")
Back to Journals » Drug Design, Development and Therapy » Volume 19
The Role of Radiosensitizing Drugs in Osteosarcoma Treatment: Mechanisms and Clinical Perspectives
Authors Zhang Y, Xie Y, Wang Y, Huang P, Lu Y
Received 16 December 2024
Accepted for publication 9 March 2025
Published 14 March 2025 Volume 2025:19 Pages 1927—1942
DOI https://doi.org/10.2147/DDDT.S512479
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Yilei Zhang,* Yuhuan Xie,* Yiwen Wang,* Panpan Huang, Yao Lu
School of Basic Medicine, Gannan Medical University, Ganzhou, Jiangxi, 341000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yao Lu; Panpan Huang, Email [email protected]; [email protected]
Abstract: Osteosarcoma is a highly malignant bone tumor that is resistant to radiotherapy and is associated with poor treatment outcomes and prognoses. Understanding the mechanisms of radioresistance and finding strategies to enhance the radiosensitivity is crucial for improving clinical efficacy. The aim of this review was to address the approaches for enhancing the efficacy of radiotherapy in osteosarcoma, thereby improving patient outcomes. Specifically, we have focused on the mechanisms of radiosensitization and the relationship between drugs that enhance radiosensitivity and cancer. These mechanisms involve a delay in DNA damage repair, promotion of apoptosis, inhibition of angiogenesis, and regulation of the tumor microenvironment. In addition, we have summarized the effects of these drugs on the proliferation, migration, invasion and apoptosis of osteosarcoma cell lines. Finally, we have discussed the therapeutic effects and adverse reactions of these drugs in other cancers, providing valuable guidance for clinical treatment strategies tailored to patients with osteosarcoma.
Keywords: osteosarcoma, radiotherapy, radiosensitization, cancer
Graphical Abstract:

Introduction
Osteosarcoma is the most common primary malignant tumor, with the total number of cases accounting for approximately 35% of all cases of primary malignant tumors.1,2 Moreover, patients with incompletely resectable or recurrent osteosarcoma have an extremely poor prognosis, which warrants an urgent clinical need for noninvasive therapies to improve the survival of patients with osteosarcoma.3,4 Radiotherapy has a positive impact on clinical outcomes in cases of poor prognosis owing to surgical margins within the lesion or unresectable lesions.5 Unlike chemotherapy and surgery, radiotherapy is used to ensure better local control, which allows precise irradiation of the tumor site and avoids impacting healthy tissues. However, the radioresistance in osteosarcoma cells greatly reduces the therapeutic benefits of traditional radiotherapy.6,7 According to experience, innovation of ways to use existing drugs is an important direction of drug research and development (R&D), as it can greatly reduce costs and shorten the time of R&D. Therefore, we envisioned that some of the existing drugs could be used to maximize the killing effect of radiotherapy on osteosarcoma cells, thereby improving the overall quality of life in patients. This approach may stimulate the potential use of existing drugs as sensitizers for radiotherapy. Indeed, several drugs target osteosarcoma cells, which can increase the sensitivity of osteosarcoma cells to radiotherapy, thereby decreasing the dose of radiotherapy and increasing its efficacy, greatly improving the survival rate in patients. Through a comprehensive review of numerous studies, we aimed to discuss in detail the current situation on classical and emerging drugs that may overcome the radioresistance of osteosarcoma and provide a valuable reference for improving the sensitivity of osteosarcoma to radiotherapy. In addition, numerous clinical trials have demonstrated the clinical value of these drugs in cancer treatment. Hence, we further discuss the clinical trials that explain the important role of these drugs in cancer treatment.
Delay in DNA Damage Repair
The main mechanism of how radiotherapy kills tumor cells is a damage it causes to their DNA, thereby inhibiting their growth and division and ultimately leading to their death. However, tumor cells rely on cell cycle checkpoints to gain time to repair DNA damage caused by radiation exposure, resulting in the resistance to radiotherapy. Fortunately, DNA damage repair and cell cycle checkpoint inhibitors can reverse this process, allowing osteosarcoma cells to skip cell cycle checkpoints and enter mitosis directly, eventually leading to apoptosis. Therefore, using drugs that target checkpoint-related regulators or DNA damage repair is a promising strategy for improving the efficacy of radiotherapy in osteosarcoma.
Decitabine was approved by the Food and Drug Administration in July 2020 for use in acute myeloid leukemia and myelodysplastic syndrome8 and has demonstrated good efficacy. Decitabine is a deoxycytidine analog and DNA methyltransferase inhibitor. It blocks DNA methylation by inhibiting DNA methyltransferase and induces cell cycle arrest in the G2/M phase through the p53-independent pathway in human cancer cells.9 Compared to irradiation alone, pretreatment of osteosarcoma cells with decitabine at the G2/M phase enhanced irradiation-induced apoptosis. This was achieved by demethylating the promoters of 14-3-3σ, CHK2 and DAPK-1 genes, promoting the expression of these genes, and then activating the G2/M checkpoint response in osteosarcoma cells, to improve the radiotherapy effect of osteosarcoma10 (Figure 1 and Table 1). Currently, this drug is widely used for the clinical treatment of several cancers. A Phase II multicenter trial of decitabine in patients with high-risk chronic granulomonocytic leukemia was conducted in Italy. Among patients with chronic myelomonocytic leukemia who received intravenous decitabine (Figure 2), 76–93% were at high or intermediate risk. Forty-two patients treated with six cycles of decitabine showed an overall efficacy of 47.6%, with seven complete remissions (16.6%), eight myelosuppressions (19%), one partial remission (2.4%), and four hematological improvements (9.5%). The median follow-up time was 51.5 months (range: 44.4–57.2), and median survival time 17 months, which was significantly longer in the response group than that in non-response group (P=0.02).11 In addition, in two retrospective analyses, the overall response rate (ORR) for decitabine was 26–68% and 2-year survival rate was 25%–48%.12,13 Related studies have added that DAC can up-regulate the expression of PD-L1 in metastatic colorectal cancer, which may make PD-1/PD-L1 inhibitors more likely to play a role, that is, the combination of DAC and anti-PD-1/anti-PD-L1 antibodies may be a potential treatment for radiosensitization of osteosarcoma.14 However, clinical trials have shown that patients may experience adverse events such as anemia, neutropenia, and thrombocytopenia11,12 (Table 2). Altogether, decitabine is a promising drug for the treatment of certain hematological cancers; however, its side effects should not be ignored. Decitabine has great potential as a radiotherapy sensitizer for osteosarcoma and may provide more effective treatment options for osteosarcoma patients.
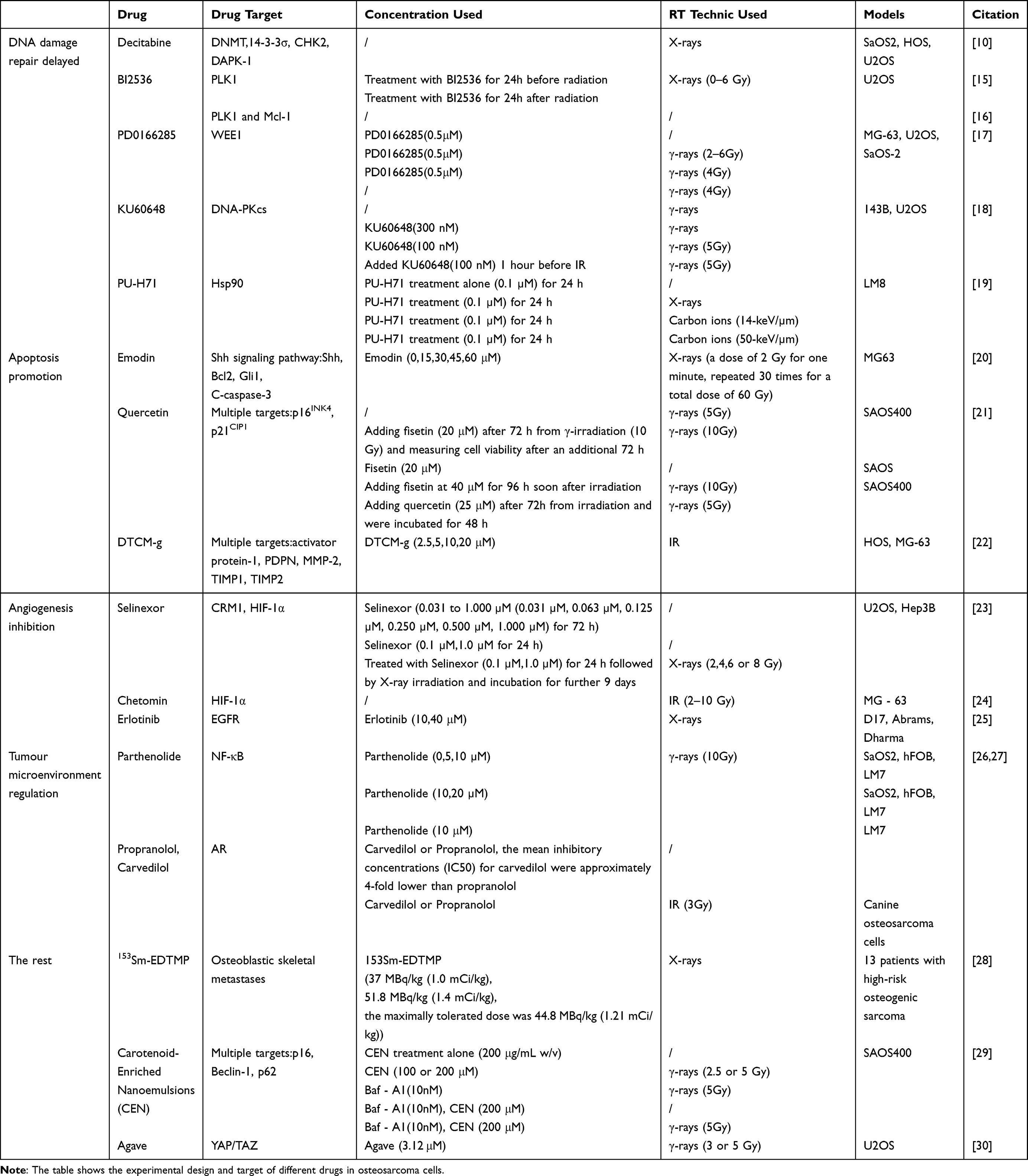 |
Table 1 Preclinical Drug Trial Record Table |
 |
Table 2 Clinical Trials of Drugs That Sensitize Osteosarcoma Radiotherapy in the Treatment of Other Cancers |
 |
Figure 1 Mechanism of decitabine, BI2536, PD0166285, KU60648, PU-H71, emodin, quercetin and DTCM-g in radiosensitization of osteosarcoma. Among them, Decitabine, BI2536, PD0166285, KU60648 and PU-H71 achieved radiosensitization mainly by delaying DNA damage repair mechanisms, while Emodin, Quercetin and DTCM-g achieved by promoting apoptosis. |
 |
Figure 2 Clinical trials of the drug in other cancers. Different drugs that can sensitize osteosarcoma to radiotherapy can affect different tumors through different routes of administration. |
BI2536, a dihydropteridinone compound that can act at low nanomolar concentrations, is a highly specific small-molecule PLK1 inhibitor. BI2536 blocked osteosarcoma cells (KHOS and U2OS) in the G2/M phase, thereby effectively inducing mitotic arrest and apoptosis.15 When BI2536 was combined with radiotherapy, it inhibited DNA damage repair of U2OS, and also blocked U2OS cells in the G2/M phase, thereby exerting a radiosensitizing effect16 (Figure 1). Notably, when canine osteosarcoma cells D17 were treated with BI2536, the number of cells was reduced from 20%±2.4% to 7.4%±2% in the G0/G1 phase and increased from 18.3%±2.4% to 47.2%±7.7% in the G2/M phase, decreasing cell viability by up to 61.2% (P<0.0001)31 (Table 1). BI2536 has a significant inhibitory effect on various cancer cells in clinical trials and is widely used in cancer treatment. A Phase II clinical trial was conducted between July 2006 and April 2008 in patients with recurrent stage IIIB/IV non-small cell lung cancer (Figure 2). Four patients had partial remission (4.2%), with a median progression-free survival (PFS) of 8.3 weeks (58-day 95% confidence interval [CI]: 48–85) and a median overall survival (OS) of 28.7 weeks (201-day 95% CI: 180–305), respectively (Table 2). During BI2536 treatment, 37% of the patients experienced grade 4 neutropenia as well as some mild adverse effects such as fatigue (31%) and nausea (27%).32 This shows that BI2536 was well-tolerated by the patients. This observation was confirmed in another Phase I clinical trial on the treatment of patients with advanced solid tumors. According to statistics, common mild to moderate adverse reactions are nausea (52%), fatigue (52%), and loss of appetite (44%), among others. Even with the dose-limiting toxicity (DLT) and the maximum tolerated dose (MTD) (200 mg), the most common adverse reaction was only reversible neutropenia (56%).33 Based on the acceptable safety and potential strong anti-tumor efficacy of BI2536, it is worthy of further clinical evaluation and research to fill the gaps in the specific progress and application of BI2536 in human metabolism research. In addition, the positive results of BI2536 in a preclinical study on osteosarcoma provided a scientific basis for its application potential as a radiotherapy sensitizer.
PD0166285 is a small pyridine pyrimidine molecule that inhibits WEE1 at nanomolar concentrations and prevents cell cycle checkpoint arrest in the S and G2 phases.34–36 By inhibiting CDK1/2 activity through tyrosine 15 site dephosphorylation,37,38 PD0166285 prevents mitotic entry, allowing time for DNA repair. In osteosarcoma cells, it eliminated the G2 checkpoint, induced mitotic catastrophe, and enhanced radiosensitivity (Figure 1). After the combined treatment of osteosarcoma cells with PD0166285 and γ-irradiation, the cell viability was decreased by 2 times, proportion of mitotic cells increased by 2–4 times, and caspase activation level increased by 11–22 times17 (Table 1). In addition, preclinical studies on PD0166285 have revealed its potential as a highly effective drug for leukemia, melanoma, and liver cancer treatment.39–41 Based on these findings, we are optimistic about its efficacy in osteosarcoma treatment in future clinical trials.
KU60648 is a newly developed, potent, small-molecule DNA-PKcs inhibitor that simultaneously targets multiple isomers of DNA-PK and PI3K. KU60648 inhibited DNA damage recognition by inhibiting DNA-PKcs expression and hindering their binding to DNA ends42 (Figure 1). In combination with radiation, KU60648 increased the radiosensitivity of osteosarcoma cells (143 B and U2OS) by 1.5- and 2.5-fold, respectively. It also increased the accumulation of 143B and U2OS cells at the G2/M transition point by 55% and 45%, and the proportion of cells with >20 γH2AX foci by 59% and 107%, respectively18 (Table 1). In summary, KU60648 significantly improved the effects of radiotherapy in osteosarcoma. It also downregulated the double-strand breaks repair ability in colorectal cancer cells and enhanced the sensitivity of cancer cells to KU60648.43 However, administration routes and vectors need to be carefully considered to minimize potential side effects in normal tissues and cells expressing DNA-PKcs.
PU-H71 is a carboxylic acid derivative that exerts radiosensitization by reducing the protein expression levels of Rad51 and Ku70 in mouse osteosarcoma cells (Figure 1). Treatment of LM8 cells with PU-H71 reduced the doses of X-rays and C-ions (D10) required to reduce their survival rate of LM8 cells to 10%, thereby increasing their sensitivity to X-rays and C-ions44 (Table 1). Rad51 and Ku70 are involved in two major double-strand breaks repair pathways (homologous recombination and non-homologous end-joining), respectively. Therefore, the radiosensitizing effect of PU-H71 may involve the regulation of these pathways. Further studies should use human osteosarcoma cell lines to elucidate the underlying mechanisms. In addition, PU-H71 is the most promising Hsp90 inhibitor because of its high solubility and specificity for the ATP-binding region of Hsp90. PU-H71 also exhibits strong antitumor activity.19 This was confirmed in a Phase I clinical trial for the treatment of solid tumors and lymphomas. The results showed that 42.9% of the patients who received intravenous injections of different PU-H71 concentrations were stable Blood test results showed that only a few patients had adverse reactions of anemia45 (Figure 3 and Table 2). Moreover, an open, single-arm, first-in-human trial of PU-H71 in patients with advanced solid tumors showed that the incidence of grade ≥2 adverse events such as common nausea, headache, and fatigue was extremely low, and only one patient exhibited a grade 3 adverse event (vomiting) that could be related to PU-H71. The human body has good tolerance to PU-H71.19,46 PU-H71 is still in the R&D stages. Due to the limited number of clinical trials, data on its therapeutic effects and possible adverse reactions in patients with cancer are insufficient, and market access has not been obtained yet. However, PU-H71 showed a radiosensitizing effect in the preclinical model of osteosarcoma, which provided a novel idea for an excellent treatment strategy of radiotherapy for osteosarcoma in the future.
 |
Figure 3 Mechanism of radiosensitization of Selinexor, chetomin, Erlotinib, Parthenolide, Propranolol, 153Sm-EDTMP and CEN on osteosarcoma. Among them, Selinexor, chetomin and Erlotinib play a role by inhibiting angiogenesis, Parthenolide and Propranolol play a role by regulating tumor microenvironment, 153Sm-EDTMP and CEN have no clear classification. |
Promotion of Apoptosis
Radiation therapy can affect the expression of various apoptosis-related proteins such as Bcl-2 and caspase family proteins, thereby inducing apoptosis. Nevertheless, osteosarcoma cells may use certain mechanisms to combat apoptosis and improve their tolerance to radiotherapy. The application of radiotherapy sensitizers that interfere with these anti-apoptotic mechanisms may improve the efficacy of radiotherapy in patients with osteosarcoma.
Emodin, an anthraquinone derivative, has shown potential in radiotherapy by attenuating radioresistance in human osteosarcoma cells. This effect was attributed to the inhibition of the Shh signaling pathway, leading to the suppression of Shh and Bcl-2 expression, inhibition of the nuclear translocation of Gli1, and increase of caspase-3 cleavage20 (Figure 1). During apoptosis, the Shh signaling pathway affects Bcl-2 expression by regulating the nuclear translocation of Gli1, thereby controlling cell survival and death. Caspase-3 cleavage plays an important role in the final stages of and promotes apoptosis. These intertwined factors determine the outcome of cell fate (Table 1). However, current clinical trial data on its application in osteosarcoma radiotherapy are limited, and the high efficacy of emodin in preclinical trials of osteosarcoma radiotherapy suggests its potential to be developed as a radiosensitizer for osteosarcoma.
Quercetin is a natural flavonoid with anti-proliferative activity in vitro. When 25 μM quercetin was administered after infrared radiation treatment (5 Gy), p21CIP1 and p16INK4 expression levels were significantly reduced compared with infrared radiation treatment alone, and cell viability of SAOS400 (an osteosarcoma cell line obtained after simulated clinical sublethal dose of γ-ray irradiation) was significantly reduced by about 29%21 (Figure 1 and Table 1). They also affect apoptosis by regulating the activity and expression of apoptosis-related proteins. Regarding clinical applications, quercetin dose levels between 60 mg / m2 and 1700 mg / m2 produced dose-limiting nephrotoxicity, but no bone marrow suppression. In a patient with cisplatin-resistant ovarian cancer, CA 125 (a tumor marker) levels were decreased from 295 to 55 units/mL after two courses of quercetin (420 mg/m2), whereas in another patient with hepatocellular carcinoma, serum alpha-fetoprotein levels decreased after several courses of treatment. In conclusion, quercetin can be safely administered by i.v. bolus at a dose injection47 (Figure 3). Additionally, the risk of extraprostatic manifestations of prostate cancer (stage III or IV tumors) was reduced significantly, which was associated with an increased intake of cruciferous vegetables rich in quercetin and sulfur (P=0.02).48,49 In addition, its combination with raphanin showed the best therapeutic effect on pancreatic cancer stem cells and was nontoxic to mice49 (Table 2). These results underscore the potential of quercetin as a radiosensitizer in osteosarcoma therapy, suggesting its promising applications in clinical settings. However, current research on the involvement of quercetin in osteosarcoma radiotherapy does not provide sufficient information. In addition, considering the synergistic effect of quercetin and raphanin, combining quercetin with other drugs may be a feasible choice for patients with osteosarcoma in the future.
DTCM-g is a novel piperidine compound that is a macrophage activation inhibitor of AP-1 (a heterodimer composed of c-Fos and c-Jun) activity (Figure 1). DTCM-g enhanced the radiosensitivity of HOS and MG-63 cells by reducing the regulation of AP-1 on target genes, and inhibiting AP-1 and the expression of PDPN, MMP-2, TIMP1, and TIMP2, which participate in the regulation of extracellular matrix degradation and reconstruction, thereby affecting the occurrence and process of apoptosis22 (Table 1). Therefore, DTCM-g can be used as an effective adjuvant therapy to improve the effects of radiotherapy. Moreover, DTCM-g demonstrated radiosensitization capabilities in glioblastoma,50,51 further emphasizing its potential as a valuable therapeutic drug in radiotherapy.
Apoptosis is essentially a process of signal transduction. In addition to the key role of Bcl-2 and caspase family proteins in apoptosis, p53 protein is also an important molecule to regulate apoptosis. Under normal circumstances, the level of p53 is strictly maintained at a low level. Activated p53 translocates into the nucleus as a transcription factor and regulates the expression of hundreds of genes, including many pro-apoptotic genes. Studies have shown that juniper berry extract can activate the ER stress pathway of human SH-SY5Y neuroblastoma cells, affect the expression of HSPA5 gene, and promote the expression and nuclear transport of p53. The activated p53 promotes the expression of specific cell cycle and cell survival regulatory genes, and ultimately leads to DNA fragmentation and apoptosis.52 This discovery provides new ideas and research directions for the development of new radiosensitizers. However, juniper berry extract has not yet entered the clinical research stage, and its safety, efficacy and mechanism of action still need to be further verified by systematic preclinical studies and clinical trials. Future research should focus on its pharmacokinetic characteristics, optimal administration regimen, and synergy with other treatment methods to evaluate its clinical application potential.
Inhibition of Angiogenesis
Malignant tumors promote angiogenesis to consume additional oxygen and nutrients, thereby facilitating their rapid growth and dissemination. The poor therapeutic outcomes and prognosis of radiotherapy in osteosarcoma can be attributed to an ongoing tumor-induced angiogenesis.23,53 Anti-angiogenic drugs effectively suppress tumor angiogenesis. When combined with radiotherapy, these drugs provide a more effective approach for tumor control (Figure 1).
Selinexor exhibits a wide range of antitumor activities and was first approved for the treatment of multiple myeloma (USA) on July 3, 2019. It is an orally administered, selective nuclear translocation inhibitor that binds specifically and reversibly to CRM1 (exportin 1, XPO1) at cysteine residue 528.51,54 CRM1 is a key nuclear export receptor and one of its cargo proteins, prolyl hydroxylase 2 (PHD2), initiates the degradation of hypoxia-inducible factors (HIFs) under normoxic conditions. In osteosarcoma radiotherapy, selinexor inhibits CRM1, affecting PHD2 and targeting HIF-1α, thereby increasing radiosensitivity23,55 (Figure 2 and Table 1). This property of selinexor that allows it to target the HIF pathway may be a potent tool for overcoming hypoxia-induced radioresistance. Regarding clinical treatment with selinexor, a phase I study reported that 189 patients with advanced solid tumors were treated with selinexor between June 2012 and October 2015. Among the 157 patients who could be evaluated for efficacy, 1 had complete remission (melanoma), and 6 patients (melanoma, colorectal cancer, ovarian cancer, prostate cancer, thymoma, and cervical cancer) had imaging partial remission, with an objective remission rate of 4%. In addition, 67 patients (43%) had stable disease, of which 27 (17%) had durable disease control (≥4 months) and 83 (53%) had disease progression to optimal remission (Figure 3). Moreover, blood test results showed severe adverse events or toxic reactions caused by selinexor, including thrombocytopenia (16%), hyponatremia (13%), anemia (9%) and neutropenia (8%). Other common grade 3 or 4 AEs were fatigue (15%) and anorexia (6%). Notably, all the patients were less tolerant to higher doses of selinexor54 (Table 2). These results indicate that selinexor exhibits monotherapeutic activity with an acceptable safety profile and demonstrates efficacy in patients with advanced solid tumors. Although the objective remission rate was low, some patients achieved durable disease control or partial remission. In addition, some studies have pointed out that compared with monotherapy, Selinexor combined with anti-PD-1, anti-PD-L1 or anti-CTLA4 antibodies can more effectively slow down the growth rate of tumors. When Selinexor combined with high-dose anti-PD-L1 antibodies, the tumors in mice completely subsided and produced lasting immune control, which is expected to become a key force in the treatment of osteosarcoma.56 This provides evidence for the future development of selinexor as a clinical radiotherapy drug for osteosarcoma treatment.
Chetomin, derived from chaetomium globosum, is a dithiobiketopiperazine metabolite that disrupts the binding of HIF-1α and HIF-2α to p300, thereby attenuating the HIF pathway24,55 (Figure 3). It affects angiogenesis by inhibiting HIF-1α, resulting in decreased CA9 and VEGF mRNA expression.25,57 In vitro studies have shown that chetomin inhibits HIF-1α to significantly reduce hypoxia-induced radioresistance in MG-63 cells24 (Table 1). Chetomin enhances the therapeutic effect in osteosarcoma by targeting angiogenesis and enhancing the radiosensitivity of tumor cells, suggesting its potential application in treatment.
Erlotinib was first approved by the Food and Drug Administration on November 18, 2004, as a monotherapy for patients with locally advanced or metastatic non-small cell lung cancer (NSCLC).58 It acts as a selective inhibitor of EGFR tyrosine kinase that significantly promotes VEGF expression when co-administered with radiation, thereby reducing the viability of osteosarcoma cells and increasing their sensitivity to radiotherapy. This was related to EGFR inhibition and VEGF pathway activation25 (Figure 2 and Table 1). Notably, the overactivation of the VEGF pathway can make tumors resistant to treatment with EGFR inhibitors; therefore, its use needs to be considered in light of its potential to cause treatment resistance. Erlotinib is a commonly used targeted therapeutic agent. A Phase II clinical trial conducted at the University of Texas MD Anderson Cancer Center, the United States of America, demonstrated that erlotinib was feasible for the treatment of most of the patients with incurable cutaneous squamous-cell carcinoma. The trial results showed evaluable efficacy in 29 of 39 treated patients. All the responses were partial responses (PR). The disease control rate (PR + disease stabilization) was 72% (21/29). Median PFS was 4.7 months (95% CI: 3.5–6.2); median OS was 13 months (95% CI: 8.4–20.5). The 1- and 3-years OS rate was 53% and 19%, respectively. Overall, erlotinib was well-tolerated by the patients, with the most common adverse reactions being acne-like rashes (64%) and malaise (46%). Most treatment-related adverse reactions were of grades 1 and 2, with none of grade 4 or 5 and the blood test results did not show any adverse reactions.59 The EURTAC trial demonstrated significant advantages over standard chemotherapy. Researchers conducted an open-label, randomly assigned Phase III clinical trial in 42 hospitals in France, Italy, and Spain and enrolled 174 patients with NSCLC and EGFR mutations; 86 patients were treated with erlotinib and 87 with standard chemotherapy. The median PFS was 9.7 months (95% CI: 8.4–12.3) in the erlotinib group, compared with 5.2 months (95% CI: 4.5–5.8) in the standard chemotherapy group (hazard ratio 0.37, 95% CI: 0.25–0.54, P<0.0001) (Figure 3). Grade 3–4 adverse reactions mainly included rashes (11 out of 84 patients; 13%) in the erlotinib group, with none reported in the chemotherapy group. Neutropenia (none vs 18 cases [22%]), anemia (1 case [1%] vs 3 cases [4%]), and elevated aminotransferase concentration (2 [2%] vs 0) were also observed. Five patients (6%) in the erlotinib group developed serious treatment-related adverse reactions, as opposed to 16 patients (20%) in the chemotherapy group60 (Table 2). From the available clinical trial data, erlotinib has shown good tolerability and some efficacy in the treatment of the cutaneous squamous-cell carcinoma and NSCLC. This provided encouraging evidence for its potential role as a radiosensitizer in the treatment of clinical osteosarcoma.
Regulation of the Tumor Microenvironment
The occurrence and development of osteosarcomas are closely related to the tumor microenvironment. Signaling pathways involving several components of the tumor microenvironment support tumor cells and provide the conditions necessary for their growth and spread.61 Therefore, some drugs achieve sensitization by affecting the osteosarcoma microenvironment during radiotherapy. Recent studies have shown that the overexpression of inflammatory markers in highly invasive osteosarcoma is significantly correlated with the decrease of radiotherapy efficacy. For such highly inflammatory cells, the effectiveness of radiosensitizers in improving radiotherapy sensitivity will also be discussed.62 Unfortunately, there is no direct study to verify that the following drugs will sensitize high inflammatory cells to radiotherapy, but their significant anti-inflammatory effects still give us great confidence, and we hope that more studies will verify this conjecture in the future.
Parthenolide is a natural terpenoid alkaloid possessing a wide range of pharmacological properties, including anti-inflammatory and antitumor activities. It inhibits NF-κB signaling (This will reduce the levels of IL-6 and TNF-α) and potentiates tumor cell responses to stimuli (such as radiation-induced DNA damage); cells treated with parthenolide or γ-irradiation alone contained an average of 31% and 12% apoptotic nuclei, respectively, whereas those treated with the combination showed 56% apoptotic subpopulations. In addition, compared with other single treatments, the cell death rate and active caspase-3 in the combined treatment were increased significantly, and the CD133 + stem cell-like cell pool in osteosarcoma was eliminated to a greater extent26 (Figure 3 and Table 1). Workers exposed to plants containing parthenolide were thought to be susceptible to contact dermatitis, but current reports state that the drug is mutagenic in vivo and in vitro due to oxidative DNA damage or other possible mechanisms, raising concerns about the genotoxicity of the drug.63 This natural drug shows great therapeutic potential owing to its dual antiproliferative and apoptosis-inducing effects, its anti-inflammatory effect also suggests its potential to sensitize high-inflammatory cells to radiotherapy, which highlights its promise in osteosarcoma radiotherapy treatment.
Propranolol was first approved for angina pectoris treatment in Japan on August 18, 1966, and carvedilol was first approved for angina pectoris, essential hypertension, and renal hypertension treatment in Japan on January 19, 1993. Both are nonselective β-blockers and AR antagonists, can reduce the secretion of IL-6, reverse the immune microenvironment,64 which may affect cell proliferation, survival, metastasis, and angiogenesis. These compounds may exert radiation resistance by inhibiting the AR-mediated pro-survival pathways and tumor cell viability in a concentration-dependent manner (Figure 2). In osteosarcoma cell lines, the two drugs reduced cell survival and colony formation after 3 Gy of radiotherapy, thereby enhancing radiosensitivity. Long-term treatment can significantly improve the effects of radiotherapy and render tumor cells more sensitive to radiotherapy than short-term treatment27 (Table 1). Studies have shown that carvedilol reduces pro-inflammatory cytokines and increases the expression of anti-inflammatory cytokines and IL-10 protein, and inhibits the occurrence of excessive inflammation.65 A preliminary study demonstrated the positive effects of these two drugs in osteosarcoma treatment. These two drugs are effective against various tumors. In a single-arm clinical study of patients with metastatic angiosarcoma, the inclusion of propranolol or carvedilol in the treatment regimen resulted in a median PFS of 9 and 36 months, respectively, which was significantly longer than the survival in patients who received chemotherapy, radiotherapy, and/or surgery66 (Table 2). In addition, these drugs exhibited preclinical efficacy against breast cancer,67 neuroblastoma,68 locally advanced NSCLC,69 melanoma,70 and hepatocellular carcinoma in patients with cirrhosis71 (Figure 3). Another study found that diarrhea with weight loss (32.9%) and irritability with reduced sleep (25.6%) were the most common adverse effects, and 4.2% of patients were even forced to stop taking the drug because of serious adverse effects.72 In addition, the skin-related side effects of these two drugs, such as contact dermatitis, have attracted attention.73 Even with the aforementioned problems, adverse reactions can be largely avoided if the appropriate dose and administration time are explored. Therefore, propranolol and carvedilol have remarkable clinical value in the radiotherapy treatment for osteosarcoma and the treatment of other diseases.
Additional Agents
The mechanism of action of some drugs remain unknown, but the excellent efficacy of these drugs in the radiosensitization of osteosarcoma deserves our attention.
153Sm-EDTMP, or Samarium-153 ethylenediaminetetramethylene phosphonic acid, is a bone-targeting radiopharmaceutical that selectively eradicates osteoblastic bone metastases, prolonging disease-free survival in high-risk patients. Thirteen patients with high-risk osteosarcoma were treated with X-ray combined with 153Sm-EDTMP; after the end of the study, 5 (38%) had stable disease (6 weeks after treatment), and the other 8 had disease progression, with the median time to disease progression of 51 days. The toxicity caused by treatment is limited to the hematopoietic system, and platelets are the most affected. Of the 12 patients, 8 had grade 3 or 4 thrombocytopenia. Three patients had grade 1 or 2 neutropenia, 5 patients had grade 3 or 4 neutropenia, and anemia symptoms were mild28 (Table 1). Upon intravenous administration, samarium-153 binds to hydroxyapatite in bone tissue with a higher affinity for tumor sites than that for normal bone.74,75 It emits low-energy β particles (average energy 233 keV) (Figure 3) by directly acting on osteosarcoma cells to control and treat lesions. Its bone-targeting property has important clinical and safety implications, and has shown significant efficacy and safety in alleviating bone metastasis pain caused by prostate, breast, and other malignant tumors. In a clinical trial, 277 patients with persistent severe pain due to bone metastases received intravenous (IV) injections of 153Sm-EDTMP. Assessment of pain intensity and relief showed 9.1 ± 0.61 units initially; 1.3 units (54%) after 4.2 ± 3 weeks; and pain reduction to 2.4 ± 1.4 units (74%) after 12 weeks76 (Table 2). Adverse effects of this drug are relatively rare, but it is contraindicated in pregnant women, patients with hepatic and renal insufficiency, and patients with low white blood cell and platelet counts. Samarium-153 can produce a toxic effect in tumor cells by emitting low-energy β-particles, but it only demonstrated preliminary efficacy and potential in the treatment of tumors. Since clinical trials on its anti-tumor efficacy are limited, further research is needed to verify its efficacy and safety. However, its performance as a radiotherapy sensitizer for osteosarcoma is encouraging, and may offer a powerful tool for osteosarcoma treatment.
CEN was launched in May 1997 as a nanoemulsion rich in carotenoids. CEN synergizes with γ-rays to scavenge reactive oxygen species in senescent SAOS400 cells, and the cell viability decreased to 55–60% in the group without treatment; this was higher than the rate in other single treatment groups (Table 1). This combination activates lethal autophagy dependent on AMPK activation (AMPK kinase is activated by a decrease in intracellular ATP and a change in the ATP:AMP ratio), enhancing the sensitivity of radiation-resistant cells to cell death and efficacy of radiotherapy29,77 (Figure 3). As a natural pigment and nutritional supplement, it is generally safe and minimizes the damage to normal tissue cells, making it a superior adjuvant for osteosarcoma treatment.
The main chemical constituents of CEN are polysaccharides, which contain antioxidants and nutrients. When osteosarcoma cells were treated with γ-rays and agave, the cell viability was more inhibited than those treated with single radiation, which indicated that agave effectively improves the killing effect of radiotherapy in osteosarcoma cells (Table 1). Agave induced the early degradation of YAP/TAZ, followed by inhibition of NF-κB, which further reduced the transcriptional functions of YAP and TAZ. This cascade of actions resulted in a reduction in the interaction between the DNA-binding transcription factors YAP and TAZ, thereby inhibiting the transcription of genes that drive oncogenesis. Modulation of these molecular mechanisms leads to a significant inhibition of cell viability, colony formation, and cell migration, ultimately contributing to apoptosis and increased sensitivity to radiation therapy30 (Figure 3). This inexpensive and low-toxicity natural drug showed promising radiosensitizing effects on osteosarcoma cells, warranting further research on its efficacy.
Prospects
In the future, osteosarcoma radiotherapy sensitizers may play an important role in osteosarcoma treatment. With a deeper understanding of cancer biology and its therapeutic mechanisms, the need to enhance the efficacy of radiotherapy is becoming increasingly urgent. Studies have shown that osteosarcoma is characterized by a wide range of mutations and genomic rearrangements, including single nucleotide variations (SNVs), small fragment insertions/deletions (indels) and copy number variations (CNVs). These genomic changes not only affect coding sequences, but also involve non-coding sequences, such as regulatory elements and repetitive sequences (such as LTR elements and HERVs), leading to gene expression disorders, which in turn drive the occurrence and development of tumors.78 In the whole exome sequencing and whole transcriptome analysis of osteosarcoma, it was found that many genes were significantly differentially expressed between osteosarcoma and normal tissues.79,80 These differentially expressed genes, both coding and non-coding sequences, can be regarded as potential targets for in-depth study of osteosarcoma, including the development of new radiotherapy sensitizers.
Some radiosensitizers (such as decitabine and selinexor) have been verified to have synergistic effects with immunotherapy by inducing the expression of PD-L1 and CTLA4 at immune checkpoints. The emerging field of combined use with immunotherapy provides new vitality and hope for the treatment of osteosarcoma.
Although most current studies focused on using these sensitizers in osteosarcoma treatment, their mechanisms of action may also be applicable to other types of tumors, which is of great significance for most patients with cancer. The use of radiotherapy sensitizers in osteosarcoma is likely to incorporate the patients’ genetic, molecular, and environmental characteristics to maximize treatment efficacy. In the future, there may be an increased focus on multi-agent combination therapy strategies, including the use of radiotherapy sensitizers in combination with chemotherapy, targeted therapy, or immunotherapy. Such combination therapies may act on tumor cells through different pathways, thereby inhibiting tumor growth and spread more effectively. A deeper understanding of cancer biology and therapeutic mechanisms will facilitate the discovery of additional radiotherapy sensitizers for osteosarcoma. These novel sensitizers may target specific receptors, molecular pathways, or tumor characteristics to improve the killing effect of radiotherapy on tumor cells, thereby enhancing therapeutic efficacy. They may also have an impact on other types of cancers.
Some of these drugs are in early stages of evaluation and require further clinical validation and translation. Clinical research and practice will continue to drive the development and application of osteosarcoma radiotherapy sensitizers, and more future clinical trials and studies are likely to assess the safety and efficacy of these agents, and ultimately, their application in clinical practice.
Acknowledgments
We would like to thank all colleagues and mentors for their assistance. We would like to thank all authors who provided information on the radiosensitivity of osteosarcoma and its related fields.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation for Young Scholars of China (Grant No.82202928 to Panpan Huang), Jiangxi Provincial Natural Science Foundation (20232BAB206111 to Panpan Huang), Gannan Medical University start-up funds (Grant No. QD202113 to Panpan Huang), and National College Students Innovation Entrepreneurship Training Plan Program of China (S202410413004 to Yuhuan Xie).
Disclosure
The authors declare that they have no competing interests.
References
1. Xue W, Zhang Z, Yu H, et al. Development of nomogram and discussion of radiotherapy effect for osteosarcoma survival. Sci Rep. 2023;13(1):223. doi:10.1038/s41598-023-27476-9
2. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3–13.
3. Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;21 Suppl 7:vii320–5. doi:10.1093/annonc/mdq276
4. Yang J, Jiang H, Fu Q, et al. Blue light photobiomodulation induced apoptosis by increasing ROS level and regulating SOCS3 and PTEN/PI3K/AKT pathway in osteosarcoma cells. J Photochem Photobiol B. 2023;249:112814. doi:10.1016/j.jphotobiol.2023.112814
5. Ozaki T, Flege S, Kevric M, et al. Osteosarcoma of the pelvis: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol. 2003;21(2):334–341. doi:10.1200/JCO.2003.01.142
6. Oertel S, Blattmann C, Rieken S, et al. Radiotherapy in the treatment of primary osteosarcoma--a single center experience. Tumori. 2010;96(4):582–588. doi:10.1177/030089161009600411
7. Spałek MJ, Poleszczuk J, Czarnecka AM, et al. Radiotherapy in the management of pediatric and adult osteosarcomas: a multi-institutional cohort analysis. Cells. 2021;10(2):366. doi:10.3390/cells10020366
8. Dhillon S. Decitabine/cedazuridine: first approval. Drugs. 2020;80(13):1373–1378. doi:10.1007/s40265-020-01389-7
9. Shin DY, Sung Kang H, Kim G-Y, et al. Decitabine, a DNA methyltransferases inhibitor, induces cell cycle arrest at G2/M phase through p53-independent pathway in human cancer cells. Biomed Pharmacother. 2013;67(4):305–311. doi:10.1016/j.biopha.2013.01.004
10. Li Y, Geng P, Jiang W, et al. Enhancement of radiosensitivity by 5-Aza-CdR through activation of G2/M checkpoint response and apoptosis in osteosarcoma cells. Tumour Biol. 2014;35(5):4831–4839. doi:10.1007/s13277-014-1634-5
11. Santini V, Allione B, Zini G, et al. A Phase II, multicentre trial of decitabine in higher-risk chronic myelomonocytic leukemia. Leukemia. 2018;32(2):413–418. doi:10.1038/leu.2017.186
12. Aribi A, Borthakur G, Ravandi F, et al. Activity of decitabine, a hypomethylating agent, in chronic myelomonocytic leukemia. Cancer. 2007;109(4):713–717. doi:10.1002/cncr.22457
13. Wijermans PW, Rüter B, Baer MR, et al. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk Res. 2008;32(4):587–591. doi:10.1016/j.leukres.2007.08.004
14. Taib N, Merhi M, Inchakalody V, et al. Treatment with decitabine induces the expression of stemness markers, PD-L1 and NY-ESO-1 in colorectal cancer: potential for combined chemoimmunotherapy. J Transl Med. 2023;21(1):235. doi:10.1186/s12967-023-04073-y
15. Lund-Andersen C, Patzke S, Nähse-Kumpf V, et al. PLK1-inhibition can cause radiosensitization or radioresistance dependent on the treatment schedule. Radiother Oncol. 2014;110(2):355–361. doi:10.1016/j.radonc.2013.12.014
16. Liu X, Choy E, Harmon D, et al. Inhibition of polo-like kinase 1 leads to the suppression of osteosarcoma cell growth in vitro and in vivo. Anticancer Drugs. 2011;22(5):444–453. doi:10.1097/CAD.0b013e32834513f4
17. PosthumaDeBoer J, Würdinger T, Graat HC, et al. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer. 2011;11(1):156. doi:10.1186/1471-2407-11-156
18. Mamo T, Mladek AC, Shogren KL, et al. Inhibiting DNA-PK(CS) radiosensitizes human osteosarcoma cells. Biochem Biophys Res Commun. 2017;486(2):307–313. doi:10.1016/j.bbrc.2017.03.033
19. Li HK, Matsumoto Y, Furusawa Y, et al. PU-H71, a novel Hsp90 inhibitor, as a potential cancer-specific sensitizer to carbon-ion beam therapy. J Radiat Res. 2016;57(5):572–575. doi:10.1093/jrr/rrw054
20. Qu W, Wang Y, Wu Q, et al. Emodin impairs radioresistance of human osteosarcoma cells by suppressing sonic hedgehog signaling. Med Sci Monit. 2017;23:5767–5773. doi:10.12659/MSM.907453
21. Russo M, Spagnuolo C, Moccia S, et al. Biochemical and cellular characterization of new radio-resistant cell lines reveals a role of natural flavonoids to bypass senescence. Int J Mol Sci. 2021;23(1):301. doi:10.3390/ijms23010301
22. Brassesco MS, Pezuk JA, de Oliveira JC, et al. Activator protein-1 inhibition by 3-[(dodecylthiocarbonyl)methyl]-glutamaride impairs invasion and radiosensitizes osteosarcoma cells in vitro. Cancer Biother Radiopharm. 2013;28(4):351–358. doi:10.1089/cbr.2012.1305
23. von Fallois M, Kosyna FK, Mandl M, et al. Selinexor decreases HIF-1α via inhibition of CRM1 in human osteosarcoma and hepatoma cells associated with an increased radiosensitivity. J Cancer Res Clin Oncol. 2021;147(7):2025–2033. doi:10.1007/s00432-021-03626-2
24. Jin Z, Aixi Y, Baiwen Q, et al. Inhibition of hypoxia-inducible factor-1 alpha radiosensitized MG-63 human osteosarcoma cells in vitro. Tumori. 2015;101(5):578–584. doi:10.5301/tj.5000243
25. Mantovani FB, Morrison JA, Mutsaers AJ. Effects of epidermal growth factor receptor kinase inhibition on radiation response in canine osteosarcoma cells. BMC Vet Res. 2016;12:82. doi:10.1186/s12917-016-0707-7
26. Zuch D, Giang A-H, Shapovalov Y, et al. Targeting radioresistant osteosarcoma cells with parthenolide. J Cell Biochem. 2012;113(4):1282–1291. doi:10.1002/jcb.24002
27. Duckett MM, Phung SK, Nguyen L, et al. The adrenergic receptor antagonists propranolol and carvedilol decrease bone sarcoma cell viability and sustained carvedilol reduces clonogenic survival and increases radiosensitivity in canine osteosarcoma cells. Vet Comp Oncol. 2020;18(1):128–140. doi:10.1111/vco.12560
28. Loeb DM, Garrett‐Mayer E, Hobbs RF, et al. Dose-finding study of 153 Sm-EDTMP in patients with poor-prognosis osteosarcoma. Cancer. 2009;115(11):2514–2522. doi:10.1002/cncr.24286
29. Russo M, Moccia S, Spagnuolo C, et al. Carotenoid-enriched nanoemulsions and γ-rays synergistically induce cell death in a novel radioresistant osteosarcoma cell line. Int J Mol Sci. 2022;23(24):15959. doi:10.3390/ijms232415959
30. Ferraiuolo M, Pulito C, Finch-Edmondson M, et al. Agave negatively regulates YAP and TAZ transcriptionally and post-translationally in osteosarcoma cell lines. Cancer Lett. 2018;433:18–32. doi:10.1016/j.canlet.2018.06.021
31. Gola C, Licenziato L, Accornero P, et al. The mitotic regulator polo-like kinase 1 as a potential therapeutic target for c-Myc-overexpressing canine osteosarcomas. Vet Comp Oncol. 2022;20(4):890–900. doi:10.1111/vco.12854
32. Sebastian M, Reck M, Waller CF, et al. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. J Thorac Oncol. 2010;5(7):1060–1067. doi:10.1097/JTO.0b013e3181d95dd4
33. Mross K, Frost A, Steinbild S, et al. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2008;26(34):5511–5517. doi:10.1200/JCO.2008.16.1547
34. Wang Y, Li J, Booher RN, et al. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61(22):8211–8217.
35. Beck H, Nähse-Kumpf V, Larsen MSY, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32(20):4226–4236. doi:10.1128/MCB.00412-12
36. Hirai H, Iwasawa Y, Okada M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8(11):2992–3000. doi:10.1158/1535-7163.MCT-09-0463
37. Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257(5078):1955–1957. doi:10.1126/science.1384126
38. Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14(9):1878–1891. doi:10.1002/j.1460-2075.1995.tb07180.x
39. Zhou Z, Zhong L, Chu X, et al. HDAC11 mediates the ubiquitin-dependent degradation of p53 and inhibits the anti-leukemia effect of PD0166285. Med Oncol. 2023;40(11):325. doi:10.1007/s12032-023-02196-2
40. Hashimoto O, Shinkawa M, Torimura T, et al. Cell cycle regulation by the Wee1 inhibitor PD0166285, pyrido [2,3-d] pyimidine, in the B16 mouse melanoma cell line. BMC Cancer. 2006;6(1):292. doi:10.1186/1471-2407-6-292
41. Hashimoto O, Ueno T, Kimura R, et al. Inhibition of proteasome-dependent degradation of Wee1 in G 2 -arrested Hep3B cells by TGFβ1. Mol Carcinog. 2003;36(4):171–182. doi:10.1002/mc.10111
42. Munck JM, Batey MA, Zhao Y, et al. Chemosensitization of cancer cells by KU-0060648, a dual inhibitor of DNA-PK and PI-3K. Mol Cancer Ther. 2012;11(8):1789–1798. doi:10.1158/1535-7163.MCT-11-0535
43. Hinrichsen I, Ackermann A, Düding T, et al. Loss of MLH1 sensitizes colon cancer cells to DNA-PKcs inhibitor KU60648. Mol Carcinog. 2017;56(7):1816–1824. doi:10.1002/mc.22640
44. Jhaveri K, Taldone T, Modi S, et al. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–755. doi:10.1016/j.bbamcr.2011.10.008
45. Phase I study of the Hsp90 inhibitor, PU-H71, in patients with refractory solid tumors and low-grade non-hodgkin’s lymphoma. 2012.
46. Speranza G, Anderson L, Chen AP, et al. First-in-human study of the epichaperome inhibitor PU-H71: clinical results and metabolic profile. Invest New Drugs. 2018;36(2):230–239. doi:10.1007/s10637-017-0495-3
47. Ferry DR, Smith A, Malkhandi J, et al. Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res. 1996;2(4):659–668.
48. Kirsh VA, Peters U, Mayne ST, et al. Prospective study of fruit and vegetable intake and risk of prostate cancer. J Natl Cancer Inst. 2007;99(15):1200–1209. doi:10.1093/jnci/djm065
49. Zhou W, Kallifatidis G, Baumann B, et al. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int J Oncol. 2010;37(3):551–561. doi:10.3892/ijo_00000704
50. Roberto GM, Paiva HH, Botelho de Souza LE, et al. DTCM-glutarimide delays growth and radiosensitizes glioblastoma. Anticancer Agents Med Chem. 2018;18(9):1323–1329. doi:10.2174/1871520618666180423105740
51. Neggers JE, Vercruysse T, Jacquemyn M, et al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol. 2015;22(1):107–116. doi:10.1016/j.chembiol.2014.11.015
52. Lantto TA, Laakso I, Dorman H, et al. Cellular stress and p53-associated apoptosis by Juniperus communis L. Berry extract treatment in the human SH-SY5Y neuroblastoma cells. Int J Mol Sci. 2016;17(7):1113. doi:10.3390/ijms17071113
53. Rani V, Prabhu A. Combining angiogenesis inhibitors with radiation: advances and challenges in cancer treatment. Curr Pharm Des. 2021;27(7):919–931. doi:10.2174/1381612826666201002145454
54. Abdul Razak AR, Mau-Soerensen M, Gabrail NY, et al. First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol. 2016;34(34):4142–4150. doi:10.1200/JCO.2015.65.3949
55. Kung AL, Zabludoff SD, France DS, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6(1):33–43. doi:10.1016/j.ccr.2004.06.009
56. Farren MR, Hennessey RC, Shakya R, et al. The exportin-1 inhibitor selinexor exerts superior antitumor activity when combined with t-cell checkpoint inhibitors. Mol Cancer Ther. 2017;16(3):417–427. doi:10.1158/1535-7163.MCT-16-0498
57. Staab A, Loeffler J, Said HM, et al. Effects of HIF-1 inhibition by chetomin on hypoxia-related transcription and radiosensitivity in HT 1080 human fibrosarcoma cells. BMC Cancer. 2007;7:213. doi:10.1186/1471-2407-7-213
58. Cohen MH, Johnson JR, Chen Y-F, et al. FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist. 2005;10(7):461–466. doi:10.1634/theoncologist.10-7-461
59. Gold KA, Kies MS, William WN, et al. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: a single-arm Phase 2 clinical trial. Cancer. 2018;124(10):2169–2173. doi:10.1002/cncr.31346
60. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised Phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi:10.1016/S1470-2045(11)70393-X
61. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi:10.1016/j.immuni.2014.06.010
62. Hashimoto K, Nishimura S, Shinyashiki Y, et al. Characterizing inflammatory markers in highly aggressive soft tissue sarcomas. Medicine. 2022;101(39):e30688. doi:10.1097/MD.0000000000030688
63. Amorim MH, Gil da Costa RM, Lopes C, et al. Sesquiterpene lactones: adverse health effects and toxicity mechanisms. Crit Rev Toxicol. 2013;43(7):559–579. doi:10.3109/10408444.2013.813905
64. Lavine JA, Farnoodian M, Wang S, et al. β2-adrenergic receptor antagonism attenuates CNV through inhibition of VEGF and IL-6 expression. Invest Ophthalmol Vis Sci. 2017;58(1):299–308. doi:10.1167/iovs.16-20204
65. Li B, Liao Y-H, Cheng X, et al. Effects of carvedilol on cardiac cytokines expression and remodeling in rat with acute myocardial infarction. Int J Cardiol. 2006;111(2):247–255. doi:10.1016/j.ijcard.2005.08.065
66. Amaya CN, Perkins M, Belmont A, et al. Non-selective beta blockers inhibit angiosarcoma cell viability and increase progression free- and overall-survival in patients diagnosed with metastatic angiosarcoma. Oncoscience. 2018;5(3–4):109–119. doi:10.18632/oncoscience.413
67. Montoya A, Amaya CN, Belmont A, et al. Use of non-selective β-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget. 2017;8(4):6446–6460. doi:10.18632/oncotarget.14119
68. Pasquier E, Street J, Pouchy C, et al. β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br J Cancer. 2013;108(12):2485–2494. doi:10.1038/bjc.2013.205
69. Chaudhary KR, Yan SX, Heilbroner SP, et al. Effects of β-adrenergic antagonists on chemoradiation therapy for locally advanced non-small cell lung cancer. J Clin Med. 2019;8(5):575. doi:10.3390/jcm8050575
70. Bustamante P, Miyamoto D, Goyeneche A, et al. Beta-blockers exert potent anti-tumor effects in cutaneous and uveal melanoma. Cancer Med. 2019;8(17):7265–7277. doi:10.1002/cam4.2594
71. Thiele M, Wiest R, Gluud LL, et al. Can non-selective beta-blockers prevent hepatocellular carcinoma in patients with cirrhosis? Med Hypotheses. 2013;81(5):871–874. doi:10.1016/j.mehy.2013.08.026
72. Pandey V, Tiwari P, Imran M, et al. Adverse drug reactions following propranolol in infantile hemangioma. Indian Pediatr. 2021;58(8):753–755. doi:10.1007/s13312-021-2286-3
73. Tatu AL, Elisei AM, Chioncel V, et al. Immunologic adverse reactions of β-blockers and the skin. Exp Ther Med. 2019;18(2):955–959. doi:10.3892/etm.2019.7504
74. Goeckeler WF, Edwards B, Volkert WA, et al. Skeletal localization of samarium-153 chelates: potential therapeutic bone agents. J Nucl Med. 1987;28(4):495–504.
75. Farhanghi M, Holmes RA, Volkert WA, et al. Samarium-153-EDTMP: pharmacokinetic, toxicity and pain response using an escalating dose schedule in treatment of metastatic bone cancer. J Nucl Med. 1992;33(8):1451–1458.
76. Correa-González L, Arteaga de Murphy C, Pichardo-Romero P, et al. (153)Sm-EDTMP for pain relief of bone metastases from prostate and breast cancer and other malignancies. Arch Med Res. 2014;45(4):301–308. doi:10.1016/j.arcmed.2014.03.006
77. Russo GL, Moccia S, Russo M, et al. Redox regulation by carotenoids: evidence and conflicts for their application in cancer. Biochem Pharmacol. 2021;194:114838. doi:10.1016/j.bcp.2021.114838
78. Ho XD, Nguyen HG, Trinh LH, et al. Analysis of the expression of repetitive DNA elements in osteosarcoma. Front Genet. 2017;8:193. doi:10.3389/fgene.2017.00193
79. Reimann E, Kõks S, Ho XD, et al. Whole exome sequencing of a single osteosarcoma case--integrative analysis with whole transcriptome RNA-seq data. Hum Genomics. 2014;8(1):20. doi:10.1186/s40246-014-0020-0
80. Ho XD, Phung P, Q Le V, et al. Whole transcriptome analysis identifies differentially regulated networks between osteosarcoma and normal bone samples. Exp Biol Med. 2017;242(18):1802–1811. doi:10.1177/1535370217736512
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.