")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 17
Targeting the Epigenetic Marks in Type 2 Diabetes Mellitus: Will Epigenetic Therapy Be a Valuable Adjunct to Pharmacotherapy?
Authors Odimegwu CL, Uwaezuoke SN, Chikani UN, Mbanefo NR, Adiele KD, Nwolisa CE, Eneh CI , Ndiokwelu CO , Okpala SC, Ogbuka FN, Odo KE, Ohuche IO, Obiora-Izuka CE
Received 20 May 2024
Accepted for publication 3 August 2024
Published 21 September 2024 Volume 2024:17 Pages 3557—3576
DOI https://doi.org/10.2147/DMSO.S479077
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Konstantinos Tziomalos
Chioma Laura Odimegwu,1 Samuel Nkachukwu Uwaezuoke,1 Ugo N Chikani,1 Ngozi Rita Mbanefo,1 Ken Daberechi Adiele,1 Charles Emeka Nwolisa,2 Chizoma Ihuarula Eneh,3 Chibuzo Obiora Ndiokwelu,1 Somkenechi C Okpala,1 Francis N Ogbuka,3 Kenneth E Odo,1 Ijeoma Onyinyechukwu Ohuche,1 Chinelo Elizabeth Obiora-Izuka1
1Department of Pediatrics, the University of Nigeria Teaching Hospital (UNTH), Ituku-Ozalla Enugu, Nigeria; 2Department of Pediatrics, Federal University Teaching Hospital, Owerri, Nigeria; 3Department of Pediatrics, Enugu State University Teaching Hospital (ESUTH), Enugu, Nigeria
Correspondence: Samuel Nkachukwu Uwaezuoke, Department of Pediatrics, UNTH, Ituku-Ozalla Enugu, Nigeria, Tel +234 8033248108, Email [email protected]
Abstract: Although genetic, environmental, and lifestyle factors largely contribute to type 2 diabetes mellitus (T2DM) risk, the role of epigenetics in its pathogenesis is now well established. The epigenetic mechanisms in T2DM mainly consist of DNA methylation, histone modifications and regulation by noncoding RNAs (ncRNAs). For instance, DNA methylation at CpG islands in the promoter regions of specific genes encoding insulin signaling and glucose metabolism suppresses these genes. Modulating the enzyme mediators of these epigenetic marks aims to restore standard gene expression patterns and improve glycemic control. In targeting these epigenetic marks, using epigenetic drugs such as DNA methyltransferase (DNAMT), histone deacetylase (HDAC) and histone acetyltransferase (HAT) inhibitors has led to variable success in humans and experimental murine models. Specifically, the United States’ Food and Drug Administration (US FDA) has approved DNAMT inhibitors like 5-azacytidine and 5-aza-2ʹ-deoxycytidine for use in diabetic retinopathy: a T2DM microvascular complication. These DNAMT inhibitors block the genes for methylation of mitochondrial superoxide dismutase 2 (SOD2) and matrix metallopeptidase 9 (MMP-9): the epigenetic marks in diabetic retinopathy. Traditional pharmacotherapy with metformin also have epigenetic effects in T2DM and positively alter disease outcomes when combined with epigenetic drugs like DNAMT and HDAC inhibitors, raising the prospect of using epigenetic therapy as a valuable adjunct to pharmacotherapy. However, introducing small interfering RNAs (siRNAs) in cells to silence specific target genes remains in the exploratory phase. Future research should focus on regulating gene expression in T2DM using long noncoding RNA (lncRNA) molecules, another type of ncRNA. This review discusses the epigenetics of T2DM and that of its macro- and microvascular complications, and the potential benefits of combining epigenetic therapy with pharmacotherapy for optimal results.
Keywords: type 2 diabetes mellitus, epigenetics, DNA methylation, histone modifications, noncoding RNA regulation, therapeutics, epigenetic therapy
Introduction
In adulthood, type 2 diabetes mellitus (T2DM) is the prevalent form of diabetes, whereas type 1 diabetes mellitus (T1DM) occurs predominantly in children. However, the incidence of T2DM has been increasing in the pediatric age group within the past four decades because of the rising trend of childhood obesity.1 The disease results from reduced insulin sensitivity at the target cells or decreased production by the pancreatic ß-cells. The putative risk factors include genetics, lifestyle factors, and environmental factors.2 More importantly, a synergy of these factors rather than a single factor contributes to T2DM risk. Lifestyle and environmental factors are modifiable, while genetics and female sex constitute non-modifiable risk factors.2 Given the relationship between T2DM and body mass index, women are reportedly more obese than men in some sub-Saharan African settings,3 underscoring the role of the female sex as a risk factor. Another possible risk factor is maternal nutritional status during fetal embryogenesis, which may play a role through DNA methylation.4 Poor maternal nutritional status is associated with intra-uterine growth retardation (IUGR) and subsequent low birth weight. A study from the United Kingdom, three decades ago, reported that individuals with a birth weight below 2.5 kg had high odds of developing T2DM later in life.5
Although the genetic mechanisms of T2DM remain unclear, single nucleotide polymorphism (SNP) is a recognized mechanism that increases T2DM risk. Genome-wide association studies (GWAS) have unraveled sixty-five distinct T2DM-associated genes and their positions on the chromosome.6 Several genes linked to T2DM within the past decade accounted for just 10% of its total genetic component.7 For instance, SNP in the transcription factor 7-like 2 (TCF7L2) gene is associated with T2DM risk in different racial groups.8–11 The gene modulates explicitly the expression of the pro-glucagon gene and, thus, glucagon-like peptide-1 (GLP-1) release, besides its involvement in glycemic control and insulin production.12 Nevertheless, genetic risk for T2DM appears dynamic with global migration, suggesting an environmental influence on the disease’s genetic basis.13
Recent developments in the field of genetics reveal this environmental influence on the complete set of genes in man and their expression. Hence, epigenetics has gained prominence with the discovery of its role in malignancy and chronic kidney disease (CKD).14,15 Epigenetics’ definitions have evolved with time. Subsequently, scientists generally described the epigenetic characteristic as “a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence”.16 Epigenetics involves chemical modifications that influence the regulation of gene expression resulting from environmental factors and persist through mitosis or meiosis.17 The epigenetic marks often include DNA methylation, histone modifications and regulation by noncoding RNAs (ncRNAs), each changing gene expression with an unaltered DNA sequence. Recently, these epigenetic marks are widely reported as contributory factors in the pathogenesis of T2DM and its macro- and microvascular complications, and now constitute novel therapeutic targets in the disease.18,19 Interestingly, conventional pharmacotherapy with metformin is noted to produce epigenetic effects in T2DM as mounting evidence suggests the drug’s impact on epigenomics, microRNA (miRNA) levels, and subsequent gene expression.20 Also, inhibitors of DNA methyltransferase (DNAMT) and histone deacetylase (HDAC), which are prototypes of epigenetic therapy, are reportedly effective in diabetic retinopathy.21,22 More importantly, these discoveries underscore the prospect of using epigenetic therapy as a valuable adjunct to pharmacotherapy.
In this review, we discuss the epigenetics of T2DM and that of its macro- and microvascular complications and the potential benefits of combining epigenetic therapy with pharmacotherapy for optimal results.
Literature Search Strategy
We retrieved the relevant information for this narrative review by using the following medical database search engines: Google, PubMed, MEDLINE, Embase, Cochrane Library, Scopus and Web of Science. Using appropriate descriptors like “Type 2 diabetes mellitus”, “epigenetics”, “epigenetic mechanisms”, “epigenetic therapy”, “risk factors” and “pharmacotherapy”, we searched the databases for primary data (research articles and clinical trials) and secondary data (systematic reviews/meta-analyses, narrative reviews, conference proceedings and book chapters) on related papers published in or translated into the English Language. Selection criteria for these papers also included publication within the past three decades. We excluded study protocols, opinions and letters to the editor.
Some Risk Factors of T2DM: Recent Perspectives
Obesity
Obesity contributes to approximately 55% of T2DM cases.23 The rising incidence of obesity in children within the last forty years may have contributed to the concomitant increase in the incidence of T2DM in childhood and adolescence.24 Obesity is identified as the most prominent risk factor for the development and progression of T2DM in all age categories.25 Obesity and T2DM form a nexus in their pathogenesis and molecular mechanisms, driven by several factors such as adipose tissue, body fat distribution, inflammation, free fatty acids (FFAs), gut microbiome, dyslipidemia and homeostatic variables like adiponectin.25 The mechanistic link between obesity and T2DM has revealed new insights on the possibility of developing multi-targeted molecules that can treat both disorders. Chronic obesity is associated with insulin resistance, a precursor of T2DM.26 Specifically, dysfunctional brown adipose tissue contributes to insulin resistance and hyperlipidemia. A variant of fibroblast growth factor 21 (FGF21), an activator of brown adipose tissue, reportedly improved insulin sensitivity and reduced triglyceride levels in obese subjects with T2DM, making it a potential therapeutic agent to induce “browning” of white adipose tissue.27 Also, some authors noted that activating brown adipose tissue in white adipose tissue-mediated thermogenesis inhibits insulin resistance and ameliorates metabolic dysfunction.28 This observation is predicated on the role of macrophages in inducing metabolic inflammation and their interactions with adipocytes. Brown adipose tissue appears to blunt the inflammatory capacity of macrophages compared to white adipose tissue.28 Truncal adiposity, or visceral fat, secretes a group of hormones called adipokines. These are cytokines (cell-signaling proteins) involved in obesity-associated inflammation, the evolution of metabolic syndrome, and T2DM. The activity of adipokines impacts adipocyte hypoxia and macrophage chemotaxis in several organs, resulting in a reduced adipose tissue production and adiponectin (an enhancer of insulin sensitivity) and an increased number of macrophages and T-cells within adipocytes, among other sequelae.29,30 Other authors have previously proposed that adipose tissue inflammation is the major propeller of insulin resistance in obesity.31 Besides, a cascade of pathophysiologic processes eventually leads to the development of metabolic inflammation, a fundamental factor involved in the pathogenesis of insulin resistance.32 Obesity triggers adenine nucleotide translocase 2 (ANT2), an inner mitochondrial protein, leading to adipocyte hypoxia. This worsens adipocyte tissue dysfunction and inflammation inducing activated macrophages and hypertrophied adipocytes to increase pro-inflammatory cytokine levels, such as tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1) and interleukin-6 (IL-6); these cytokines result in a chronic inflammatory state.32 On the other hand, some investigators recently demonstrated that reduced macrophage recruitment in obesity potentially ameliorates insulin resistance in experimental animals.33 More importantly, this finding underscores the fact that adipose tissue macrophages also contribute to obesity-related insulin resistance. Finally, TNF-α may activate the nuclear factor kappa-light-chain-enhancer of the activated B-cell (NF-kB) pathway that is associated with insulin resistance.34 Altered levels of adipokines such as retinol-binding protein 4 (RBP4) and adiponectin in obesity are documented risk factors for T2DM.35 RBP4 decreases insulin sensitivity in skeletal muscles and the liver, whereas adiponectin enhances insulin sensitivity (as previously noted).36
Lifestyle Factors
Dietary composition (especially a calorie-dense westernized diet) and sedentary lifestyle are thought to be the primary risk factors of T2DM.37,38 Findings from a recent mixed-methods study corroborated that lifestyle changes, particularly with regard to dietary quality and physical activity, are essential in managing T2DM.39 The concept of diabetes-protective lifestyle factors is now supported by evidence from several studies and underlying molecular mechanisms. However, T2DM-associated lifestyle factors do not appear to act alone, as an increased disease risk occurs in individuals with a high genetic or epigenetic risk exposed to these lifestyle factors.40 Firstly, dietary types vary in their association with the disease. Recent systematic reviews and meta-analyses show that unlike refined grains and sugar-sweetened beverages that appear to increase T2DM risk, low calorie-dense plant food (such as vegetables and fruits) and fermented dairy products are all associated with lower disease risk.41–43 In contrast, nuts offer some protection against T2DM although they represent a high calorie-density food.44 Thus, the United States’ Department of Health and Human Services now recommends Mediterranean or vegetarian diet for T2DM rather than an emphasis on limiting the quantity of dietary carbohydrates, fat and protein.45 The underlying molecular mechanisms of diabetes-protective diets are now well described. For instance, a westernized diet comprising refined sugars and saturated fat enhances the upregulation of pro-inflammatory mediators: leading to sustained low-grade inflammation.46 Phytochemicals from plant food counteract this process by activating the anti-inflammatory and cell-defense genes,47 whereas a fiber-rich diet enhances the production of short-chain fatty acids (SFAs) by gut bacteria.48 These SFAs directly bind to free fatty acid receptor 2 on pancreatic ß-cells, improving cellular growth and function and resulting in reduced T2DM risk.49 Thus, the receptor is speculated as a novel therapeutic target site for diabetes. Secondly, the influence of physical activity on disease risk has been documented in several epidemiologic studies.37,50–52 A comparative analysis of high versus low total physical activity showed a reduction in relative diabetes risk by about 30% while these activities were negatively correlated with disease risk.37,50 Also, other investigators showed that exercise positively affected insulin sensitivity and glycemic control in non-diabetic subjects.51,52 Similarly, sedentary lifestyle is strongly linked to incident diabetes or obesity, irrespective of the degree of physical activity.53,54 The underlying molecular mechanism appears to be in tandem with that of dietary factors as individuals with sedentary lifestyle reportedly have higher levels of circulating pro-inflammatory mediators.55 Importantly, physical activity has also been shown to reduce low-grade inflammation.56 Finally, sleep deprivation and chronic stress increase susceptibility to T2DM because both lifestyle factors can affect insulin sensitivity and lead to poor glucose tolerance. For instance, a meta-analytical study showed that a sleep duration of 7–8 hours per day was associated with the lowest diabetes risk, whereas the risk increased by 9% for each one-hour shorter duration of sleep.57 In contrast, longer sleep duration may also be a risk factor for future T2DM or metabolic syndrome although these findings appear inconsistent.58,59 Nevertheless, some researchers demonstrated that a five-day sleep deprivation resulted in a reduced whole-body insulin sensitivity,60 as well as a decreased glucose excretion rate following a night of four hour-sleep.61
Gut Microbiota
New developments in diabetology show that gut microbiota may be involved in the development of T2DM. Patients with T2DM have a different composition of gut bacteria than those without the disease.62 T2DM patients tend to harbor lower colonies of beneficial bacteria and higher colonies of harmful bacteria. For instance, the genera of Bifidobacterium, Bacteroides, Faecalibacterium, Akkermansia, and Roseburia were negatively associated with T2DM (ie, protective against T2DM) whereas the genera of Ruminococcus, Fusobacterium, and Blautia were positively associated with the disease (ie, predisposes to T2DM).62 Gut microbiota contributes to metabolic disease and T2DM through multiple molecular mechanisms: regulation of inflammation, interaction with dietary constituents, and influence on gut permeability, glucose and lipid metabolism, insulin sensitivity and overall energy homeostasis.62 Generally, T2DM is associated with high levels of pro-inflammatory cytokines, chemokines and inflammatory proteins. Thus, Bacteroides and Akkermansia reportedly suppress TNF-α while butyrate-producing bacteria like Roseburia and Faecalibacterium inhibit the activity of NF-kB via the action of butyrate.62 Again, a species of Akkermansia has a strong α-glucosidase inhibitory activity that prevents the breakdown of complex carbohydrates, thus decreasing post-prandial hyperglycemia. Although the link between the gut bacterial microbiome and T2DM is still a subject of ongoing research, these findings suggest that targeting the gut microbiota through probiotic supplementation may be a promising strategy for preventing and treating the disease.
Epigenetic Effects of T2DM Risk Factors
Obesity potentially affects DNA methylation patterns (hypermethylation and hypomethylation) in several tissues, including adipose tissue and skeletal muscle.63 These DNA methylation alterations can influence gene expression for insulin signaling, glucose metabolism, and inflammation. Lifestyle factors such as diet and physical activity can also impact the epigenetic regulation of T2DM. High-fat diets induce epigenetic mechanisms that disrupt insulin signaling, fat metabolism, and inflammation.64 Inflammatory signaling plays a fundamental role in insulin resistance, as macrophages activated in adipose tissue and the liver are associated with metabolic disorders and secretion of inflammatory mediators, leading to systemic insulin resistance.65,66 Thus, hypermethylation on pro-inflammatory genes potentially suppresses these genes, attenuates the inflammatory response and increases insulin sensitivity.
On the other hand, hypomethylation and histone modification (hyperacetylation) are associated with the overexpression of candidate genes, leading to increased fat metabolism and inflammation; the resultant obesity precedes insulin resistance.67 In contrast, regular exercise leads to beneficial epigenetic modifications like DNA demethylation or hypomethylation that may enhance insulin sensitivity.68 Microbiota-sensitive epigenetic changes include DNA methylation, histone modifications, and ncRNA regulation. Epigenetic-modifying enzymes, DNAMT/histone methyltransferases (HMTs), and histone acetyltransferase (HATs) depend on appropriate substrates (methyl and acetyl donors) to catalyze changes to the chromatin.69 The microbiota can synthesize several epigenetic substrates, co-factors, or regulators of epigenetic enzyme activity.70 The gut microbiota can also produce metabolites that potentially affect epigenetic mechanisms. For instance, SFAs derived from dietary fiber fermentation can inhibit HDAC, resulting in histone hyperacetylation and increased expression of target genes.69
Pathogenesis of T2DM and the Conventional Pharmacologic Options
Although our understanding of T2DM as a complex multifactorial disease is still evolving, dysregulated secretory functions of pancreatic islets and insulin resistance at the target tissues are central in disease pathogenesis. It is believed that an inadequate ß-cell compensatory mechanism to counteract insulin resistance at these target sites is crucial to the deranged glycemic levels and lipid metabolism. Individuals with T2DM vary in their phenotypic features of insulin resistance or dysfunctional ß-cells. In some patients, insulin resistance predominates over insulinopenia, and vice versa in others, probably due to some reported determinants of insulin resistance such as obesity, high triglycerides, low high-density lipoprotein-cholesterol (HDL-C) and alcohol intake.70 New concepts on the pathogenesis of T2DM are emerging. Firstly, there is a focus on the role of the interplay between several metabolic pathways involving insulin’s major target tissues, and an inter-organ crosstalk.71 Apart from hormones and organokines such as hepatokines (eg, fetuin and selenoprotein P), adipokines (eg, adiponectin), and myokines (eg, IL-6), dysglycemia is associated with metabolic changes driven by tissue-derived metabolites (eg, FFAs and amino acids) which contribute to T2DM onset and play important roles in the inter-organ crosstalk during the development of the disease.71 Thus, diabetes is now seen as multifactorial disease in which insulin resistance and elevated FFAs collaborate with hyperglycemia, hyperinsulinemia, fat accumulation, oxidative stress, and inflammation in the liver and other tissues.72
Secondly, unraveling the pathogenesis of T2DM has revealed other perspectives although aging-related visceral adiposity, oxidative stress, and altered mitochondrial function are the recognized mechanistic propellers of insulin resistance.73,74 According to the “portal hypothesis”, there is direct hepatic exposure to released FFAs and pro-inflammatory adipokines into the portal vein in obesity, resulting in hepatic insulin resistance and T2DM.75 However, more potential contributors to insulin resistance include the consequences of intracellular lipid metabolism and ATP generation in the liver and skeletal muscle76 and the role of the hypothalamus.77
Emerging evidence indicates that dual-specificity phosphatase 8 (Dusp8), encoded by the Dusp8 gene, controls neuronal signaling in the hypothalamus.77 The Dusp8 gene is responsible for increased susceptibility to T2DM. Leptin (one of the adipokines) activates the hypothalamic cells with leptin receptors to maintain normoglycemia. The fundamental role of leptin involves regulating long-term caloric balance as its circulating level correlates with the number of caloric reserves stored in adipose tissue as triglycerides.78 Thus, leptin level influences appetite and satiety: the brain’s adaptive mechanisms to regulate the body’s caloric reserves. The hypothalamic cells modulate glycemic levels through autonomic interconnections as the autonomic innervation of the liver and skeletal muscles drives cellular glucose uptake. In T2DM, this autonomic regulation of glycemic level appears abnormal.79 Increasing age and dietary fats are associated with leptin and insulin resistance. Neurons that mediate glucose regulation and leptin sensitivity become blunted to the effect of leptin, highlighting the potential role of leptin in the pathogenesis of T2DM.80 Dysfunctional hypothalamic astrocytes that synthesize fatty-acid binding protein 7 (FABP7) are suggested as the contributory factor to leptin and insulin resistance because these cells influence the function of contiguous leptin-sensitive neurons and regulate leptin sensitivity.81 Interestingly, infusing a single dose of intra-hypothalamic fibroblast growth factor 1 (FGF1) in diabetic animal models resulted in normoglycemia, an outcome attributed to the stimulation of the hypothalamic astrocytes which are also targets of FGF1.82,83
The traditional pharmacotherapy for T2DM comprises the sulfonylureas (eg, chlorpropamide and glibenclamide), biguanides (eg, metformin), inhibitors of dipeptidyl peptidase-4 (DPP-4) (eg, sitagliptin, vildagliptin, and linagliptin), and inhibitors of sodium-glucose co-transporter 2 (SGLT2) or gliflozins (eg, canagliflozin). Among these classes of anti-diabetic drugs, metformin stands out as the only drug that exert additional pleiotropic effects, mediated by several epigenetic modifications.84 These modifications that have been reported in various organs, tissues and cellular compartments are responsible for the effects of the drug on glycemic control, local and systemic inflammation, oxidative stress and fibrosis.84 The influence of metformin on T2DM epigenetics is subsequently discussed in details in this paper.
Epigenetic Marks in T2DM
Epigenetic marks are currently regarded as essential mediators of gene–environment interactions in T2DM, leading to several disease variations. They contribute to the development of T2DM by changing gene expression in response to environmental and lifestyle factors related to diet, exercise, and stress. They may reveal possible targets for developing new approaches to treat or prevent T2DM. Epigenetic therapy involves the application of medications or other epigenome-influencing techniques for therapeutic purposes in diseases affected by epigenetic changes. Unlike gene therapy, it is reversible, meaning it is “druggable” for targeted therapies.85 In other words, the chemical bonding of the medication to a “druggable” target should be therapeutically beneficial to the patient by changing the function of the target. Among the prominent epigenetic marks in T2DM, DNA methylation and histone modifications alter gene expression at the transcription level, while ncRNAs affect the gene expression at the translation level. However, DNA methylation and histone modifications play significant roles in the epigenetics of the disease (Table 1 and Figure 1).
 |
Table 1 The Major Epigenetic Modifications Involved in Type 2 Diabetes Mellitus (T2DM) |
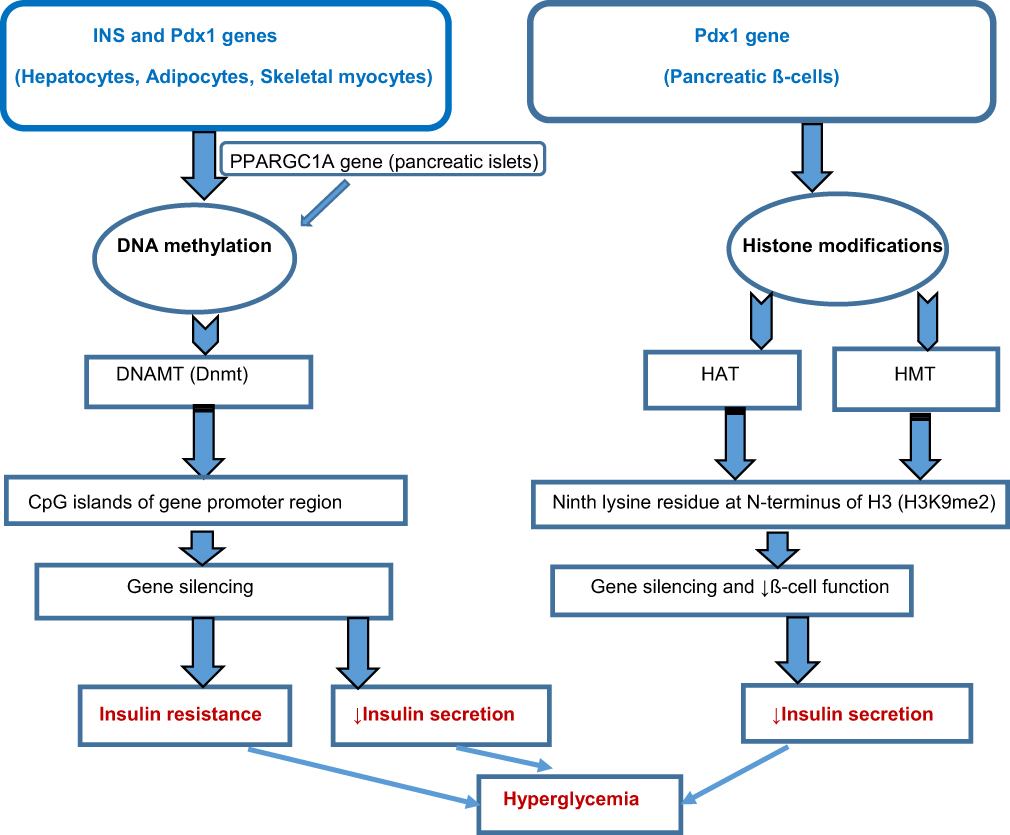 |
Figure 1 Schematic diagram indicating the primary epigenetic mechanisms in type 2 diabetes mellitus (T2DM). Abbreviations: SOD2, superoxide dismutase 2 mmP-9, matrix metallopeptidase 9 SIRT 1, Sirtuin 1 PTP 1B, protein tyrosine phosphatase 1B PPARγ, peroxisome proliferator-activated receptor gamma; INS, insulin signaling Pdx1, pancreatic and duodenal homeobox 1 PPARGC1A, peroxisome proliferator-activated receptor gamma coactivator 1-alpha DNAMT, DNA methyltransferase DNA, deoxyribonucleic acid CpG, cytosine-phosphate-guanine HAT, histone acetyltransferase HMT, histone methyltransferase. |
DNA Methylation
It involves adding a methyl group to the cytosine-phosphate-guanine (CpG) islands at gene promoter regions. This mechanism attracts proteins involved in chromatin folding and gene silencing.95 It is the most extensively studied modification in T2DM, as genome-wide DNA methylation studies have identified differentially methylated regions (DMRs) in affected patients, many of which are associated with genes responsible for insulin signaling (INS), glucose metabolism, and inflammation. For instance, DNA methylation of CpG islands at the promoter regions of specific genes in hepatocytes, adipocytes, and skeletal myocytes encoding insulin signaling and glucose metabolism suppresses these genes. It contributes significantly to insulin resistance, inhibition of pancreatic ß-cells, and, subsequently, the development of T2DM.96 The type of epigenetic mark, the degree of modification (mono-methylation [me], di-methylation [me2], and tri-methylation [me3]), and the position of the modification determine the nature of the epigenetic effects. Furthermore, gene expression depends on the modification site (ie, gene body and CpG islands at promoter regions), combination, and modification pattern.97 Hypermethylation and hypomethylation at the promoter regions of specific genes in pancreatic ß-cells result in gene silencing and increased gene expression, respectively.97 For instance, DNA methylation occurs in the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1A) gene; it is the energy metabolism-regulating gene as it interacts with other transcription factors and co-activates their functions, thus influencing gene expression for energy metabolism, oxidative phosphorylation, and gluconeogenesis.98,99 PPARGC1A, the protein encoded by the PPARGC1A gene (primarily expressed in the liver, skeletal muscles, heart, and brain), can regulate glucose-dependent insulin production in the pancreatic islets by influencing ATP synthesis via oxidative phosphorylation. The PPARGC1A gene is downregulated in T2DM through DNA methylation with a marked reduction in messenger RNA (mRNA) expression of this gene in the islets of T2DM donors compared to that of donors without the disease.86 The data showed that PPARGC1A gene expression in pancreatic ß-cells was reduced in human and experimental animal models of T2DM and was associated with reduced insulin secretion.86 The vital role of this epigenetic mark in sustaining dysfunctional islets is also well reported.100 For instance, genes involved in islet function, such as pancreatic islet-specific transcription factor (Pdx1), PPARGC1A, INS, and glucagon-like peptide-1 receptor (GLP1R), were hypermethylated in T2DM.87–89 Also, hypermethylation drives the suppression of defined genes in the pancreatic islets of T2DM patients, leading to elevated HbA1C due to ß-cell’s functional distortion. The hyperglycemic state, in turn, results in DNA hypermethylation at Pdx1 and INS genes.87,88
Histone Modifications
Histones are proteins that mediate chromatin folding and compaction. Their chemical changes can occur through several mechanisms involving the main types: H2A, H2B, H3, and H4.97 Besides other chemical modifications such as phosphorylation, ubiquitination, and sumoylation, the primary histone modifications are methylation and acetylation at the lysine residue of the histone proteins’ amino-terminus (or the N-terminus).101 These histone proteins’ positively charged amino acids naturally interact with the negatively charged DNA phosphate groups. Acetylation of histone tails weakens these interactions between the histone proteins and the DNA involved in gene expression. For instance, there is a reduction in histone acetylation at the promoter region of the peroxisome proliferator-activated receptor gamma (PPARγ) gene in the adipocytes and adipose tissue-resident immune cells of patients with T2DM. This gene encodes for the nuclear protein PPARγ, which plays a role in modulating gene expression. PPARγ is expressed in adipocytes, where it regulates adipocyte differentiation and functions as a transcription factor (ie, it can bind to specific DNA sequences and influence gene expression). It governs the gene’s expression in insulin sensitivity, inflammation, and lipid metabolism. Hyperacetylation at the N-terminus triggers increased transcription, while hypoacetylation results in transcriptional gene silencing.97 Despite the fundamental concept regarding the alteration of chromatin structure via modifications to histone tails, the exact mechanisms by which these changes to histone tails affect DNA–histone interactions are yet to be fully understood.102
Nevertheless, in the hyperglycemic milieu, insulin promoter factor or Pdx1 (Pancreatic and duodenal homeobox 1) stimulates the expression of an insulin-coding gene by enlisting these coactivators or transcriptional co-activating proteins viz HAT, p300, and SET Domain-containing protein 7 (SETD7), one of the HMTs; these coactivators respectively result in hyperacetylation and di-methylation at the fourth lysine residue within the N-terminus of histone H3, ie, histone H3 lysine 4 di-methylation (H3K4me2) and the formation of euchromatin at the promoter region of the insulin-coding gene.103 Pdx1 also brings about the ß-cell-related expression of the SETD7 gene, thus controlling the genes that encode glucose-dependent insulin release.103 Some investigators reported that the subsequent development of T2DM after IUGR in experimental murine models led to gradual and sustained epigenetic silencing of the Pdx1 gene in ß-cells.90 Di-methylation and acetylation on the ninth lysine residue within the N-terminus of histone H3, ie, histone H3 lysine 9 di-methylation (H3K9me2) in ß-cell of these animal models resulted in Pdx1 gene repression and reduced ß-cell function. These epigenetic mechanisms were associated with chemical attraction and upstream transcription factor 1 (USF1) binding to the Pdx1 promoter region.
Given that USF1 is essential for activating Pdx1 transcription, Pdx1 transcription silencing follows the reduced binding of USF1 to Pdx1.104,105 Similarly, increased di-methylation on the ninth lysine residue within the N-terminus of histone H3 (H3K9me2) and decreased tri-methylation on the fourth lysine residue within the N-terminus of histone H3 (H3K4me3) are associated with increased and repressed transcription, respectively. Notably, the lysine residues of the N-terminus of histone proteins may undergo various degrees of N-methylation (me, me2, and me3), thus amplifying the level of gene regulation.106
Regulation by Non-Coding RNAs
The ncRNAs, such as miRNAs and long-noncoding RNAs (lncRNAs), have also been implicated in T2DM epigenetics. The miRNAs are small, single-stranded, ncRNA molecules with twenty-one to twenty-three nucleotides.107 They play a role in silencing the mRNA (preventing their translation into proteins) and post-transcriptional gene regulation.108 Although there are myriads of miRNAs, the scale of their role in epigenetic regulation constitutes one of the current research gaps in epigenetics.109 However, dysregulation of several miRNAs occurs in individuals with T2DM. Some regulate insulinotropic genes and glucose homeostasis, while lncRNAs may be involved in developing insulin resistance. Recent evidence shows that differentiation of the pancreatic cells and a hyperglycemic state frequently dysregulate lncRNAs.97 Some authors suggest that the location of several T2DM-related SNPs within the lncRNA loci underscores the role of lncRNA in T2DM and its complications.110 For instance, lncRNA-p3134 is linked to glucose metabolism and insulin signaling in ß-cells with a high expression in T2DM.111 Its high expression appears in tandem with the upregulation of Pdx1, one of the transcription factors of insulin. A study shows that hyperglycemia stimulates the insulin gene promoter by Pdx1, which eventually amplifies insulin production and release.112
Furthermore, a fraction of miRNAs maintain normoglycemia and T2DM epigenetics.113,114 For instance, upregulation of miR-375 in the pancreatic islets potentially inhibits glucose-mediated insulin production, whereas downregulation of this miRNA enhances insulin production.91 Another report suggests a different miRNA (miR-9) upregulation markedly reduces glucose-induced insulin release.115
Role of microRNAs in T2DM Pathogenesis
Pancreatic ß-cells and insulin target tissues express a specific set of miRNAs, ubiquitous in human tissues and not cell-specific. However, miR-375 is highly concentrated in pancreatic islets, where it modulates the expression of target genes involved in insulin secretion and ß-cell hypertrophy in response to insulin resistance.91 Dysregulated miRNAs are involved in pancreatic ß-cell function, insulin signaling, adipocyte differentiation, and inflammation, which play critical roles in T2DM pathogenesis. The miRNA signature of ß-cells and insulin target tissues is altered in T2DM, contributing to their functional impairment under hyperglycemic states.116,117 Several miRNAs, such as miR-21, miR-34a, and miR-146a, are highly expressed in the islets of diabetic murine models; these miRNAs disrupt the function of ß-cells.118,119 Similarly, expression of miR-29 and miR-34a is increased in insulin target tissues, possibly driving insulin resistance.120 Additionally, dysregulated miR-143, miR-802, miR-103, and miR-107 occur in dietary murine models of obesity and T2DM.121–123 Interestingly, these miRNAs contribute to the development of insulin resistance in these animal models. Biopsies of human skeletal muscle from T2DM patients revealed more than sixty differentially expressed miRNAs, including the upregulation of miR-143 and downregulation of miR-206 and miR-133a.124 Besides the dysregulation in insulin target tissues, T2DM leads to changes in vascular, cardiac, retinal, and renal expressions of miRNAs, suggesting the role of these ncRNAs in long-term T2DM complications.125 Given the stability of miRNAs in biological samples (such as blood, saliva, and urine), they are becoming attractive diagnostic biomarkers in T2DM as they are easily detectable with quantitative polymerase-chain reaction (qPCR). Following the first identification of T2DM-related blood miRNA profile more than a decade ago,126 other investigators detected an increase in the expression of other diabetes-related miRNAs in T2DM patients compared to prediabetic subjects; these include miR-9, miR-29a, miR-30d, miR-34a, miR-124a, miR-146a and miR-375.92 Subsequently, other researchers identified upregulation of miR-27a, miR-150, miR-192, miR-320a, and miR-375 in T2DM patients and established a significant direct correlation between fasting blood glucose and altered miR-27a and miR-320a levels.93
Long Noncoding RNAs and T2DM Pathogenesis
Studies have identified several lncRNAs as potential regulators of pancreatic ß-cell function and insulin secretion central to T2DM pathogenesis. Dysregulated lncRNA profiles play essential roles in the pathogenesis of metabolic diseases like T2DM and obesity, as upregulated expression of several lncRNAs occurs in these disorders.127 For instance, some researchers observed that lncRNA profile in peripheral blood mononuclear cells of T2DM patients showed significantly elevated levels of these lncRNAs: HOTAIR (HOX transcript antisense RNA), MEG3 (maternally expressed gene 3), LET (low expression in tumor), MALAT1 (metastasis-associated lung adenocarcinoma transcript 1), MIAT (myocardial infarction-associated transcript), GAS5 (growth arrest-specific transcript 5), PLUTO (pilus-long noncoding RNA), NBR2 (neighbor of BRAC1 gene 2), Linc-p21 (long intergenic noncoding RNA p21), XIST (X–inactive-specific transcript), PANDA (P-21-associated ncRNA DNA damage-activated), ENST00000550337.1 (Ensembl transcript identifier00000550337.1), and CDKN2BAS1 (CDKN2B antisense RNA 1) compared to that of their non-diabetic controls.94 Again, other researchers showed that the lncRNA NEAT1 (nuclear-enriched abundant transcript 1) regulated insulin gene expression and ß-cell function.127 Its downregulation leads to impaired glucose-dependent insulin secretion and reduced expression of genes responsible for the hormone’s synthesis and secretion.
The Role of Epigenetic Marks in T2DM Complications
Emerging evidence suggests that epigenetic marks contribute to the pathogenesis of several micro-and macrovascular complications of T2DM.128 Endothelial dysfunction appears crucial to developing these vascular complications, which are associated with elevated endothelin 1 (EDN-1) production, a peptide that causes vasoconstriction and increased fibrosis, and is abundant in T2DM vascular complications.129 The epigenetic changes reported in endothelial cells in T2DM, which lead to endothelial dysfunction, include methylation defects (hypermethylation) in the CpG islands of the EDN 1 gene (resulting in its overexpression), hypomethylation and hyperacetylation of histone proteins in the promoter region of the NF-кB gene (leading to its overexpression) and overexpression of miRNAs such as miR-126.126,130,131 A macrovascular complication like T2DM-related coronary artery disease is associated with two essential miRNAs: miR-1 and miR-133.132,133 The epigenetic changes in the two common microvascular complications of T2DM (diabetic nephropathy and diabetic neuropathy) are well-documented. GWAS indicates a direct correlation between DNA hypermethylation and the inflammatory process in diabetic nephropathy.134,135 For instance, hypermethylation of the promoter region of the Ras protein activator-like 1 (RASAL1) gene occurred in animal models of this T2DM complication.136 Other authors also reported that hypomethylation of the myo-inositol oxygenase (MIOX) gene results in oxidative stress and fibrosis, which drive the progression of diabetic nephropathy.137 Also, miRNAs such as miR-192 and miR-215 are highly expressed in the urine of T2DM patients with diabetic nephropathy.138 Similarly, diabetic neuropathy is associated with DNA methylation defects. Hypermethylation of the NINJ2 gene in Schwann cells leads to decreased protein (ninjurin2) expression, vital for post-injury peripheral nerve regeneration.139 In animal models of diabetic neuropathy, reduced expression of miR-25 and miR-146 also occurs.140,141
Epigenetic Therapy and Drugs with Epigenetic Effects
Therapeutic strategies targeting epigenetic marks in T2DM are still in the early phase, but several promising approaches now exist. Interestingly, the reversible nature of the disease’s epigenetic changes raises the prospects of novel treatments. Several small molecules with epigenetic activity (epidrugs) are undergoing clinical trials.142 One potential therapeutic target is modulating the enzyme mediators of epigenetic marks (eg, inhibiting DNAMTs, HATs, and HDAC) to restore standard gene expression patterns and improve glycemic control (Figure 2).
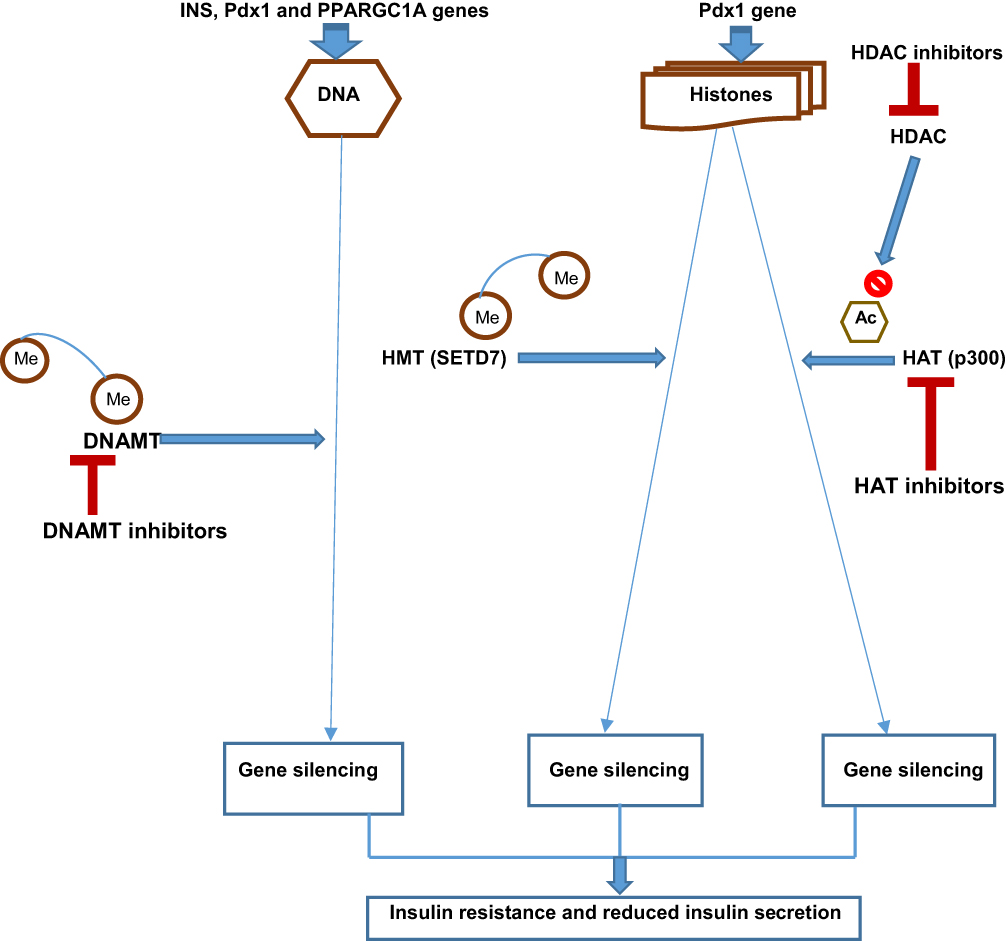 |
Figure 2 Schematic diagram showing the target sites of some epigenetic drugs in type 2 diabetes mellitus. Abbreviations: INS, insulin signaling Pdx1, pancreatic and duodenal homeobox 1 PPARGC1A, peroxisome proliferator-activated receptor gamma coactivator 1-alpha DNAMT, DNA methyltransferase HMT, histone methyltransferase HAT, histone acetyltransferase, HDAC, histone deacetylase SETD7, SET Domain-containing protein 7 Me, methyl group Ac, acetyl group. Notes: DNAMT inhibitors: 5-azacytidine, 5-aza-2ʹ-deoxycytidine. HAT inhibitors: Epigallocatechin-3-gallate, Vorinostat, Romidepsin. HDAC inhibitors: Trichostatin A, Sodium butyrate, Givinostat. |
HDAC Inhibitors
An established link exists between insulin signaling and the regulation of HAT and HDAC.143 For instance, hyperglycemia increases the binding of Pdx1 with HAT p300, resulting in hyperacetylation and increased insulin transcription. In contrast, hypoglycemia enhances Pdx1 binding with HDAC, decreasing insulin transcription.144 Thus, this HDAC’ role in the epigenetics of T2DM makes it a novel therapeutic target and has led to the discovery of its inhibitors (eg, trichostatin A, sodium butyrate, and givinostat) for the modulation of insulin signaling and ß-cell function.22,144
DNAMT Inhibitors
DNAMT (Dnmt) inhibitors are another group of small molecules that can modify epigenetic marks in T2DM. Three major types of DNAMTs are biologically active: Dnmt1, Dnmt3a, and Dnmt3b.97 Pharmacologic therapeutic strategies for certain malignancies now involve targeting DNAMTs to block DNA methylation. For instance, first-generation non-selective DNAMT inhibitors (5-azacytidine and 5-aza-2ʹ-deoxycytidine) are currently approved by the United States’ Food and Drug Administration (US FDA) for myeloid cancers and cutaneous T-cell lymphoma.145 They block all the three types of DNAMTs. In T2DM, hypermethylation of the genes responsible for ß-cell function, such as INS, PDX1, GLP1R, and PPARGC1A, results in their suppression. Again, a common complication of T2DM, like diabetic retinopathy, is characterized by some epigenetic marks such as methylation of mitochondrial superoxide dismutase 2 (SOD2) and matrix metallopeptidase 9 (MMP-9), increased transcription of lysine-specific histone demethylase 1(LSD1), DNAMTs, highly expressed miRNAs and vascular endothelial growth factor (VEGF) in diabetic murine models.146 Defective retinal mitochondrial activity leads to progressive retinal damage in diabetic retinopathy. Micro-aneurysms, vasculopathy, hemorrhages, neo-vascularization, and scarring characterize this diabetic microvascular complication. The SOD2 gene encodes the enzyme (SOD2) that removes free radicals and circumvents cellular oxidative injury. Cellular oxidative injury results from the action of LSD1 in diabetic retinopathy because of the SOD2 gene downregulation in blood vessels. The MMP-9 gene is similarly downregulated with the involvement of MMP-9 in cellular apoptosis, thereby worsening the effects of diabetic retinopathy.147 Thus, its epigenetic therapy involves blocking the methylation of SOD2 and MMP-9 genes. The therapeutic efficacy of DNAMT inhibitors like 5-azacytidine and 5-aza-2ʹ-deoxycytidine in this diabetic complication is promising as these epigenetic drugs successfully stopped the genes’ methylation, leading to symptom reduction in both human and murine diabetic retina.21,147,148 Interestingly, the US FDA has approved the use of these drugs in diabetic retinopathy. However, another DNAMT inhibitor, such as zebularine, yielded inconclusive results149 (Table 2).
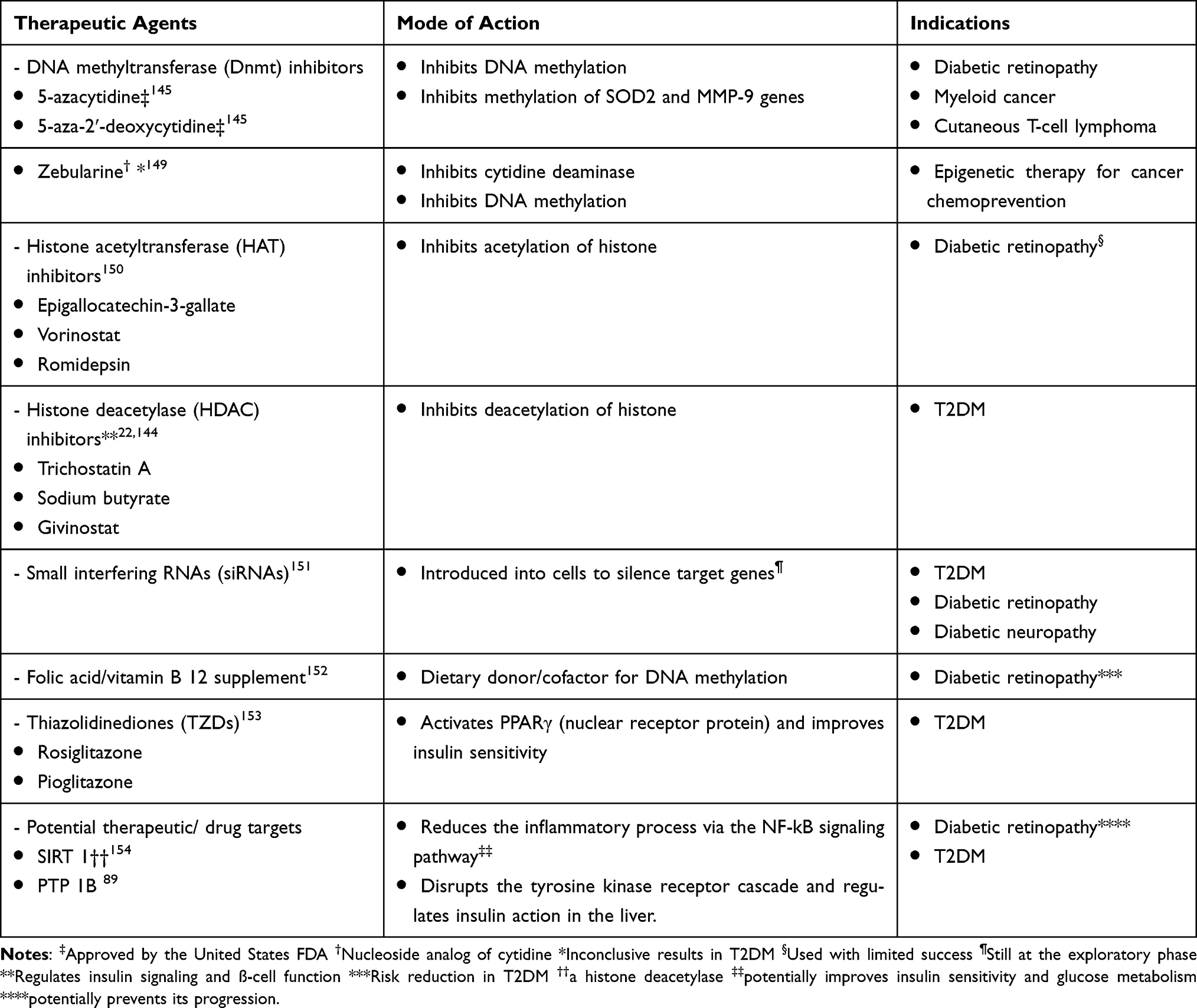 |
Table 2 Some Epigenetic Drugs and Drugs with Potential Epigenetic Effects in Type 2 Diabetes Mellitus (T2DM) |
Targeting Methyl Donors for DNA Methylation
Given the role of folic acid as a dietary donor for DNA methylation155 and the role of vitamin B12 as a cofactor involved in transferring the methyl group from folic acid to DNA,156 it is not surprising that low folic acid levels may lead to abnormal DNA methylation.150,152 More importantly, low serum levels of folic acid and vitamin B12 can increase diabetic retinopathy risk,157 whereas their supplementation may reduce disease risk.152
Sirtuin 1 (SIRT 1) as a Therapeutic Target
SIRT 1 is an HDAC that regulates gene transcription, cell proliferation, and apoptosis.158 Highly expressed SIRT 1 can prevent the progression of diabetic retinopathy.154 Hypermethylation of SIRT 1 promoter in T2DM results in transcription silencing,159,160 leading to pathologic changes of diabetic retinopathy. Furthermore, some researchers suggest that the deacetylation activity of SIRT 1 in adipocytes and macrophages reduces the inflammatory process via the NF-kB signaling pathways, resulting in improved insulin sensitivity and glucose metabolism.161–163 This modulatory role in insulin sensitivity portrays SIRT 1 as a potential therapeutic target in T2DM.
Protein Tyrosine Phosphatase 1B (PTP-1B) as a Drug Target
PTP-1B plays a role in the INS pathway (INS cascade), a cascade of molecular processes resulting from insulin binding to its receptors. Activated insulin receptor (IR) then phosphorylates IR substrates (IRS) on tyrosine residues, which serve as docking sites for activating downstream signaling molecules such as phosphoinositide 3-kinase (PI3K). The subsequent activation of the Akt pathway, also known as the PI3K pathway, modulates several downstream signaling pathways involved in glucose metabolism. For instance, Akt, also known as protein kinase B (PKB), promotes the translocation of GLUT4 (glucose transporter 4) to the cell surface, ensuring cellular glucose uptake.164 Thus, dysregulation of the INS pathway potentially results in insulin resistance. PTP-1B dephosphorylates the tyrosine residues, disrupting the tyrosine kinase receptor cascade. Also, it regulates insulin action in the liver and several tissues by modulating the interplay between the two subunits of IRS (IRS 1 and IRS 2), underscoring its vital role in insulin resistance and as a drug target in managing T2DM.97
Using Noncoding RNA (ncRNA)
An additional approach in epigenetic therapy is to use ncRNA molecules, such as miRNAs or lncRNAs, to regulate gene expression. The strategy involves attempting to reduce the highly expressed miRNAs in diabetic retinopathy despite their unresolved role in the disease. Again, it is possible to target cells using small interfering RNAs (siRNAs), a type of ncRNA, even though no known methods exist to achieve this novel intervention.115 Worse still, the delivery of siRNAs to the affected tissues is complex. Nevertheless, circular RNAs (circRNAs), another type of ncRNA molecule, now have established roles in T2DM and diabetic retinopathy. CircRNAs represent a diverse class of ncRNAs with potential regulatory functions within cells as they play critical regulatory roles in gene expression and contribute to several pathological processes. For instance, a highly expressed circRNA-TFRC (transferring receptor, TFRC) potentially increases T2DM risk because circRNA is generally linked with insulin resistance.97 Again, a few circRNAs (such as circRNA_0054633, circ-HIPK3, circANKRD36, and circRNA11783-2) play a significant role in the evolution of T2DM. Some authors reported that circ-HIPK3 is significantly raised in diabetic retinopathy as it mediates retinal vascular dysfunction in T2DM.153 A previous observation showed that four circRNAs in human pancreatic islets (circCIRBP, circZKSCAN, circRPH3AL, and circCAMSAP1) were explicitly associated with T2DM status.97 For instance, circCIRBP and circCAMSAP1 played roles in insulin secretory index in isolated human pancreatic islets and T2DM status in human peripheral blood, respectively.97
Other Therapeutic Targets
The nuclear receptor protein PPARγ is also a target for pharmacologic intervention in T2DM. Some authors reported that it was highly expressed in insulin resistance.165,166 Synthetic medications called thiazolidinediones (TZDs) or glitazones potentially activate PPARγ by attaching to PPARγ receptors and forming a complex with a transcription factor (retinoid-X receptor).153 This complex binds to a specific DNA motif at the target gene’s promoter region and increases insulin sensitivity and the expression of GLUT4, which aids glucose uptake, thus improving glycemic control in T2DM.167,168 Common examples of these drugs include rosiglitazone and pioglitazone. They are grouped as oral hypoglycemic drugs along with biguanides. Additionally, glitazones help reduce blood pressure and improve lipid metabolism by increasing HDL-cholesterol levels. Adverse effects include edema, congestive heart failure, weight gain, fractures, and hepatotoxicity.
Furthermore, HAT inhibitors such as epigallocatechin-3-gallate, vorinostat, and romidepsin hold prospects as possible therapeutic agents, although the outcomes are still unsatisfactory.150 However, therapies targeting specific genes of the mammalian genome with methylation and acetylation inhibitors remain a subject of research and debate.
Combining Epigenetic Therapy with Other Treatment Modalities
Combining epigenetic therapy with other treatment modalities may be potentially helpful in managing T2DM. Firstly, lifestyle interventions such as diet and exercise can influence epigenetic modifications, thus improving the effects of epigenetic therapies. For example, a healthy diet and regular exercise can reduce DNA methylation and histone modifications associated with T2DM. Interestingly, some authors in a recent review showed that complementing phytochemicals (from the Mediterranean diet) with a DNA methylation-based epigenetic intervention can improve glycemic control in T2DM patients.169 Secondly, combining stem cell-based therapy with epigenetic therapy holds prospects for managing the disease. Whereas the former regenerates pancreatic ß-cells, the latter may improve stem cells’ therapeutic potential in T2DM by optimizing their differentiation into functional cells or enhancing their survival through epigenetic modulation approaches.170 Thirdly, given the chronic low-grade inflammation associated with the development of T2DM, immunotherapy (such as targeting pro-inflammatory cytokines) is a possible treatment option. Because epigenetic modifications modulate immune cell function and inflammation, combining immunotherapy with epigenetic therapy potentially regulates these processes, enhancing insulin sensitivity and glycemic control in T2DM.171
Finally, mounting evidence suggests the impact of metformin on epigenomics, microRNA levels, and subsequent gene expression. Adding metformin to DNAMT or HDAC inhibitors may synergistically modulate gene expression and enhance insulin sensitivity, as metformin affects DNA methylation patterns and histone modifications.172 For instance, metformin leads to hypomethylation of specific genes responsible for glucose metabolism and insulin signaling pathways. Additionally, the drug can trigger changes in histone modifications, resulting in altered gene expression patterns associated with T2DM. It also activates adenosine monophosphate-activated protein kinase (AMPK), leading to the modulation of epigenetic mechanisms by influencing the activity of DNAMTs, class II HDACs, and HATs that mediate DNA and histone modifications. Metformin inhibits these enzymes by phosphorylation, although the action of HAT1 may be increased. The drug also reduces the expression of multiple HMTs, increases the class III HDAC and SIRT1 activities, and decreases the influence of DNAMT inhibitors.171 These alterations influence the epigenome and gene expression and may contribute to the antidiabetic properties of metformin. Furthermore, some investigators noted that metformin significantly downregulated serum miR-21, miR-126, and miR-223 levels in T2DM patients, making them potential biomarkers for disease diagnosis and providing a possible miRNA-based therapeutic strategy.130,173 These findings derive from the influence of metformin on miRNA expression, which is involved in insulin signaling pathways.
Conclusions
The epigenetic modifications involved in the pathogenesis of T2DM and its micro- and macrovascular complications are now extensively documented in the literature. Generally, hypermethylation (mediated by DNAMTs) at specific sites of candidate genes involved in insulin signaling and glucose metabolism results in gene silencing and reduced insulin production, and insulin resistance. Similarly, acetylation (mediated by HATs) and di-methylation within the N-terminus of histone 3 in ß-cells leads to gene repression and reduced glucose-dependent insulin release. Additionally, upregulation of specific miRNAs and lncRNAs in pancreatic islets (out of myriads of ncRNAs documented) also results in insulinopenia. Although more studies are essential to understand the epigenetic changes contributing to disease pathogenesis fully, there is mounting evidence that targeting the major epigenetic marks like DNA methylation, histone modifications, and ncRNA regulation may provide novel approaches for preventing and treating the disease. Several epigenetic drugs have been identified and used with variable success in humans and experimental animal models, especially in T2DM macro- and microvascular complications. Inhibitors of DNAMT, HDAC, and HAT are thus prototypes of the disease’s epigenetic therapy. Whereas HDAC and HAT inhibitors are yet to be universally applied in clinical practice, DNAMT inhibitors like 5-azacytidine and 5-aza-2ʹ-deoxycytidine have been approved by the US FDA for use in diabetic retinopathy. The traditional pharmacotherapy of T2DM with metformin is well reported to have epigenetic effects and positively alter disease outcomes when combined with epigenetic therapy typified by epi-drugs like DNAMT and HDAC inhibitors. These developments raise the prospect of using epigenetic therapy as a useful adjunct to pharmacotherapy. Better still, giving a combination of the traditional anti-diabetic drugs and epi-drugs or drugs with epigenetic effects may become the norm in managing T2DM and its micro- and macrovascular complications in the future. However, utilizing ncRNAs by introducing siRNAs into cells to silence specific target genes holds promise as a therapeutic tool for diabetes complications although this novel strategy remains in the exploratory phase.151 Decoding the role of lncRNA in T2DM pathogenesis and the underlying mechanisms is an essential area for further studies. Given that the extent of miRNAs’ role (an example of ncRNA) in epigenetic regulation is one of the current research gaps in T2DM epigenetics, we recommend that future research should focus on how to regulate gene expression in the disease using lncRNA molecules, another example of ncRNA.
Abbreviations
AMPK, AMP-activated protein kinase; CircRNA, Circular RNA; CpG, Cytosine-phosphate-guanine; DNAMT, DNA methyltransferase; DMR, Differentially methylated region; DPP-4, Dipeptidyl peptidase-4; Dusp8, Dual-specificity phosphatase 8; EDN1, Endothelin-1; FABP7: Fatty-acid binding protein 7; FGF1: Fibroblast growth factor 1; GDF15: Growth differentiation factor 15; GIP: Gastrointestinal peptide; GLP1: Glucagon-like peptide 1; GLP-1RA, GLP-1 receptor agonist; GLUT4: Glucose transporter 4; GWAS: Genome-wide association studies; HAT: Histone acetyltransferase; HbA1C: Glycated hemoglobin; HDAC, Histone deacetylase; HMT: Histone methyltransferase; INS: Insulin signaling; IR: Insulin receptor; IRS, Insulin receptor substrate; IUGR: Intrauterine growth retardation; LncRNA: Long-coding RNA; LSD1: Lysine-specific demethylase 1; MIOX: Mixed oxidant solution; MiRNA: MicroRNA; mRNA: Messenger RNA; MMP-9: Matrix metalloproteinase 9; MODY: Maturity-onset diabetes of the young; NcRNA: Noncoding RNA; NF-kB: Nuclear factor kappa-light-chain-enhancer of activated B cells; NINJ2: Nerve injury-induced protein 2; PDX1: Pancreatic and duodenal homeobox 1; PI3K: Phosphoinositide 3-kinase; PKB: Protein kinase B; PPARGC1A: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PPARγ: Peroxisome proliferator-activated receptor gamma; RASAL1: Ras protein activator-like 1; RBP4: Retinol-binding protein 4; SETD7: SET Domain-containing protein 7; SGLT2, Sodium-glucose cotransporter 2; SIRT 1: Sirtuin 1; SNP: Single nucleotide polymorphism; SOD2, Superoxide dismutase 2; SUR1: Sulfonylurea receptor 1; TCF7L2: Transcript factor 7-like 2; TNF-α: Tumor necrosis factor-α; TZD: Thiazolidinedione; USF1: Upstream transcription factor 1; VEGF: Vascular endothelial growth factor.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors disclose that there are no conflicts of interests.
References
1. Valaiyapathi B, Gower B, Ashraf AP. Pathophysiology of type 2 diabetes in children and adolescents. Curr Diabetes Rev. 2020;16(3):220–229. doi:10.2174/1573399814666180608074510
2. Bellou V, Belbasis L, Tzoulaki I, Evangelou E. Risk factors for type 2 diabetes mellitus: an exposure-wide umbrella review of meta-analyses. PLoS One. 2018;13(3):e0194127. doi:10.1371/journal.pone.0194127
3. Hilawe EH, Yatsuya H, Kawaguchi L, Aoyama A. Differences by sex in the prevalence of diabetes mellitus, impaired fasting glycemia and impaired glucose tolerance in sub-Saharan Africa: a systematic review and meta-analysis. Bull WHO. 2013;91(9):671–682. doi:10.2471/BLT.12.113415
4. Bellver J, Mariani G. Impact of parental over- and underweight on the health of offspring. Fertil Steril. 2019;111(6):1054–1064. doi:10.1016/j.fertnstert.2019.02.128
5. Hales CN, Barker DJ, Clark PM, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303(6809):1019–1022. doi:10.1136/bmj.303.6809.1019
6. Ayub Q. Revisiting the thrifty gene hypothesis via 65 loci associated with susceptibility to type 2 diabetes. Amer J Human Genet. 2014;94(2):176–185. doi:10.1016/j.ajhg.2013.12.010
7. Herder C, Roden M. Genetics of type 2 diabetes: pathophysiologic and clinical relevance. Eur J Clin Invest. 2011;41(6):679–692. doi:10.1111/j.1365-2362.2010.02454.x
8. Horikoshi M, Hara K, Ito C, Nagai R, Froguel P, Kadowaki T. A genetic variation of the transcription factor 7-like 2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia. 2007;50(4):747–751. doi:10.1007/s00125-006-0588-6
9. van Vliet-Ostaptchouk JV, Shiri-Sverdlov R, Zhernakova A, et al. Association of variants of transcription factor 7-like 2 (TCF7L2) with susceptibility to type 2 diabetes in the Dutch Breda cohort. Diabetologia. 2007;50(1):59–62. doi:10.1007/s00125-006-0477-z
10. Chang YC, Chang TJ, Jiang YD, et al. Association study of the genetic polymorphisms of the transcription factor 7-like 2 (TCF7L2) gene and type 2 diabetes in the Chinese population. Diabetes. 2007;56(10):2631–2637. doi:10.2337/db07-0421
11. Danquah I, Othmer T, Frank LK, Bedu-Addo G, Schulze MB, Mockenhaupt FP. The TCF7L2 rs7903146 (T) allele is associated with type 2 diabetes in urban Ghana: a hospital-based case-control study. BMC Med Genet. 2013;14:96. doi:10.1186/1471-2350-14-96
12. Del Bosque-Plata L, Martínez-Martínez E, Espinoza-Camacho MÁ, Gragnoli C. The Role of TCF7L2 in type 2 diabetes. Diabetes. 2021;70(6):1220–1228. doi:10.2337/db20-0573
13. Gibbons A. 12th International Congress of Human Genetics. Diabetes genes decline out of Africa. Science. 2011;334(6056):583. doi:10.1126/science.334.6056.583
14. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi:10.1093/carcin/bgp220
15. Uwaezuoke SN, Okafor HU, Muoneke VN, Odetunde IO, Odimegwu CL. Chronic kidney disease in children and epigenetics: future therapeutic trajectories. Biomed Reports. 2016;5(6):660–664. doi:10.3892/br.2016.781
16. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Develop. 2009;23(7):781–783. doi:10.1101/gad.1787609
17. Deans C, Maggert KA. What do you mean ‘epigenetic’? Genetics. 2015;199(4):887–896. doi:10.1534/genetics.114.173492
18. Ling C. Epigenetic regulation of insulin action and secretion - role in the pathogenesis of type 2 diabetes. J Intern Med. 2020;288(2):158–167. doi:10.1111/joim.13049
19. Roy A. Type 2 Diabetes and epigenetics: a comprehensive review. Adv New Under Medi Scien. 2024;9:56–67. doi:10.9734/bpi/anums/v9/7458B
20. Bridgeman SC, Ellison GC, Melton PE, Newsholme P, Mamotte CDS. Epigenetic effects of metformin: from molecular mechanisms to clinical implications. Diabetes Obes Metab. 2018;20(7):1553–1562. doi:10.1111/dom.13262
21. Xie MY, Yang Y, Liu P, Luo Y, Tang SB. 5-aza-2ʹ-deoxycytidine in the regulation of antioxidant enzymes in retinal endothelial cells and rat diabetic retina. Int J Ophthalmol. 2019;12(1):1–7. doi:10.18240/ijo.2019.01.01
22. Chunyang C, Chunren M, Shuai H, et al. DNA methylation in diabetic retinopathy: pathogenetic role and potential therapeutic targets. Cell Biosci. 2022;12:186. doi:10.1186/s13578-022-00927-y
23. Eberhart MS, Ogden C, Engelgau M, Cadwell B, Hedley AA, Saydah SH. Prevalence of overweight and obesity among adults with diagnosed diabetes- United States, 1988-1994 and 1999-2002. Morbid Mortal Wkly Report. 2004;53:1066–1068. PMID: 15549021.
24. Rosenbloom A, Silverstein JH, Eds. Type 2 Diabetes in Children and Adolescents: A Clinician’s Guide to Diagnosis, Epidemiology, Pathogenesis, Prevention, and Treatment. American Diabetes Association; 2003.
25. Chandrasekaran P, Weiskirchen R. The role of obesity in type 2 diabetes mellitus- an overview. Int J Mol Sci. 2024;25(3):1882. doi:10.3390/ijms25031882
26. Brooks NA. Type 2 diabetes: lifestyle changes and drug treatment. Virtual Mentor. 2009;11(3):237–241. doi:10.1001/virtualmentor.2009.11.3.cprl1-0903
27. Cuevas-Ramos D, Mehta R, Aguilar-Salinas CA. Fibroblast growth factor 21 and browning of white adipose tissue. Front Physiol. 2019;10:37. doi:10.3389/fphys.2019.00037
28. Bertola A, Gallerand A, Ivanov S. Immune cell involvement in brown adipose tissue functions. Disc Immunol. 2022;1(1):kyac007. doi:10.1093/discim/kyac007
29. Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis and, impaired angiogenesis. J Clin Invest. 2017;127:74–82. doi:10.1172/JCI88883
30. Straub LG, Scherer PE. Metabolic messengers: adiponectin. Nat Metab. 2019;1:334–339. doi:10.1038/s42255-019-0041-z
31. Weisberg SP, McCann D, Desai M, Rosenbaun M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi:10.1172/JCI19246
32. Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature. 2019;576:51–60. doi:10.1038/s41586-019-1797-8
33. Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi:10.1172/JCI26498
34. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi:10.1172/JCI29069
35. Graham TE, Yang Q, Bluher M, et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. New Engl J Med. 2006;354:2552–2563. doi:10.1056/NEJMoa054862
36. Yang Q, Graham TE, Mody N, et al. Serum retinol-binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436(7049):356–362. doi:10.1038/nature03711
37. Smith AD, Crippa A, Woodcock J, Brage S. Physical activity and incident type 2 diabetes mellitus: a systematic review and dose-response meta-analysis of prospective cohort studies. Diabetologia. 2016;59(12):2527–2545. doi:10.1007/s00125-016-4079-0
38. Chatterjee S, Khunti K, Davies MJ. Type 2 diabetes. Lancet. 2017;389(10085):2239–2251. doi:10.1016/S0140-6736(17)30058-2
39. van den Burg EL, Schoonakker MP, Korpershoek B, et al. Self-initiated lifestyle changes during a fasting-mimicking diet programme in patients with type 2 diabetes: a mixed-methods study. BMC Prim Care. 2024;25(1):148. doi:10.1186/s12875-024-02405-5
40. Kolb H, Martin S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017;15:131. doi:10.1186/s12916-017-0901-x
41. Jannasch F, Kroger J, Schulze MB. Dietary patterns and type 2 diabetes: a systematic literature review and meta-analysis of prospective studies. J Nutr. 2017;147(6):1174–1182. doi:10.3945/jn.116.242552
42. Schwingshackl L, Hoffmann G, Lampousi AM, et al. Food groups and risk of type 2 diabetes mellitus: a systematic review and meta-analysis of prospective studies. Eur J Epidemiol. 2017. doi:10.1007/s10654-017-0246-y
43. Fardet A, Boirie Y. Associations between food and beverage groups and major diet-related chronic diseases: an exhaustive review of pooled/meta-analyses and systematic reviews. Nutr Rev. 2014;72:741–762. doi:10.1111/nure.12153
44. Bao Y, Han J, Hu FB, et al. Association of nut consumption with total and cause-specific mortality. N Engl J Med. 2013;369:2001–2011. doi:10.1056/NEJMoa1307352
45. U.S. Department of Health and Human Services. 2015-2020 Dietary Guidelines for Americans. Available from: https://health.gov/dietaryguidelines/2015/.
46. Herieka M, Erridge C. High-fat meal induced postprandial inflammation. Mol Nutr Food Res. 2014;58:136–146. doi:10.1002/mnfr.201300104
47. Stefanson AL, Bakovic M. Dietary regulation of Keap1/Nrf2/ARE pathway: focus on plant-derived compounds and trace minerals. Nutrients. 2014;6:3777–3801. doi:10.3390/nu6093777
48. Boets E, Gomand SV, Deroover L, et al. Systemic availability and metabolism of colonic-derived short-term fatty acids in healthy subjects: a stable isotope study. J Physiol. 2017;595:541–555. doi:10.1113/JP272613
49. Priyadarshini M, Wicksteed B, Schiltz GE. SCFA receptors in pancreatic beta cells: novel diabetes targets? Trends Endocrinol Metab. 2016;27:653–664. doi:10.1016/j.tem.2016.03.011
50. Aune D, Norat T, Leitzmann M, Tonstad S, Vatten LJ. Physical activity and the risk of type 2 diabetes: a systematic review and dose-response meta-analysis. Eur J Epidemiol. 2015;30:529–542. doi:10.1007/s10654-015-0056-z
51. DiPietro L, Gribok A, Stevens MS, Hamm LF, Rumpler W. Three 15-min bouts of moderate post-meal walking significantly improves 24-h glycemic control in older people at risk for impaired glucose tolerance. Diabetes Care. 2013;36:3262–3268. doi:10.2337/dc13-0084
52. Parker L, Shaw CS, Banting L, et al. Acute low-volume high-intensity interval exercise and continuous moderate-intensity exercise elicit a similar improvement in 24-h glycemic control in overweight and obese adults. Front Physiol. 2016;7:661. doi:10.3389/fphys.2016.00661
53. de Rezende LF, Rey-Lopez JP, Matsudo VK, Do Carmo LO. Sedentary behavior and health outcomes among older adults: a systematic review. BMC Public Health. 2014;14:333. doi:10.1186/1471-2458-14-333
54. Biswas A, Oh PI, Faulkner GE, et al. Sedentary time and its association with risk for disease incidence, mortality, and hospitalization in adults: a systematic review and meta-analysis. Ann Intern Med. 2015;162(2):123–132. doi:10.7326/M14-1651
55. Gabel L, Ridgers ND, Della Gatta PA, et al. Associations of sedentary time patterns and TV viewing time with inflammatory and endothelial function biomarkers in children. Pediatr Obes. 2015;11:194–201. doi:10.1111/ijpo.12045
56. Gjevestad GO, Holven KB, Ulven SM. Effects of exercise on gene expression of inflammatory markers in human peripheral blood cells: a systematic review. Curr Cardiovasc Risk Rep. 2015;9:34. doi:10.1007/s12170-015-0463-4
57. Shan Z, Ma H, Xie M, et al. Sleep duration and risk of type 2 diabetes: a meta-analysis of prospective studies. Diabetes Care. 2015;38:529–537.
58. Xi B, He D, Zhang M, Xue J, Zhou D. Shorter sleep duration predicts risk of metabolic syndrome: a systematic review and meta-analysis. Sleep Med Rev. 2014;18:293–297. doi:10.1016/j.smrv.2013.06.001
59. Leng Y, Cappuccio FP, Surtees PG, Luben R, Brayne C, Khaw KT. Daytime napping, sleep duration and increased 8-year risk of type 2 diabetes in a British population. Nutr, Metab Cardiovasc Dis. 2016;26:996–1003. doi:10.1016/j.numecd.2016.06.006
60. Rao MN, Neylan TC, Grunfeld C, Mulligan K, Schambelan M, Schwartz JM. Sub-chronic sleep restriction causes tissue-specific insulin resistance. J Clin Endocrinol Metab. 2015;100:1664–1671. doi:10.1210/jc.2014-3911
61. Donga E, van Dijk M, van Dijk JG, et al. A single night of partial sleep deprivation induces insulin resistance in multiple metabolic pathways in healthy subjects. J Clin Endocrinol Metab. 2010;95:2963–2968. doi:10.1210/jc.2009-2430
62. Gurung M, Li Z, You H, et al. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine. 2020;51:102590. doi:10.1016/j.ebiom.2019.11.051
63. Wu FY, Yin RX. Recent progress in epigenetics of obesity. Diabetol Metab Syndr. 2022;14(1):171. doi:10.1186/s13098-022-00947-1
64. Prasad M, Rajagopal P, Devarajan N, et al. A comprehensive review on high-fat diet-induced diabetes: an epigenetic view. J Nutr Biochem. 2022;107:109037. doi:10.1016/j.jnutbio.2022.109037
65. Lackey DE, Olefsky IJM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. 2016;12(1):15–28. doi:10.1038/nrendo.2016.189
66. Olefsky JM, Glass ICK. Macrophages, inflammation and insulin resistance. Annu Rev Physiol. 2010;219–246. doi:10.1146/annurev-physiol-021909-135846
67. Barrón-Cabrera E, Ramos-Lopez O, González-Becerra K, et al. Epigenetic modifications as outcomes of exercise interventions related to specific metabolic alterations: a systematic review. Lifestyle Genom. 2019;12(1–6):25–44. doi:10.1159/000503289
68. Woo V, Alenghat T. Epigenetic regulation by gut microbiota. Gut Microbes. 2022;14(1):2022407. doi:10.1080/19490976.2021.2022407
69. Miro-Blanch J, Yanes O. Epigenetic regulation at the interplay between gut microbiota and host metabolism. Front Genet. 2019;10:638. doi:10.3389/fgene.2019.00638
70. Bermudez V, Salazar J, Martinez M, et al. Prevalence and associated factors of insulin resistance in adults from Maracaibo City, Venezuela. Adv Prev Med. 2016;9405105:13. doi:10.1155/2016/9405105
71. Sanches JM, Zhao LN, Salehi A, Wollheim CB, Kaldis P. Pathophysiology of type 2 diabetes and the impact of altered metabolic inter-organ crosstalk. FEBS J. 2021;290(3):620–648. doi:10.1111/febs.16306
72. Bedi O, Aggarwal S, Trehanpati N, Ramakrishna G, Krishan P. Molecular and pathological events involved in the pathogenesis of diabetes-associated nonalcoholic fatty liver disease. J Clin Exp Hepatol. 2019;9(5):607–618. doi:10.1016/j.jceh.2018.10.004
73. Gabriely I, Ma XH, Yang XM, et al. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes. 2002;51(10):2951–2958. doi:10.2337/diabetes.51.10.2951
74. Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300(5622):1140–1142. doi:10.1126/science.1082889
75. Rytka JM, Wueest S, Schoenle EJ, Konrad D. The portal theory supported by venous drainage-selective fat transplantation. Diabetes. 2011;60(1):56–63. doi:10.2337/db10-0697
76. Reed J, Bain S, Kanamarlapudi V. A review of current trends with type 2 diabetes epidemiology, etiology, pathogenesis, treatments, and future perspectives. Diabet Metabol Syndr Obes. 2021;14:3567–3602. doi:10.2147/DMSO.S319895
77. Schriever SC, Kabra DG, Pfuhlmann K, et al. Type 2 diabetes risk gene Dusp8 regulates hypothalamic Jnk signaling and insulin sensitivity. J Clin Invest. 2020;130(11):6093–6108. doi:10.1172/JCI136363
78. Al-Hussaniy HA, Alburghaif AH, Naji MA. Leptin hormone and its effectiveness in reproduction, metabolism, immunity, diabetes, hopes, and ambitions. J Med Life. 2021;14(5):600–605. doi:10.25122/jml-2021-0153
79. Lundqvist MH, Almby K, Wiklund U, et al. Altered hormonal and autonomic nerve responses to hypo- and hyperglycemia are found in overweight and insulin-resistant individuals and may contribute to the development of type 2 diabetes. Diabetologia. 2021;64(3):641–655. doi:10.1007/s00125-020-05332-z
80. Salazar J, Chávez-Castillo M, Rojas J, et al. Is ‘leptin resistance’ another key resistance to manage type 2 diabetes? Curr Diabet Rev. 2020;16(7):733–749. doi:10.2174/1573399816666191230111838
81. Adlanmerini M, Nguyen HC, Krusen BM, et al. Hypothalamic REV-ERB nuclear receptors control diurnal food intake and leptin sensitivity in diet-induced obese mice. J Clin Invest. 2021;131(1):e140424. doi:10.1172/JCI140424
82. Alonge KM, D’Alessio DA, Schwartz MW. Brain control of blood glucose levels: implications for the pathogenesis of type 2 diabetes. Diabetologia. 2021;64(1):5–14. doi:10.1007/s00125-020-05293-3
83. Bentsen MA, Rausch DM, Mirzadeh Z, et al. Transcriptomic analysis links diverse hypothalamic cell types to fibroblast growth factor 1-induced sustained diabetes remission. Nat Commun. 2020;11(1):4458. doi:10.1038/s41467-020-17720-5
84. Giordo R, Posadino AM, Mangoni AA, Pintus G. Metformin-mediated epigenetic modifications in diabetes and associated conditions: biological and clinical relevance. Biochem Pharmacol. 2023;215:115732. doi:10.1016/j.bcp.2023.115732
85. Huang H, Weng H, Deng X, Chen J. RNA modifications in cancer: functions, mechanisms, and therapeutic implications. Annu Rev Cancer Biol. 2020;4:221–224. doi:10.1146/annurev-cancerbio-030419-033357
86. Ling C, Del Guerra S, Lupi R, et al. Epigenetic regulation of PPARGC1A in human type 2 diabetes islets and effect on insulin secretion. Diabetologia. 2008;51(4):615–622. doi:10.1007/s00125-007-0916-5
87. Yang BT, Dayeh TA, Kirkpatrick CL, et al. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA 1c levels in human pancreatic islets. Diabetologia. 2011;54(2):360–367. doi:10.1007/s00125-0101967-6
88. Yang BT, Dayeh TA, Volkov PA, et al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol. 2012;26(7):1203–1212. doi:10.1210/me.2012-1004
89. Hall E, Dayeh T, Kirkpatrick CL, Wollheim CB, Nitert MD, Ling C. DNA methylation of the glucagon-like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Med Genet. 2013;14:76. doi:10.1186/1471-2350-14-76
90. Jin QIAN, Kaytor EN, Towle HC, Olson LK. Upstream stimulatory factor regulates Pdx-1 gene expression in differentiated pancreatic β-cells. Biochem J. 1999;341(Pt 2):315–322. PMID: 10393088.
91. Poy MN, Eliasson L, Krutzfeldt J, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432(7014):226–230. doi:10.1038/nature03076
92. Kong L, Zhu J, Han W, et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol. 2011;48(1):61–69. doi:10.1007/s00592-010-0226-0
93. Karolina DS, Tavintharan S, Armugam A, et al. Circulating miRNA profiles in patients with metabolic syndrome. J Clin Endocrinol Metab. 2012;97(12):E2271–2276. doi:10.1210/jc.2012-1996
94. Sathishkumar C, Prabu P, Mohan V, Balasubramanyam M. Linking a role of lncRNAs (long noncoding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum Genomics. 2018;12(1):41. doi:10.1186/s40246-018-0173-3
95. Razin A. CpG methylation, chromatin structure, and gene silencing: a three-way connection. EMBO J. 1998;17(17):4905–4908. doi:10.1093/emboj/17.17.4905
96. Zhou Z, Sun B, Li X, Zhu C. DNA methylation landscapes in the pathogenesis of type 2 diabetes mellitus. Nutr Metab. 2018;15:47. doi:10.1186/s12986-018-0283-x
97. Singh R, Chandel S, Dey D, et al. Epigenetic modification and therapeutic targets of diabetes mellitus. Biosci Rep. 2020;40(9):BSR20202160. doi:10.1042/BSR20202160
98. Sanchis-Gomar F, Garcia-Gimenez JL, Pallardo FV. Mitochondrial biogenesis in health and disease: molecular and therapeutic approaches. Curr Pharm Des. 2014;20(35):5619–5633. doi:10.2174/1381612820666140306095106
99. Liang H, Ward WF. PGC-1α: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30(4):145–151. doi:10.1152/advan.00052.2006
100. Dayeh T, Ling C. Does epigenetic dysregulation of pancreatic islets contribute to impaired insulin secretion and type 2 diabetes? Biochem Cell Biol. 2015;93(5):511–521. doi:10.1139/bcb-2015-0057
101. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi:10.1038/47412
102. Marpadga A, Reddy RN. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res. 2011;90(3):421–429. doi:10.1093/cvr/cvr024
103. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008;118(6):2316–2324. doi:10.1172/JCI33655
104. Sharma S, Leonard J, Lee S, Chapman HD, Leiter EH, Montminy MR. Pancreatic islet expression of the homeobox factor STF-1 relies on an E-box motif that binds USF. J Biol Chem. 1996;271(4):2294–2299. doi:10.1074/jbc.271.4.2294
105. Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. 2005;436(7054):1103. doi:10.1038/nature04048
106. Green D, Dalmany T, Chapman T. Micro-guards and micro-messengers of the genome. Heredity. 2016;116(2):125–134. doi:10.1038/hdy.2015.84
107. Bartel DP. Metazoan microRNAs. Cell. 2018;173:20–51. doi:10.1016/j.cell.2018.03.006
108. Prasanth KV, Spector DL. Eukaryotic regulatory RNAs: an answer to the ‘genome complexity’ conundrum. Genes Dev. 2007;21(1):11–42. doi:10.1101/gad.1484207
109. Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32. 2 alters susceptibility to type 1 diabetes. Nat Genet. 2010;42(1):68–71. doi:10.1038/ng.493
110. Yin DD, Zhang EB, You LH, et al. Downregulation of lncRNA TUG1 affects apoptosis and insulin secretion in mouse pancreatic β cells. Cell Physiol Biochem. 2015;35(5):1892–1904. doi:10.1159/000373999
111. Ruan Y, Lin N, Ma Q, et al. Circulating LncRNAs analysis in patients with type 2 diabetes reveals novel genes influencing glucose metabolism and islet β-cell function. Cell Physiol Biochem. 2018;46(1):335–350. doi:10.1159/000488434
112. Yan L, Zhang X, Xu F, et al. Four-microRNA signature for detection of type 2 diabetes. World J Clin Cases. 2020;8(10):1923–1931. doi:10.12998/wjcc.v8.i10.1923
113. Jiménez-Lucena R, Camargo A, Alcalá-Diaz JF, et al. A plasma circulating miRNAs profile predicts type 2 diabetes mellitus and prediabetes: from the CORDIOPREV study. Exp Mol Med. 2018;50(12):1–12. doi:10.1038/s12276-018-0194-y
114. Deng J, Guo F. MicroRNAs and type 2 diabetes. ExRNA. 2019;1:36. doi:10.1186/s41544-019-0038-5
115. Muhonen P, Holthofer H. Epigenetic and microRNA-mediated regulation in diabetes. Nephrol Dial Transplant. 2009;24(4):1088–1096. doi:10.1093/ndt/gfn728
116. Kumar M, Nath S, Prasad HK, Sharma GD, Li Y. Micro-RNAs: a new ray of hope for diabetes mellitus. Protein Cell. 2012;3(10):726–738. doi:10.1007/s13238-012-20550
117. Guay C, Roggli E, Nesca V, Jacovetti C, Regazzi R. Diabetes mellitus, a micro-RNA-related disease? Transl Res. 2011;157(4):253–264. doi:10.1016/j.trsl.2011.01.009
118. Roggli E, Britan A, Gattesco S, et al. Involvement of microRNAs in the cytotoxic effects exerted by pro-inflammatory cytokines on pancreatic beta-cells. Diabetes. 2010;59(4):978–986. doi:10.2337/db09-0881
119. Roggli E, Gattesco S, Caille D, et al. Changes in microRNA expression contribute to pancreatic β-cell dysfunction in prediabetic NOD mice. Diabetes. 2012;61(7):1742–1751. doi:10.2337/db11-1086
120. Herrera BM, Lockstone HE, Taylor JM, et al. Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia. 2010;53(6):1099–1109. doi:10.1007/s00125-010-1667-2
121. Trajkovski M, Hausser J, Soutschek J, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474(7353):649–653. doi:10.1038/nature10112
122. Jordan SD, Krüger M, Willmes DM, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol. 2011;13(4):434–446. doi:10.1038/ncb2211
123. Kornfeld JW, Baitzel C, Könner AC, et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of Hnf1b. Nature. 2013;494(7435):111–115. doi:10.1038/nature11793
124. Gallagher IJ, Scheele C, Keller P, et al. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Med. 2010;2(2):9. doi:10.1186/gm130
125. Kantharidis P, Wang B, Carew RM, Lan HY. Diabetes complications: the microRNA perspective. Diabetes. 2011;60(7):1832–1837. doi:10.2337/db11-0082
126. Zampetaki A, Kiechl S, Drozdov I, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. 2010;107(6):810–817. doi:10.1161/CIRCRESAHA.110.226357
127. Yang W, Lyu Y, Xiang R, Yang J. Long noncoding RNAs in the pathogenesis of insulin resistance. Int J Mol Sci. 2022;23(24):16054. doi:10.3390/ijms232416054
128. Mannar V, Boro H, Patel D, Agstam S, Dalvi M, Bundela V. Epigenetics of the pathogenesis and complications of type 2 diabetes mellitus. touchREV Endocrinol. 2023;19(1):46–53. doi:10.17925/EE.2023.19.1.46
129. Ergul A. Endothelin-1 and diabetic complications: focus on the vasculature. Pharmacol Res. 2011;63(6):477–482. doi:10.1016/j.phrs.2011.01.012
130. Jin J, Wang X, Zhi X, Meng D. Epigenetic regulation in diabetic vascular complications. J Mol Endocrinol. 2019;63(4):R103–R115. doi:10.1530/JME-19-0170
131. Prattichizzo F, Giuliani A, Ceka A, et al. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin Clin Epigenet. 2015;7(1):56. doi:10.1186/s13148-015-0090-4
132. Liu N, Bezprozvannaya S, Williams AH, et al. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Gene Dev. 2008;22(23):3242–3254. doi:10.1101/gad.1738708
133. Ikeda S, He A, Kong SW, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29(8):2193–2204. doi:10.1128/MCB.01222-08
134. Reddy MA, Natarajan R. Epigenetics in diabetic kidney disease. J Am Soc Nephrol. 2011;22:2182–2185. doi:10.1681/ASN.2011060629
135. Deng Y, Li N, Wu Y, et al. Global, Regional, and National Burden of Diabetes-Related Chronic Kidney Disease from 1990 to 2019. Front Endocrinol. 2021;12:672350. doi:10.3389/fendo.2021.672350
136. Rashid F, Ramakrishnan A, Fields C, Irudayaraj J. Acute PFOA exposure promotes epigenomics alterations in mouse kidney tissues. Toxicol Rep. 2020;7:125–132. doi:10.1016/j.toxrep.2019.12.010
137. Sharma I, Dutta RK, Singh NK, Kanwar YS. High glucose-induced hypomethylation promotes binding of sp-1 to myo-inositol oxygenase: implications in the pathobiology of diabetic tubulopathy. Am J Pathol. 2017;187(4):724–739. doi:10.1016/j.ajpath.2016.12.011
138. Jia Y, Guan M, Zheng Z, et al. MicroRNAs in urine extracellular vesicles as predictors of early-stage diabetic nephropathy. J Diabetes Res. 2016;2016:7932765. doi:10.1155/2016/7932765
139. Araki T, Milbrandt J. Ninjurin2, a novel hemophilic adhesion molecule, is expressed in mature sensory and enteric neurons and promotes neurite outgrowth. J Neurosci. 2000;20(1):187–195. doi:10.1523/JNEUROSCI.20-01-00187.2000
140. Zhang Y, Song C, Liu J, Bi Y, Li H. Inhibition of miR-25 aggravates diabetic peripheral neuropathy. Neuroreport. 2018;29(11):945–953. doi:10.1097/WNR.0000000000001058
141. Feng Y, Chen L, Luo Q, Wu M, Chen Y, Shi X. Involvement of microRNA-146a in diabetic peripheral neuropathy through regulation of inflammation. Drug Des Devel Ther. 2018;12:171–177. doi:10.2147/DDDT.S157109
142. Arguelles AO, Meruvu S, Bowman JD, Choudhury M. Are epigenetic drugs for diabetes and obesity at our doorstep? Drug Discov Today. 2016;21(3):499–509. doi:10.1016/j.drudis.2015.12.001
143. Sharma S, Taliyan R. Histone deacetylase inhibitors: future therapeutics for insulin resistance and type 2 diabetes. Pharmacol Res. 2016;113(Pt A):320–326. doi:10.1016/j.phrs.2016.09.009
144. Christensen DP, Dahllöf M, Lundh M, et al. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med. 2011;17(5–6):378–390. doi:10.2119/molmed.2011.00021
145. Kowluru RA, Santos JM, Mishra M. Epigenetic modifications and diabetic retinopathy. BioMed Res Int. 2013;2013:635284. doi:10.1155/2013/635284
146. Xie M, Tian J, Luo Y, Wei L, Lin S, Tang S. Effects of 5-aza-2ʹ-deoxycytidine and trichostatin A on high glucose-and interleukin-1ß-induced secretory mediators from human retinal endothelial cells and retinal pigment epithelial cells. Mol Vis. 2014;20:1411–1421. PMID: 25352747.
147. Kowluru RA, Mohammad G. Epigenetics and mitochondrial stability in the metabolic memory phenomenon associated with continued progression of diabetic retinopathy. Sci Rep. 2020;10(1):6655. doi:10.1038/s41598-020-63527-1
148. Bestry M, Symons M, Larcombe A, et al. Association of prenatal alcohol exposure with offspring DNA methylation in mammals: a systematic review of the evidence. Clin Clin Epigenet. 2022;14(1):12. doi:10.1186/s13148-022-01231-9
149. Zhang X, Zhao L, Hambly B, et al. Diabetic retinopathy: reversibility of epigenetic modifications and new therapeutic targets. Cell Biosci. 2017;7:42. doi:10.1186/s13578-017-0167-1
150. Malaguarnera G, Gagliano C, Salomone S, et al. Folate status in type 2 diabetic patients with and without retinopathy. Clin Ophthalmol. 2015;9:1437–1442. doi:10.2147/OPTH.S77538
151. Waghode P, Quadir SS, Choudhary D, et al. Small interfering RNA (siRNA) as a potential gene silencing strategy for diabetes and associated complications: challenges and future perspectives. J Diabetes Metab Disord. 2024;23:365–383. doi:10.1007/s40200-024-01405-7
152. Crider KS, Yang TP, Berry RJ, Bailey LP. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr. 2012;3(1):21–38. doi:10.3945/an.111.000992
153. Lebovitz HE. Thiazolidinediones: the forgotten diabetes medications. Curr Diab Rep. 2019;19(12):151. doi:10.1007/s11892-019-1270-y
154. Chen Z, Gong L, Zhang P, et al. Epigenetic down-regulation of SIRT 1 via DNA methylation and oxidative stress signaling contributes to the gestational diabetes mellitus-induced fetal programming of heart ischemia-sensitive phenotype in late life. Int J Biol Sci. 2019;15(6):1240–1251. doi:10.7150/ijbs.33044
155. Pennington KL, DeAngelis MM. Epigenetic mechanisms of the aging human retina. J Exp Neurosci. 2015;9(Suppl 2):51–79. doi:10.4137/JEN.S25513
156. Liu X, Cui H. The palliative effects of folic acid on retinal microvessels in diabetic retinopathy via regulating the metabolism of DNA methylation and hydroxymethylation. Bioengineered. 2021;12(2):10766–10774. doi:10.1080/21655979.2021.2003924
157. Pan Q, Gao Z, Zhu C, Peng Z, Song M, Li L. Overexpression of histone deacetylase SIRT 1 exerts an antiangiogenic role in diabetic retinopathy via miR-20a elevation and YAP/HIF1a/VEGFA depletion. Am J Physiol Endocrinol Metab. 2020;319(5):E932–E943. doi:10.1152/ajpendo.00051.2020
158. Mishra M, Duraisamy AJ, Kowluru RA. SIRT 1: a guardian of the development of diabetic retinopathy. Diabetes. 2018;67(4):745–754. doi:10.2337/db17-0996
159. Kowluru RA, Santos JM, Zhong Q. SIRT 1, a negative regulator of matrix metalloproteinase-9 in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2014;55(9):5653–5660. doi:10.1167/iovs.14-14383
160. Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kB-dependent transcription and cell survival by the SIRT 1 deacetylase. EMBO J. 2004;23(12):2369–2380. doi:10.1038/sj.emboj.7600244
161. Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10(7–12):65–71. doi:10.2119/2005-00029
162. Shan K, Liu C, Liu BH, et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation. 2017;136(17):1629–1642. doi:10.1161/CIRCULATIONAHA.117.029004
163. Park KS, Ciaraldi TP, Abrams-Carter L, Mudaliar S, Nikoulina SE, Henry RR. PPAR-gamma gene expression is elevated in skeletal muscle of obese and type II diabetic subjects. Diabetes. 1997;46(7):1230–1234. doi:10.2337/diab.46.7.1230
164. Loviscach M, Rehman N, Carter L, et al. Distribution of peroxisome proliferator-activated receptors (PPARs) in human skeletal muscle and adipose tissue: relation to insulin action. Diabetologia. 2000;43(3):304–311. doi:10.1007/s001250050048
165. Balasubramanyam M, Mohan V. Current concepts of PPAR-gamma signaling in diabetes. Curr Sci. 2000;79:1440–1446. Corpus ID: 86576611.
166. Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases. Epigenet Chromatin. 2017;10:23. doi:10.1186/s13072-017-0130-8
167. Toperoff G, Aran D, Kark JD, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Human Mol Genet. 2012;21(2):371–383. doi:10.1093/hmg/ddr472
168. Jinying Z. Global DNA methylation is associated with insulin resistance: a monozygotic twin study. Diabetes. 2012;61(2):542–546. doi:10.2337/db11-1048
169. Ramírez-Alarcón K, Victoriano M, Mardones L, et al. Phytochemicals as Potential Epidrugs in Type 2 Diabetes Mellitus. Front Endocrinol. 2021;12:656978. doi:10.3389/fendo.2021.656978
170. Weir GC, Cavelti-Weder C, Bonner-Weir S. Stem cell approaches for diabetes: towards beta cell replacement. Genome Med. 2011;3(9):61. doi:10.1186/gm277
171. Ding Q, Gao Z, Chen K, Zhang Q, Hu S, Zhao L. Inflammation-related epigenetic modification: the bridge between immune and metabolism in type 2 diabetes. Front Immunol. 2022;13:883410. doi:10.3389/fimmu.2022.883410
172. Yu X, Mao W, Zhai Y, et al. Anti-tumor activity of metformin: from metabolic and epigenetic perspectives. Oncotarget. 2017;8(3):5619–5628. doi:10.18632/oncotarget.13639
173. Demirsoy IH, Ertural DY, Balci S, et al. Profiles of circulating metformin treatment in patients with type 2 diabetes. J Med Biochem. 2018;37(4):499–506. doi:10.2478/jomb-2018-0009
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.