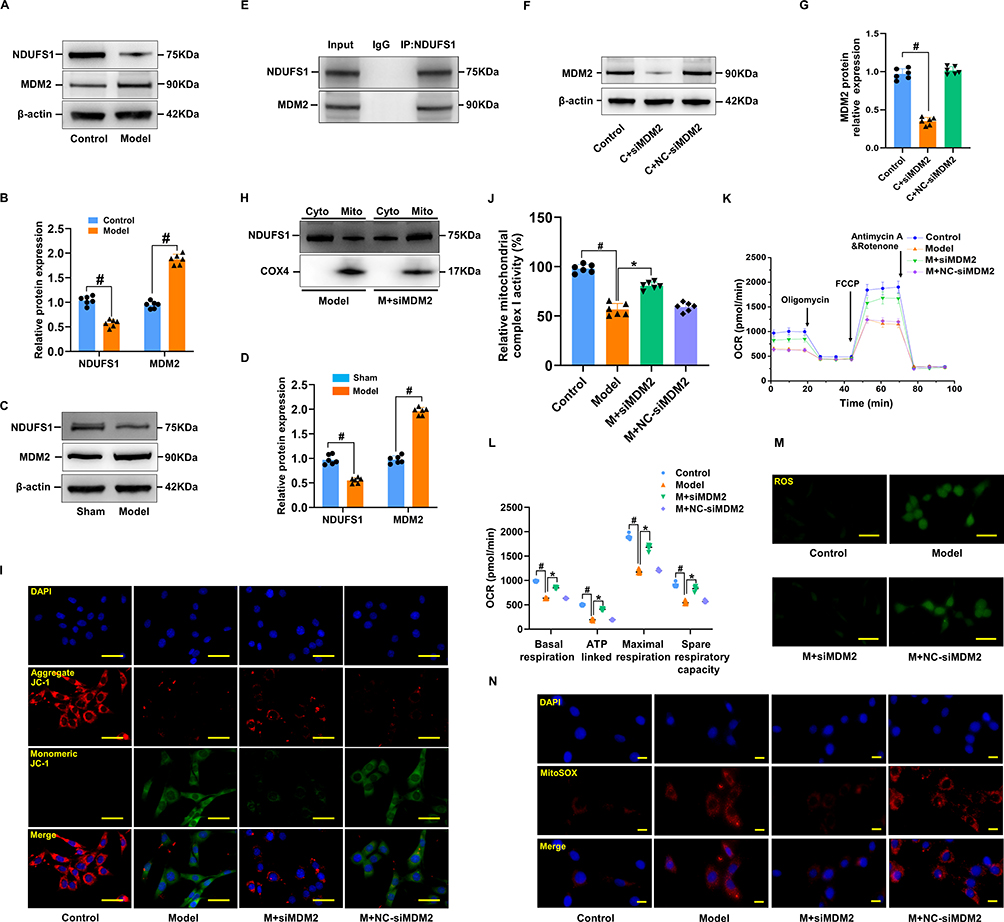 |
Figure 8 MDM2 deficiency improved mitochondrial function. (A and B) Representative Western blot images of NDUFS1 and MDM2, together with quantification in cardiomyocytes. (C and D) Immunoblot analysis of NDUFS1 and MDM in myocardial tissue. Data are presented as mean ± SD (n=6). #P < 0.05 vs Sham group. (E) Co-IP was used to illustrate the interplay between NDUFS1 and MDM2. (F and G) Immunoblot analysis of MDM2 in cardiomyocytes. (H) Western blotting illustrating the variation between the Model and Model+siMDM2 groups in terms of the cellular distribution of NDUFS1. (I) Illustrative images showed the fluorescence of JC-1 in cardiomyocytes induced with H/R and those treated with MDM2 knockdown. (J) Comparative analysis of the mitochondrial complex I activity of cardiomyocytes in various groups. (K and L) Cardiomyocytes were subjected to OCR and quantitative analysis. (M and N) Detection of intracellular and mitochondrial ROS production in the cardiomyocytes. Data are presented as mean ± SD (n=6). #P < 0.05 vs Control group; *P < 0.05 vs Model group. Scale bars: (M and I) 25 μm; (N) 10 μm. |
MDM2 Knockdown Inhibited the Ferroptosis of H/R-Induced Cardiomyocytes
To assess whether MDM2 knockdown protects the heart from MI/RI via its antioxidant mechanism, oxidative stress biomarkers such as MDA and GSH were measured. Our results indicated that MDM2 knockdown did indeed inhibit oxidative stress (Figure 9A and B). Immunofluorescence staining further indicated reduced expression of Gpx4 and a notably higher level of ptgs2 in the MDM2 knockdown group compared to the control (Figure 9C and D). Additionally, γH2AX immunofluorescence and CCK-8 assays showed that silencing MDM2 mitigated H/R-induced cardiomyocyte injury (Figure 9E and F). Collectively, these findings suggest that MDM2 silencing inhibits cardiomyocyte ferroptosis, offering protection against H/R-induced injury through its antioxidative effects.
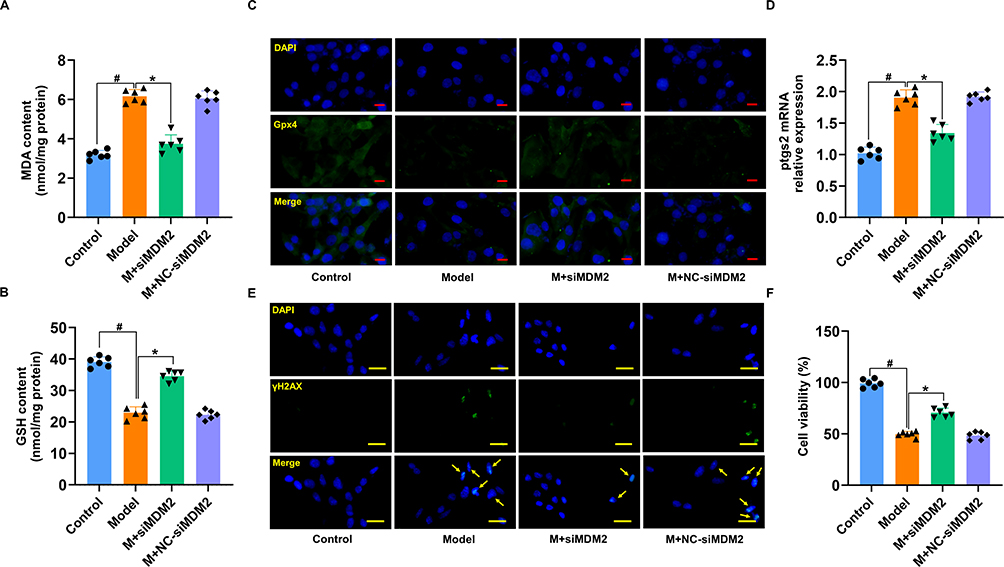 |
Figure 9 MDM2 deficiency suppressed cardiomyocytes ferroptosis. (A) MDA levels in H/R-treated cardiomyocytes induced by MDM2 knockdown. (B) GSH levels in H/R-treated cardiomyocytes induced by MDM2 knockdown. (C) Fluorescence imaging of Gpx4 in H/R-injured cardiomyocytes treated by silencing MDM2. (D) Relative mRNA expression of ptgs2. (E) Fluorescence imaging of γH2AX in H/R-injured cardiomyocytes treated by silencing MDM2. (F) The CCK-8 outcomes of H/R-damaged cardiomyocytes after MDM2-silencing therapy. Data are presented as mean ± SD (n=6). #P < 0.05 vs Control group; *P < 0.05 vs Model group. Scale bars: (C) 10 μm; (E) 25 μm. |
Lutein Facilitates Mitochondrial Translocation of NDUFS1 by Directly Binding and Sequestering MDM2
Western blotting revealed that the overall NDUFS1 expression remained unchanged in siMDM2-treated cardiomyocytes exposed to H/R (Figure 10A and B). Similarly, MDM2 expression remained unchanged in lutein@DTPP-treated cardiomyocytes (Figure 10C and D). To determine whether lutein could improve mitochondrial function by disrupting the interaction between MDM2 and NDUFS1, we conducted molecular docking studies. The docking score of lutein binding to MDM2 was −8.5 kcal/mol (Figure 10E). To further refine the binding patterns of MDM2 to lutein, we performed detailed MD simulations of MDM2-lutein complexes using the Desmond program. After 100 ns of MD simulation, we conducted an in-depth analysis of the simulated trajectories. First, we extracted RMSD (Root mean square deviation) data of MDM2 and lutein during the simulation. As shown in Figure 10F, the RMSD curve showed that the RMSD values of MDM2 and lutein stabilized after 50 ns of simulation, which indicates that the MDM2-lutein complex had reached a relatively stable state. Based on this observation, we selected a simulated trajectory of 50–100 ns for subsequent sampling analysis. As shown in Figure 10G, these conformations showed good consistency after superposition, indicating that the conformations of MDM2 and lutein maintained high stability during the simulation. At the same time, lutein could also stably bind to the active site of MDM2. Through in-depth analysis of the simulated trajectory, we extracted the RMSF data of MDM2 and lutein and calculated the corresponding B factors. The RMSF and B-factor diagrams indicated that the overall structure of the protein molecule shows low flexibility (the RMSF value of most regions is lower than 2.0 Å), suggesting that the protein structure maintains high stability during the simulation process (Figure 10H and I). Moreover, it can be seen from the RMSF diagram of lutein that most of the atoms are in a relatively stable state during the simulation, and the RMSF value is less than 2 Å (Figure 10J). This indicates that lutein can be firmly bound to the active site of MDM2, and the overall structure has a high stability. By exploring the interaction patterns within the stable interval of the dynamic locus (50–100 ns), we found that some specific amino acids play a crucial role in promoting the binding of lutein. As shown in the figure, these key amino acids mainly include Phe 97, Tyr 101, Arg 103, Phe 105, Asp 108, Leu 130, and Leu 194. They significantly enhance the binding stability and affinity by forming hydrophobic interactions and water bridge interactions with lutein (Figure 10K). As shown in Figure 10L, after statistical analysis of the interaction modes in the 50–100 ns dynamic simulation trajectory, we obtained the share of each interaction formation. The trajectory of the last 10 ns of the molecular dynamics of MDM2 and lutein complex (1000 frames in total) was extracted, and the binding free energy between MDM2 and lutein complex was calculated for each frame. As shown in Figure 10M, the MMGBSA of the lutein and MDM2 complex is stable at about −136.73 and relatively stable. Furthermore, lutein@DTPP promoted NDUFS1 translocation to mitochondria in cardiomyocytes subjected to H/R, but this effect was reduced with MDM2 overexpression (Figure 10N and O). These findings indicate that lutein enhances the mitochondrial translocation of NDUFS1 by directly binding to and sequestering MDM2.
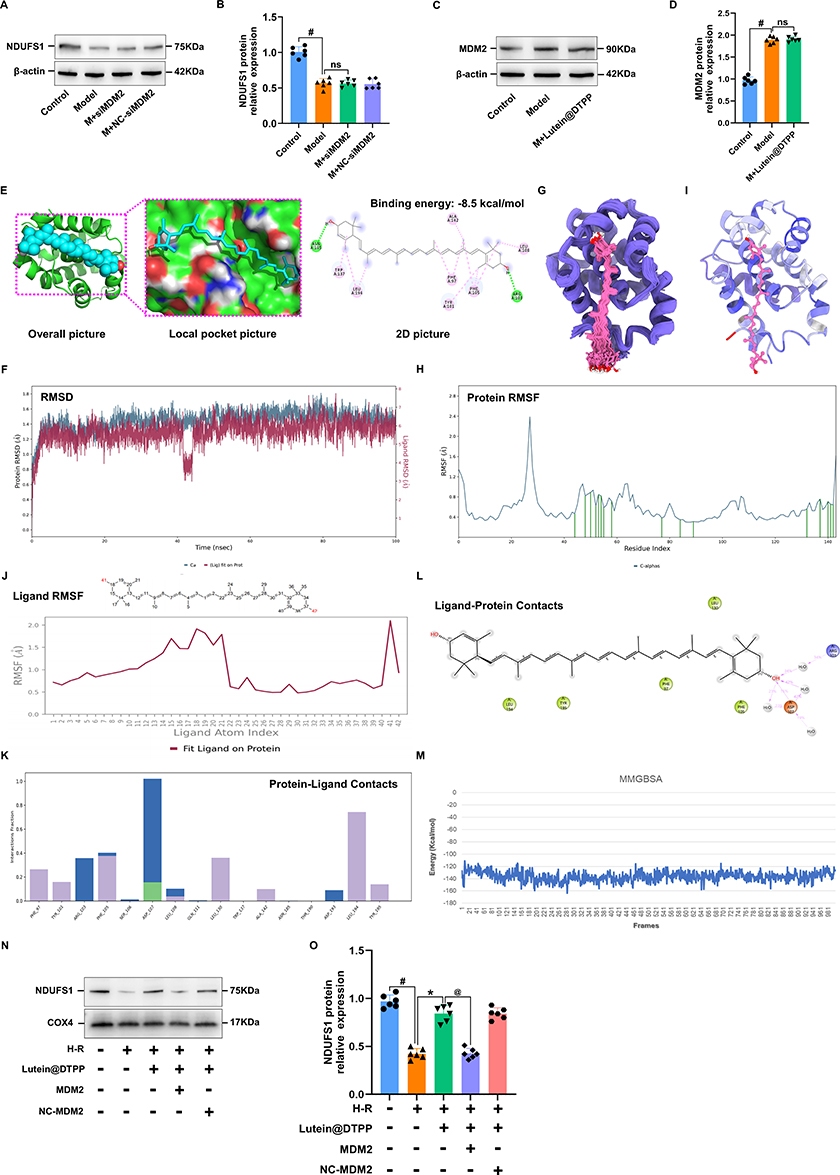 |
Figure 10 Molecular dynamics simulation results of lutein and MDM2 protein. (A and B) Representative Western blot images of NDUFS1. (C and D) Immunoblot analysis of MDM2 in cardiomyocytes. (E) Predicted binding mode between lutein and MDM2. (F) RMSD curves of MDM2 (blue) and lutein (red) in 100 ns molecular dynamics simulation. (G) 100 conformational superpositions saved per 1 ns in a 100 ns molecular dynamics simulation. (H) RMSF analysis diagram of MDM2 during 50–100 ns molecular dynamics simulation (the green marked part represents the location of amino acids interacting with the molecule). (I) B-factor distribution map based on MD molecular dynamics trajectory calculation. (J) RMSF (root-mean-square fluctuation) analysis of lutein during 50–100 ns molecular dynamics simulation. (K) Analysis of the contribution of key amino acids to lutein binding at binding sites. (L) A two-dimensional diagram of the binding model of lutein and MDM2 during molecular dynamics simulation. (M) The binding free energy of the last 10 ns of lutein and MDM2 is 1000 frames. (N and O) NDUFS1 protein expression in mitochondria of H/R-induced cardiomyocytes upon lutein@DTPP treatment under the condition of MDM2 overexpression. Data are displayed as mean ± SD (n=6). #P < 0.05 vs Control group; *P < 0.05 vs Model group; @P < 0.05 vs M+lutein@DTPP group. |
Overexpression of MDM2 Reversed the Improvement of Mitochondrial Function by lutein@DTPP
To further investigate the myocardial protective effect of lutein@DTPP through the regulation of the level of NDUFS1 in mitochondria, we detected mitochondrial ROS, mitochondrial membrane potential, MCI activity, and mitochondrial respiratory function after overexpression of MDM2. The data indicated that overexpression of MDM2 counteracted the lutein@DTPP-induced reduction in mitochondrial ROS and JC-1 monomer levels, while it also enhanced MCI activity and restored mitochondrial respiratory function in H/R-exposed cardiomyocytes treated with lutein@DTPP (Figure 11A–E). Additionally, CCK-8 analysis showed that MDM2 overexpression abrogates the increase in cell viability of H/R-exposed cardiomyocytes treated with lutein@DTPP (Figure 11F). These findings collectively suggest that lutein@DTPP supports mitochondrial function recovery through the NDUFS1 pathway, a process that is modulated by MDM2.
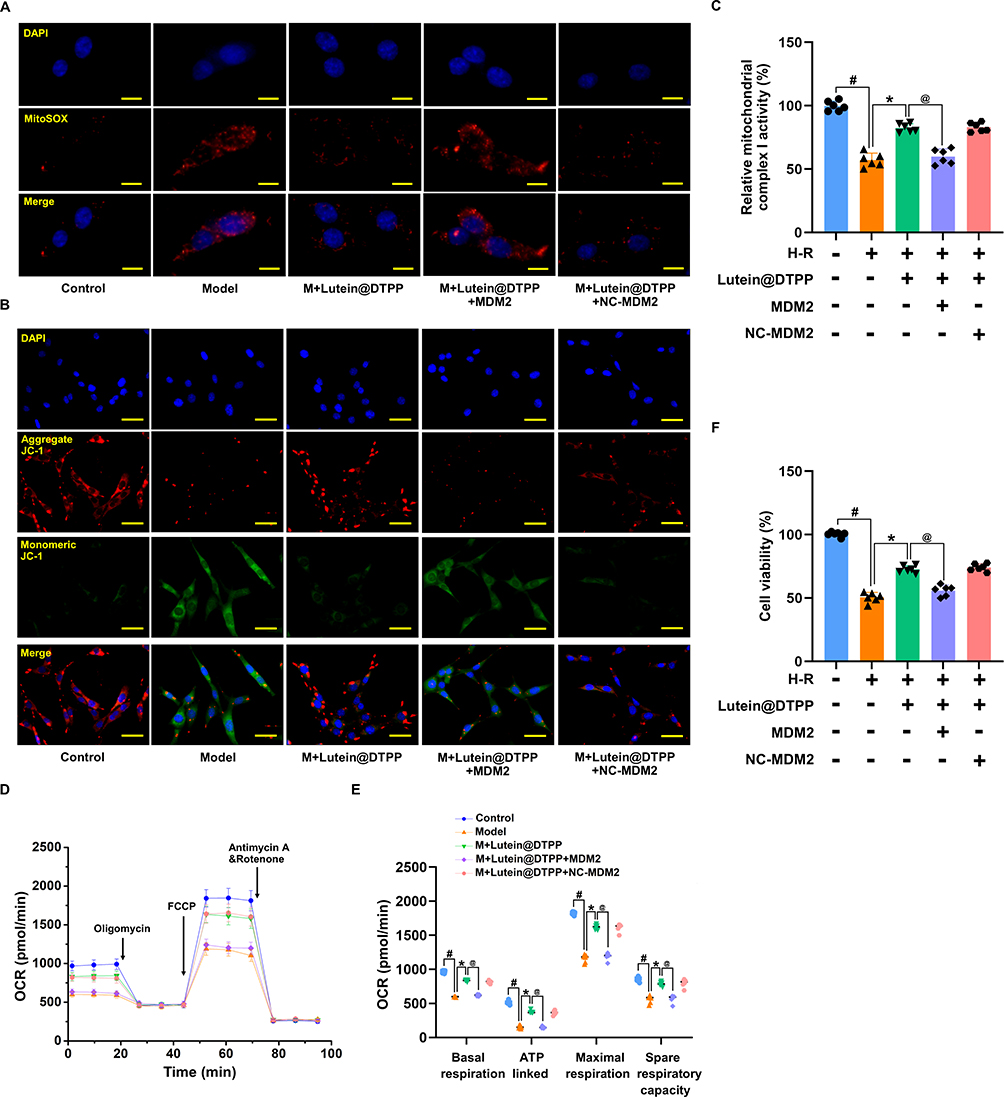 |
Figure 11 Overexpression of MDM2 abrogated the protective effect of lutein@DTPP on mitochondria. (A) Representative fluorescence images of mitochondrial ROS production. (B) Images that exemplify the fluorescence of JC-1 in cardiomyocytes. (C) Analysis of mitochondrial complex I activity in cardiomyocytes. (D and E) Measurement of OCR in cardiomyocytes and the corresponding quantitative analysis. (F) Cardiomyocyte viability is assessed via CCK-8. Data are displayed as mean ± SD (n=6). #P < 0.05 vs Control group; *P < 0.05 vs Model group; @P < 0.05 vs M+lutein@DTPP group. Scale bars: (A) 10 μm; (B) 25 μm. |
Discussion
Despite advances in the treatment of acute myocardial infarction (AMI) and increased rates of survival, remains the leading cause of mortality worldwide.41 Developing innovative strategies for cardiac protection is critical to mitigate the damaging effects of AMI. One essential aspect of post-ischemic event recovery, such as after a heart attack, is the preservation of tissues and maintaining a healthy mitochondrial population, which is key to the normal function of cells and tissues.42 It has been observed that after I/R injury, the mitochondria in cardiomyocytes are severely damaged due to high oxidative stress, leading to excessive production of ROS.43 In this study, a PCM-SH-coated lutein nanocomplex (lutein@DTPP) was developed as a noninvasive, targeted therapy for myocardial ischemia/reperfusion injury (MI/RI). The lutein@DTPP accumulated in the injured myocardium, where it was taken up by cardiomyocytes, demonstrating therapeutic efficacy against MI/RI. Furthermore, the research delved into the underlying molecular mechanisms through which lutein@DTPP exerts its protective effects against MI/RI. Initially, the targeted delivery of lutein@DTPP to cardiomyocytes was successfully achieved. Once internalized by the cells, the high levels of ROS in the myocardial tissue triggered the decomposition of lutein@DTPP, leading to the release of lutein into the cytoplasm. A key mechanism uncovered in this study was the role of MDM2 in binding to NDUFS1, a mitochondrial protein essential for cellular respiration. This binding inhibited the transport of NDUFS1 to mitochondria, contributing to mitochondrial dysfunction and increased oxidative stress. In contrast, the translocation of NDUFS1 from the cytosol to mitochondria was facilitated by MDM2 deficiency, which improved mitochondrial function, decreased mitochondrial ROS production, and restrained cardiomyocyte ferroptosis. Moreover, lutein@DTPP enhanced the function of mitochondria and decreased the rate of cardiomyocyte ferroptosis by promoting NDUFS1 translocation from the cytosol to mitochondria by binding to MDM2. These findings collectively suggest that lutein@DTPP exerts cardioprotective effects by inhibiting MDM2-mediated suppression of NDUFS1 translocation, thus preventing mitochondrial dysfunction and ferroptosis. This approach provides a promising framework for the therapeutic application of lutein in the treatment of MI/RI.
Mitochondria are essential for cellular energy production, generating ATP and serving as the primary source of intracellular ROS. Mitochondrial dysfunction and oxidative stress are key contributors to the pathogenesis of MI/RI.44,45 Nevertheless, the origins of mitochondrial ROS and the factors that induce mitochondrial dysfunction are still unclear. NDUFS1, a key member of MCI, regulates oxidative phosphorylation and the generation of ROS in the mitochondrion.46,47 Studies have shown that NDUFS1 is involved in mitochondrial ROS production and apoptosis.48
Considering that enhanced mitochondrial ROS generation and impaired mitochondrial activity are the primary causes of cardiac dysfunction in several CVDs,49,50 a therapeutic approach that targets NDUFS1 might potentially be used to treat MI/RI and other CVDs. According to our findings, lutein@DTPP facilitates the movement of NDUFS1 from the cytosol to the mitochondria, where it subsequently suppresses the formation of ROS inside the mitochondria in MI/RI, thus enhancing cardiac function.
In addition to acting as a ubiquitin ligase, the MDM2 protein identifies and degrades the N-terminal trans-activation domain of p53. Moreover, preliminary reports on MDM2 underscored the key finding that levels of MDM2 in human tumor samples vary between 5 and 50 times those of basal expression, revealing an oncogenic effect via blocking p53 function.51,52 However, the exact nature of MDM2’s involvement in the development of CVDs remains unexplained. Recent studies have found that hypertrophic cardiomyopathy is characterized by microvascular dysfunction, a condition mediated by MDM2’s regulation of hypoxia-inducible factor (HIF) signaling.53 Similarly, our research observed a significant elevation of MDM2 expression in both rats with MI/RI and cardiomyocytes exposed to H/R. The knockdown of MDM2 in these models led to a notable increase in mitochondrial levels of NDUFS1, which restored mitochondrial membrane potential, improved oxidative respiratory chain efficiency, and reduced mitochondrial ROS production. Furthermore, our findings demonstrated that MDM2 directly binds to and sequesters NDUFS1, blocking its translocation to the mitochondria. This interaction disrupts oxidative phosphorylation and contributes to mitochondrial dysfunction. Our molecular docking analysis further supported these findings by demonstrating a binding interaction between lutein and MDM2.
Lutein, a naturally occurring carotenoid found in dark green leafy vegetables such as kale and spinach, is widely known for its role in preventing macular degeneration and cataracts, as well as its impact on visual function.54 Moreover, the possible therapeutic benefits of lutein on CVDs have been attributed to several biological mechanisms, including vascular alterations, antioxidant properties, immune responses, and inflammatory function. These combined mechanisms highlight the potential positive influence of lutein on both the overall health and particular organ systems. Recently, lutein was shown to attenuate angiotensin II-mediated cardiac remodeling via the suppression of AP-1/IL-11 signaling.55 Another study on rats demonstrated that lutein ameliorates streptozotocin-mediated cardiac oxidative stress.56 In addition, a previous study indicated that lutein demonstrates cardioprotective properties via the MIAT/miR-200a/Nrf2/TXINP pathway in an experimental animal model of isoprenaline-mediated myocardial infarction.57 However, the possible protective properties of lutein on MI/RI are yet to be reported. Our findings demonstrate that targeted delivery of lutein to damaged cardiomyocytes enhances mitochondrial function and mitigates the effects of MI/RI. Mechanistically, lutein binds to MDM2, promoting the translocation of NDUFS1 from the cytosol to mitochondria in cardiomyocytes, thereby improving mitochondrial function and reducing mitochondrial dysfunction. These insights underscore lutein’s potential as a therapeutic agent in the management of MI/RI.
In recent years, research on bioimaging and targeted delivery systems has become a hot spot in the treatment of diseases.58–60 Despite the tremendous progress made in developing stimuli-sensitive drug delivery methods to improve therapeutic efficacy,61 their use in MI/RI is still in the nascent stage. Our work focused on investigating ROS, one of the numerous microenvironments observed in MI/RI. ROS is overproduced and has been recognized as a promoter of myocardial damage in MI/RI.30,62 The therapeutic applicability of lutein administered systemically to treat IR injury is strongly constrained by its biological instability and poor targeting capacity. Thus, a ROS-responsive lutein delivery platform was devised in this study by integrating the natural inflammatory response and excessive ROS release following reperfusion injury. By using the TK functional group, we were able to achieve ROS-responsive linkage. Surface modification using a ROS-sensitive linker with a ROS-sensitive TK functional group was effective in the preparation of DSPE-TK-PEG2K. This formulation enabled the targeted delivery of lutein to the damaged myocardium. In a rat model of MI/RI, intravenous administration of our formulation resulted in a significant increase in the concentration of lutein within the injured cardiac region. Consequently, this targeted delivery improved mitochondrial function and suppressed ferroptosis in cardiomyocytes, effectively enhancing overall cardiac function. This delivery method incorporates several advantages, including drug protection, targeted distribution, and controlled release, thus demonstrating substantial clinical translational potential for the treatment of MI/RI.
However, this study also has some limitations. At present, there are predictable instances of MI/RI in clinical practice. For example, hemodynamic abnormalities during open-heart surgery may lead to severe myocardial ischemia and arrhythmia. Therefore, prophylactic drug administration before surgery may reduce surgical risks. Additionally, myocardial ischemia can occur during anesthesia, often due to allergic reactions to anesthetic agents, improper administration of these drugs, pre-existing cardiovascular conditions, surgical traction, or hypothermia. Therefore, our experimental design included the prophylactic administration of lutein@DTPP to mitigate MI/RI. However, due to the inherent uncertainties associated with prophylactic administration, it has not been widely adopted in clinical practice for treating MI/RI. Consequently, the clinical use of prophylactic drug administration to address MI/RI poses new challenges for researchers.
In summary, active targeting was achieved through the development of PCM-SH chimeric ROS-sensitive liposomes, which enabled the targeted and responsive delivery of lutein. Once lutein was intravenously injected, it could reach the cardiac injury site through the chemotaxis of the myocardial-specific targeting peptide PCM-SH was released explosively in response to elevated ROS levels in damaged cardiomyocytes. Mechanistically, we demonstrated that MDM2 was upregulated in the hearts of rats exposed to MI/RI. Moreover, our finding revealed that NDUFS1, a crucial Fe/S cluster in complex I that facilitates effective mitochondrial respiration, is sequestered due to the cytosolic accumulation of MDM2. The interaction between MDM2 and NDUFS1 can lead to mitochondrial dysfunction, increased ROS production, and the subsequent activation of ferroptosis in cardiomyocytes. Notably, lutein promoted the mitochondrial translocation of NDUFS1, thereby rescuing mitochondrial function and inhibiting ferroptosis by directly binding to and sequestering MDM2. Given the reduction of NDUFS1 in mitochondria observed in MI/RI, the restoration of NDUFS1 utilizing the DSPE-TK-PEG2K-PCM-SH approach substantiates the potential for a novel therapeutic solution for MI/RI and associated CVDs based on robust experimental data.
Conclusion
In summary, active targeting was achieved by the development of PCM-SH chimeric ROS-sensitive liposomes, which enabled the targeted and responsive delivery of lutein. Mechanistically, we demonstrated that lutein@DTPP promoted the mitochondrial translocation of NDUFS1 to rescue mitochondrial function and inhibited the ferroptosis of cardiomyocytes by directly binding and sequestering MDM2.
Abbreviations
MI/RI, myocardial ischemia-reperfusion injury; H/R, hypoxia-reoxygenation; ROS, reactive oxygen species; ATP, adenosine triphosphate; NDUFS1, NADH-ubiquinone oxidoreductase 75 kDa Fe-S protein 1; NADH, nicotinamide adenine dinucleotide; CMECs, cardiac microvascular endothelial cells; Lutein@DTPP, DSPE-PEG2K-PCM-SH and DSPE-TK-PEG2K coated lutein; Lutein@DTP, DSPE-PEG2K and DSPE-TK-PEG2K coated lutein; 4-HNE, 4-Hydroxynonenal; OCR, oxygen consumption rate; MDM2, mouse double minute-2.
Data Sharing Statement
All data used during the current study are available from the corresponding author upon reasonable request.
Ethics Statement
The procedures for the use of animals (male Wistar rat; 200-250 g) in this study were in accordance with the regulations of the Ethics Committees of Harbin Medical University (ethics approval number: HMUDQ20240306001).
Acknowledgment
This work was supported by the Scientific Research Project of the Provincial Scientific Research Institute of Heilongjiang Province (CZKYF2022-1-B007), the Post-doctoral Research Start-up Fund Project of Heilongjiang Province (LBH-Q19155) and Key research and development project of Lishui (2023zdyf21).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Hao Y, Zhao D, Liu J, et al. Performance of management strategies with class i recommendations among patients hospitalized with st-segment elevation myocardial infarction in China. JAMA Cardiol. 2022;7(5):484–491. doi:10.1001/jamacardio.2022.0117
2. Zhang Z, Dalan R, Hu Z, et al. Reactive oxygen species scavenging nanomedicine for the treatment of ischemic heart disease. Adv Mater. 2022;34(35):e2202169. doi:10.1002/adma.202202169
3. Huang Q, Yao Y, Wang Y, et al. Ginsenoside Rb2 inhibits p300-mediated SF3A2 acetylation at lysine 10 to promote Fscn1 alternative splicing against myocardial ischemic/reperfusion injury. J Adv Res. 2023;13:S2090–1232(23)00399–5.
4. Ale-Agha N, Jakobs P, Goy C, et al. Mitochondrial telomerase reverse transcriptase protects from myocardial ischemia/reperfusion injury by improving complex i composition and function. Circulation. 2021;144(23):1876–1890. doi:10.1161/CIRCULATIONAHA.120.051923
5. Song R, Dasgupta C, Mulder C, Zhang L. MicroRNA-210 controls mitochondrial metabolism and protects heart function in myocardial infarction. Circulation. 2022;145(15):1140–1153. doi:10.1161/CIRCULATIONAHA.121.056929
6. Ong SB, Kalkhoran SB, Cabrera-Fuentes HA, Hausenloy DJ. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur J Pharmacol. 2015;763:104–114. doi:10.1016/j.ejphar.2015.04.056
7. Dongworth RK, Hall AR, Burke N, Hausenloy DJ. Targeting mitochondria for cardioprotection: examining the benefit for patients. Future Cardiol. 2014;10(2):255–272. doi:10.2217/fca.14.6
8. Mao Y, Zhang J, Zhou Q, et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell Res. 2024;34(1):13–30. doi:10.1038/s41422-023-00864-6
9. Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166(3):555–566. doi:10.1016/j.cell.2016.07.002
10. Ni Y, Hagras MA, Konstantopoulou V, Mayr JA, Stuchebrukhov AA, Meierhofer D. Mutations in NDUFS1 cause metabolic reprogramming and disruption of the electron transfer. Cells. 2019;8(10):1149. doi:10.3390/cells8101149
11. Letts JA, Degliesposti G, Fiedorczuk K, Skehel M, Sazanov LA. Purification of ovine respiratory complex I results in a highly active and stable preparation. J Biol Chem. 2016;29(47):24657–24675. doi:10.1074/jbc.M116.735142
12. Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281(43):32724–32727. doi:10.1074/jbc.M607135200
13. Hirst J. Mitochondrial complex I. Annu Rev Biochem. 2013;82(1):551–575. doi:10.1146/annurev-biochem-070511-103700
14. Dunham-Snary KJ, Wu D, Potus F, et al. Ndufs2, a core subunit of mitochondrial complex i, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ Res. 2013;124(12):1727–1746. doi:10.1161/CIRCRESAHA.118.314284
15. Sharanek A, Burban A, Laaper M, et al. OSMR controls glioma stem cell respiration and confers resistance of glioblastoma to ionizing radiation. Nat Commun. 2020;11(1):4116. doi:10.1038/s41467-020-17885-z
16. Hoefs SJ, Skjeldal OH, Rodenburg RJ, et al. Novel mutations in the NDUFS1 gene cause low residual activities in human complex I deficiencies. Mol Genet Metab. 2010;100(3):251–256. doi:10.1016/j.ymgme.2010.03.015
17. Zou R, Tao J, Qiu J, et al. Ndufs1 deficiency aggravates the mitochondrial membrane potential dysfunction in pressure overload-induced myocardial hypertrophy. Oxid Med Cell Longev. 2021;2021(1):5545261. doi:10.1155/2021/5545261
18. Qi B, He L, Zhao Y, et al. Akap1 deficiency exacerbates diabetic cardiomyopathy in mice by NDUFS1-mediated mitochondrial dysfunction and apoptosis. Diabetologia. 2020;63(5):1072–1087. doi:10.1007/s00125-020-05103-w
19. Qi B, Song L, Hu L, et al. Cardiac-specific overexpression of Ndufs1 ameliorates cardiac dysfunction after myocardial infarction by alleviating mitochondrial dysfunction and apoptosis. Exp Mol Med. 2022;54(7):946–960. doi:10.1038/s12276-022-00800-5
20. Gong X, Rubin LP. Role of macular xanthophylls in prevention of common neovascular retinopathies: retinopathy of prematurity and diabetic retinopathy. Arch Biochem Biophys. 2015;572:40–48. doi:10.1016/j.abb.2015.02.004
21. Nwachukwu ID, Udenigwe CC, Aluko RE. Lutein and zeaxanthin: production technology, bioavailability, mechanisms of action, visual function, and health claim status. Trends Food Sci Technol. 2016;49:74–84. doi:10.1016/j.tifs.2015.12.005
22. Hwang JS, Han SG, Lee CH, Seo HG. Lutein suppresses hyperglycemia induced premature senescence of retinal pigment epithelial cells by upregulating SIRT1. J Food Biochem. 2018;42(3):e12495. doi:10.1111/jfbc.12495
23. Choudhary R, Tandon RV. Consumption of functional food and our health concerns. Pak J Physiol. 2009;5:76–83.
24. Han H, Cui W, Wang L, et al. Lutein prevents high fat diet-induced atherosclerosis in ApoE-deficient mice by inhibiting NADPH oxidase and increasing PPAR expression. Lipids. 2015;50(3):261–273. doi:10.1007/s11745-015-3992-1
25. Liu Y, Yang G, Huo S, et al. Lutein suppresses ferroptosis of cardiac microvascular endothelial cells via positive regulation of IRF in cardiac hypertrophy. Eur J Pharmacol. 2023;959:176081. doi:10.1016/j.ejphar.2023.176081
26. Sharavana G, Joseph GS, Baskaran V. Lutein attenuates oxidative stress markers and ameliorates glucose homeostasis through polyol pathway in heart and kidney of STZ-induced hyperglycemic rat model. Eur J Nutr. 2017;56(8):2475–2485. doi:10.1007/s00394-016-1283-0
27. Wei Y, Zhu M, Li S, et al. Engineered biomimetic nanoplatform protects the myocardium against ischemia/reperfusion injury by inhibiting pyroptosis. ACS Appl Mater Interfaces. 2021;13(29):33756–33766. doi:10.1021/acsami.1c03421
28. Shi P, Li M, Song C, et al. Neutrophil-like cell membrane-coated siRNA of lncRNA AABR07017145.1 therapy for cardiac hypertrophy via inhibiting ferroptosis of CMECs. Mol Ther Nucleic Acids. 2021;27:16–36. doi:10.1016/j.omtn.2021.10.024
29. Shi P, Wu J, Li M, et al. Upregulation of Hsp27 via further inhibition of histone H2A ubiquitination confers protection against myocardial ischemia/reperfusion injury by promoting glycolysis and enhancing mitochondrial function. Cell Death Discov. 2023;9(1):466. doi:10.1038/s41420-023-01762-x
30. Chakouri N, Farah C, Matecki S, et al. Screening for in-vivo regional contractile defaults to predict the delayed doxorubicin cardiotoxicity in juvenile rat. Theranostics. 2020;10(18):8130–8142. doi:10.7150/thno.47407
31. Wang R, Wang M, Zhou J, Dai Z, Sun G, Sun X. Calenduloside E suppresses calcium overload by promoting the interaction between L-type calcium channels and Bcl2-associated athanogene 3 to alleviate myocardial ischemia/reperfusion injury. J Adv Res. 2020;34:173–186. doi:10.1016/j.jare.2020.10.005
32. Xu L, Shao Y, Ren L, et al. IQGAP2 inhibits migration and invasion of gastric cancer cells via elevating SHIP2 phosphatase activity. Int J Mol Sci. 2020;21(6):1968. doi:10.3390/ijms21061968
33. Ye Y, Qian XY, Xiao MM, et al. Decreased Sp1 expression mediates downregulation of SHIP2 in gastric cancer cells. Int J Mol Sci. 2017;18(1):220. doi:10.3390/ijms18010220
34. Zhou H, Li D, Zhu P, et al. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J Pineal Res. 2018;65(3):e12503. doi:10.1111/jpi.12503
35. Zheng Y, Wan G, Yang B, Gu X, Lin J. Cardioprotective natural compound pinocembrin attenuates acute ischemic myocardial injury via enhancing glycolysis. Oxid Med Cell Longev. 2020;2020:4850328. doi:10.1155/2020/4850328
36. Wang S, Chen Y, Wu C, Wang Y, Lin W, Bu R. Trehalose alleviates myocardial ischemia/reperfusion injury by inhibiting nlrp3-mediated pyroptosis. Appl Biochem Biotechnol. 2024;6:1194–1210. doi:10.1007/s12010-023-04613-8
37. Ertracht O, Malka A, Atar S, Binah O. The mitochondria as a target for cardioprotection in acute myocardial ischemia. Pharmacol Ther. 2014;142(1):33–40. doi:10.1016/j.pharmthera.2013.11.003
38. van der Pol A, van Gilst WH, Voors AA, van der Meer P, van Gilst WH. Treating oxidative stress in heart failure: past, present and future. Eur J Heart Failure. 2019;21(4):425–435. doi:10.1002/ejhf.1320
39. Guo J, Wang S, Wan X, et al. Mitochondria-derived methylmalonic acid aggravates ischemia-reperfusion injury by activating reactive oxygen species-dependent ferroptosis. Cell Commun Signal. 2024;22(1):53. doi:10.1186/s12964-024-01479-z
40. Yan J, Li Z, Liang Y, et al. Fucoxanthin alleviated myocardial ischemia and reperfusion injury through inhibition of ferroptosis via the NRF2 signaling pathway. Food Funct. 2023;14(22):10052–10068. doi:10.1039/D3FO02633G
41. Wang X, Pu J. Recent advances in cardiac magnetic resonance for imaging of acute myocardial infarction. Small Methods. 2023;22:e2301170.
42. Gumpper-Fedus K, Park KH, Ma H, et al. MG53 preserves mitochondrial integrity of cardiomyocytes during ischemia reperfusion-induced oxidative stress. Redox Biol. 2022;54:102357. doi:10.1016/j.redox.2022.102357
43. Luan Y, Luan Y, Feng Q, Chen X, Ren KD, Yang Y. Emerging role of mitophagy in the heart: therapeutic potentials to modulate mitophagy in cardiac diseases. Oxid Med Cell Longev. 2021;2021(1):3259963. doi:10.1155/2021/3259963
44. Li S, Li F, Wang Y, et al. Multiple delivery strategies of nanocarriers for myocardial ischemia-reperfusion injury: current strategies and future prospective. Drug Deliv. 2023;31(1):2298514. doi:10.1080/10717544.2023.2298514
45. Liu G, Lv Y, Wang Y, et al. Remote ischemic preconditioning reduces mitochondrial apoptosis mediated by calpain 1 activation in myocardial ischemia-reperfusion injury through calcium channel subunit Cacna2d3. Free Radic Biol Med. 2023;212:80–93. doi:10.1016/j.freeradbiomed.2023.12.030
46. Kussmaul L, Hirst J. The mechanism of superoxide production by NADH: ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci U S A. 2006;103(20):7607–7612. doi:10.1073/pnas.0510977103
47. Lopez-Fabuel I, Le Douce J, Logan A, et al. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc Natl Acad Sci U S A. 2016;113(46):13063–13068. doi:10.1073/pnas.1613701113
48. Sulaiman D, Li J, Devarajan A, et al. Paraoxonase 2 protects against acute myocardial ischemia-reperfusion injury by modulating mitochondrial function and oxidative stress via the PI3K/Akt/GSK-3β RISK pathway. J Mol Cell Cardiol. 2019;129:154–164. doi:10.1016/j.yjmcc.2019.02.008
49. Dey S, DeMazumder D, Sidor A, Foster DB, O’Rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. 2018;123(3):356–371. doi:10.1161/CIRCRESAHA.118.312708
50. Bueso-Ramos CE, Manshouri T, Haidar MA, Huh YO, Keating MJ, Albitar M. Multiple patterns of MDM-2 deregulation in human leukemias: implications in leukemogenesis and prognosis. Leuk Lymphoma. 1995;17(1–2):13–18. doi:10.3109/10428199509051698
51. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358(6381):80–83. doi:10.1038/358080a0
52. Shridhar P, Glennon MS, Pal S, et al. MDM2 regulation of HIF signaling causes microvascular dysfunction in hypertrophic cardiomyopathy. Circulation. 2023;148(23):1870–1886. doi:10.1161/CIRCULATIONAHA.123.064332
53. Kijlstra A, Tian Y, Kelly ER, Berendschot TT. Lutein: more than just a filter for blue light. Prog Retin Eye Res. 2012;31(4):303–315. doi:10.1016/j.preteyeres.2012.03.002
54. Chen Y, Wang L, Huang S, et al. Lutein attenuates angiotensin II- induced cardiac remodeling by inhibiting AP-1/IL-11 signaling. Redox Biol. 2021;44:102020. doi:10.1016/j.redox.2021.102020
55. Alrejaie S, Alhusaini K. Lutein ameliorates streptozotocin-induced cardiac oxidative stress in rats. J Immunol. 2016;196(1_Supplement):216. doi:10.4049/jimmunol.196.Supp.216.12
56. Abdelmonem M, Ibrahim SM, Essam RM, Amin HAA, Abd-Elmawla MA. Lutein exerts its cardioprotective effect against the experimental model of isoprenaline-induced myocardial infarction via MIAT/miR-200a/Nrf2/TXINP pathway. J Biochem Mol Toxicol. 2021;35(11):e22899. doi:10.1002/jbt.22899
57. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12(11):991–1003. doi:10.1038/nmat3776
58. Li C, Tu L, Yang J, et al. Acceptor engineering of metallacycles with high phototoxicity indices for safe and effective photodynamic therapy. Chem Sci. 2023;14(11):2901–2909. doi:10.1039/D2SC06936A
59. Xu Y, Li C, An J, et al. Construction of a 980 nm laser-activated Pt(II) metallacycle nanosystem for efficient and safe photo-induced bacteria sterilization. Sci China Chem. 2023;66(1):155–163. doi:10.1007/s11426-022-1440-2
60. Tu L, Li C, Xiong X, et al. Engineered metallacycle-based supramolecular photosensitizers for effective photodynamic therapy. Angew Chem Int Ed Engl. 2023;62(15):e202301560. doi:10.1002/anie.202301560
61. Li X, Jin Y. Inhibition of miR-182-5p attenuates ROS and protects against myocardial ischemia-reperfusion injury by targeting STK17A. Cell Cycle. 2022;21(15):1639–1650. doi:10.1080/15384101.2022.2060640
62. Shen S, He F, Cheng C, Xu B, Sheng J. Uric acid aggravates myocardial ischemia-reperfusion injury via ROS/NLRP3 pyroptosis pathway. Biomed Pharmacother. 2021;133:110990. doi:10.1016/j.biopha.2020.110990
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.