")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Unveiling the Novel Anti - Tumor Potential of Digitonin, a Steroidal Saponin, in Gastric Cancer: A Network Pharmacology and Experimental Validation Study
Authors Lu D, Huang L , Weng C
Received 4 December 2024
Accepted for publication 15 March 2025
Published 5 April 2025 Volume 2025:19 Pages 2653—2666
DOI https://doi.org/10.2147/DDDT.S504671
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Tamer Ibrahim
Dongdong Lu,1,2 Leijie Huang,1 Chunyan Weng2
1Department of Gastroenterology, Ningbo No. 2 Hospital, Ningbo, Zhejiang Province, 315000, People’s Republic of China; 2Department of Gastroenterology, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Hangzhou, Zhejiang Province, 310000, People’s Republic of China
Correspondence: Chunyan Weng, Email [email protected]
Background: Gastric cancer (GC) remains a leading cause of cancer-related mortality, with limited effective treatment options for advanced stages. As a steroidal saponin with documented anti-neoplastic properties in multiple cancers, digitonin’s mode of action in GC pathogenesis has yet to be fully elucidated. This research focused on exploring the potential of Digitonin in GC treatment using a combination of network pharmacology and experimental validation.
Methods: The inhibitory effects of Digitonin on the proliferation, invasion, and migration of gastric cancer cells were evaluated using CCK-8, colony formation, wound healing, and transwell assays. Key targets of Digitonin were identified through network pharmacology. Molecular docking and various experiments, including Western blot, immunofluorescence, and a subcutaneous xenograft model, were used for validation.
Results: Digitonin exhibited stronger cytotoxicity against GC cells and significantly inhibited GC cell proliferation, migration, and invasion. Network pharmacology analysis revealed that the core targets of Digitonin are involved in key cancer-related signaling pathways, including HIF-1α, Ras, and PI3K-Akt pathways, with HSP90AA1 and NFKB1 identified as central targets. Further molecular docking, Western blotting, and immunofluorescence experiments confirmed that Digitonin significantly suppressed the expression of HSP90AA1 and inhibited the nuclear translocation of NFKB1, inducing cell apoptosis. Additionally, a subcutaneous xenograft model of GC further validated that Digitonin effectively inhibited tumor growth.
Conclusion: Digitonin serves as a promising multi-target therapeutic agent for GC. This study underscores the potential of combining network pharmacology with traditional Chinese medicine to identify novel therapeutic targets and develop effective anti-cancer strategies. In addition, these findings suggest that digitonin could be a promising candidate for future clinical trials in GC treatment.
Keywords: gastric cancer, digitonin, network pharmacology, HSP90AA1, NFKB1
Introduction
As reported in the 2022 Global Cancer Statistics, gastric cancer (GC) is the 5th globally in both incidence and mortality.1 Due to its often late-stage diagnosis, the 5-year survival rate remains low.2 Despite recent advancements in treatment modalities, the prognosis for patients with advanced GC remains poor.3 Among them, targeted therapies have emerged as a promising approach to improve outcomes in GC by inhibiting specific molecular pathways involved in tumor growth and progression.4,5
As a commonly utilized branch of integrative medicine, traditional Chinese medicine (TCM) is appreciated by cancer patients for its therapeutic benefits and minimal severe side effects. Saponins, a diverse group of natural compounds found in various medicinal herbs, have attracted attention for their broad bioactivities, including anticancer properties.6–9 Digitonin, a steroidal saponin derived from the Digitalis genus, has shown potential in cancer therapy by inducing apoptosis, disrupting cell cycle progression, and enhancing the uptake of chemotherapeutic agents in various cancers such as liver, breast, lung, and colorectal cancers.9,10 Its ability to modulate multiple signaling pathways and sensitize cancer cells to traditional treatments highlights its potential as a therapeutic agent.11,12 Notably, recent mechanistic studies by Yang et al demonstrated digitonin-mediated suppression of KRAS/BRAF-mutant colorectal cancer progression through selective targeting of CD82, a tetraspanin family protein implicated in metastasis.13 Furthermore, digitonin reverses multidrug resistance via competitive inhibition of P-glycoprotein (P-gp)-mediated drug efflux, as evidenced by enhanced intracellular doxorubicin retention in resistant cell models.14–16 However, the deeper and more detailed mechanisms of Digitonin in the treatment of GC remain largely unknown. Further research is needed to elucidate its molecular mechanisms and evaluate its clinical applicability, paving the way for the integration of traditional compounds into modern cancer therapy.
Network pharmacology plays a unique role in cancer therapy by systematically uncovering the complex interactions between drugs, targets, and pathways. It has been instrumental in identifying multi-target mechanisms of traditional Chinese medicines, such as revealing how Huangqin Tang exerts anti-cancer effects through the STAT3, AKT1, MAPK3 pathways.17 Similarly, it has highlighted the potential of small molecules like berberine to modulate multiple cancer-related targets, addressing challenges like drug resistance and tumor heterogeneity.18 Additionally, network pharmacology has facilitated the repurposing of existing drugs, such as metformin.19 These applications underscore the importance of network pharmacology in developing more effective, multi-target therapeutic strategies for cancer.
In this study, we first evaluated the cytotoxicity, proliferation, migration, and invasion of Digitonin in GC cells. Network pharmacology and molecular docking were then used to identify the potential targets of Digitonin, followed by cell-based experiments and a subcutaneous xenograft model. The results provide a foundation for developing Digitonin as a multi-target anti-cancer agent.
Methods
Cells and Animal
The cell lines MKN1, NUGC3, HGC27, and GES1 were purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai) and maintained in RPMI-1640 medium with 10% fetal bovine serum (FBS, Gibco, USA). Male BALB/c nude mice (4 weeks old) were obtained from BK Company (Shanghai, China) and housed under specific pathogen-free conditions (25°C with 55% humidity and 12/12 h of light/dark cycle). All animal studies were conducted following institutional guidelines and received approval from the Animal Care and Use Committee of Zhejiang Chinese Medical University, adhering to the ethical guidelines outlined in the Guide for the Care and Use of Laboratory Animals.
Cell Proliferation Assay (CCK-8 Assay)
CCK-8 assay was performed (Beyotime, China) according to the protocol. 5000 MKN1, NUGC3, HGC27, and GES-1 cells were plated in 96-well plates and then treated with Digitonin (0, 0.3125, 0.625, 1.25, 2.5, 5, 10, and 20 μM) for 24, 48, and 72 hours. At the specified time point, the medium in each well was replaced with fresh medium containing 10% CCK-8 solution and incubated until the absorbance of the control cells at 450 nm reached a range of 0.8 to 1.2. GraphPad Prism (version 9.0) was used to determine the IC50.
Wound Healing
MKN1 and NUGC3 cells were plated in each chamber of wound scratch assay kit (Culture-Insert, Ibidi, Germany) and cultured until reaching 90–100% confluence. The inserts were then carefully removed, and the cells were treated with serum-free medium containing Digitonin at concentrations of 0, 0.5, and 1 μM. Wound area pictures recorded at 0 and 36 hours and calculated by migrated distance.
Colony Formation, Transwell Migration and Invasion Assays
For the colony formation assay, 1000 cells were seeded in 35 mm dishes and exposed to digitonin (0, 0.5, and 1 μM) for 14 days. For transwell assays, 5 × 10⁴ cells suspended in serum-free medium with Digitonin (0, 0.5, and 1 μM) were placed in the upper chamber, either pre-coated with Matrigel for invasion assays or left uncoated for migration assays. The lower chamber contained complete medium with 10% FBS. 48 hours later, cells on the upper surface of the membrane were removed. All cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The data were counted using ImageJ software (version 1.52a, USA).
Network Pharmacology Analysis
Potential GC-related targets were identified from three databases: GeneCards, OMIM and DisGeNET. From GeneCards, the top 2000 targets were selected based on relevance scores. Digitonin-associated targets were retrieved from the TCMSID database, which integrates data from multiple sources including SwissTargetPrediction, SEA, ChEMBL, HitPickV2, Polypharmacology Browser, and Polypharmacology Browser 2. The keywords used included “GC”, “stomach neoplasms”, “digitonin”, and “steroidal saponin”. The intersection of GC-related and Digitonin-related targets, visualized using a Venn diagram. These common targets were analyzed for KEGG pathway and GO enrichment analysis using the DAVID tool. Further comprehensive pathway analysis was performed using Metascape. All data were analyzed and visualized using appropriate bioinformatics tools (https://www.bioinformatics.com.cn/).
Identification of Core Targets Using Cytoscape
The intersecting genes between Digitonin-associated targets and GC-related targets were analyzed using Cytoscape software (version 3.8.0). Core targets were identified based on the centrality measures: degree, betweenness, and closeness. The median values for each parameter across all intersecting genes were calculated (degree > 5.18, betweenness > 22.73, and closeness > 0.024), and genes with values above these thresholds were considered core targets. The selected core genes were visualized using Cytoscape, with node size representing degree centrality.
Expression and Survival Analysis of Core Targets Using TIMER2.0 and Kaplan-Meier Plotter Database
The TIMER2.0 database was used to assess the expression patterns of ESR1, HSP90AA1, and NFKB1 across various cancer types. The gene expression profiles in both tumor and normal tissues were analyzed across different cancer types. The prognostic value of HSP90AA1 and NFKB1 in GC was assessed using the Kaplan-Meier Plotter database. Patients were divided into high and low expression groups based on Auto select best cutoff. Statistical significance was defined as a p-value less than 0.05.
Molecular Docking of Digitonin with HSP90AA1 and NFKB1
The 3D conformations of HSP90AA1 (ID: 5H22) and NFKB1 (ID: 1U36) were retrieved from the Protein Data Bank. The model of Digitonin, Alvespimycin, and Tryptanthrine were obtained from the PubChem database and regenerated by MOE (version 2022). Docking simulations were carried out with AutoDock Vina (version 1.5.7). Preparation of both proteins and ligands involved adding hydrogen atoms, removing water molecules, and calculating Gasteiger charges. The grid box was positioned to encompass each protein’s domain and allow for free molecular movement. It was set to a size of 30 Å × 30 Å × 30 Å, with a grid point distance of 0.05 nm. Docking results were evaluated based on the binding affinity. The top-ranked binding poses were visualized using PyMOL software (2.3.3 version) to analyze the interaction sites and hydrogen bonding patterns between Digitonin and the amino acid residues of the proteins.
Western Blot, Nuclear/Cytoplasmic Fractionation, and Immunofluorescence Staining
Proteins are extracted using RIPA lysis buffer, separating by SDS-PAGE and transferring to PVDF membranes. They were then incubated with specific primary antibodies, HSP90AA1 (13171-1-AP, Proteintech), NFKB1 (p50, 14220-1-AP, Proteintech), and GAPDH (60004-1-Ig, Proteintech), followed by incubation with the appropriate secondary antibodies. Final results were visualized using an ECL system and quantified with ImageJ software, normalized to GAPDH. Nuclear and cytoplasmic proteins were isolated using a Nuclear and Cytoplasmic Extraction Kit (Beyotime, China) following Digitonin treatment. Protein levels of NFKB1 (p50) in both fractions were assessed by Western blot and quantified with ImageJ, using vinculin (26520-1-AP, Proteintech) and H3 (17168-1-AP, Proteintech) as controls for cytoplasmic and nuclear fractions, respectively. For immunofluorescence, cells treated with Digitonin for 24 hours were fixed, permeabilized, and blocked, following the incubation with primary antibody and Alexa Fluor-conjugated secondary antibodies. Nuclei were counterstained with DAPI.
Apoptosis Detection by Flow Cytometry
After Digitonin treatment, the apoptosis of cells was detected by Apoptosis Detection Kit (Beyotime, China) following the protocol. Apoptotic cells were measured by flow cytometry (NovoCyte, USA) and processed with FlowJo software.
Establishment of Subcutaneous GC Xenograft Model
Male, 4-week-old BALB/c nude mice were sourced from the Experimental Animal Department of Zhejiang Chinese Medical University and housed under specific pathogen-free conditions. The room temperature was maintained at 22 ± 2°C, with a relative humidity of 50 ± 10%, and a 12-hour light/dark cycle. 5 × 10⁶ MKN1 cells were injected into the lateral side of each mouse. Tumor volume was determined weekly by using the formula (Volume = long diameter × short diameter²/2). 10 days later, mice were randomly assigned to different treatment groups and received Digitonin, 5-FU, or PBS control via intraperitoneal injection every two days for 4 weeks. The body weight of the mice was monitored. 4 weeks later, mice were humanely sacrificed, and tumors were collected for further analysis.
Measurement of Serum ALT and AST Levels in Mice
Blood samples of mice were collected via the orbital sinus technique. After allowing the whole blood to clot at room temperature for 1 hour, centrifugation was performed at 3000 ×g for 5 min. The serum fraction was collected and stored at −20°C until analysis. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured using commercial assay kits (Aspartate Aminotransferase Assay Kit, C009-2-1, Nanjing Jiancheng Bioengineering Institute; Alanine Aminotransferase Assay Kit, C010-2-1, Nanjing Jiancheng Bioengineering Institute) according to the manufacturer’s instructions.
Ethics Statement
The human public data were approved by the Biomedical Ethics Committee of Ningbo No.2 hospital (no. PJ-NBEY-KY-2025-002-01).
Statistical Analysis
All data are presented as the mean ± SD, with n = 5 for cell experiments and n = 6 for animal experiments. All datasets underwent Shapiro–Wilk normality testing, confirming parametric assumptions. Statistical comparisons among multiple groups were conducted using one-way analysis of variance (ANOVA) followed by LSD or Tamhane’s T2 tests, while two group t-test. Differences were considered statistically significant at p<0.05. * means p<0.05, ** means p<0.01, and *** means p<0.001. Statistical analyses were performed using SPSS software (version 25).
Results
Anticancer Effects of Digitonin on GC
Three GC cell lines (MKN1, HGC27, and NUGC3) and a human gastric epithelial cell line (GES-1) were treated with Digitonin (0–20 μM) for 24, 48, and 72 hours. The study revealed that Digitonin significantly curtailed the proliferation of GC cells, with mean IC50 values recorded at 3.875 μM (24h), 2.004 μM (48h), and 1.185 μM (72h) (Figure 1A–C). Conversely, the effect on GES-1 was less pronounced, with IC50 values of 15.26, 13.54, and 11.13 μM at the corresponding time points (Figure 1D). These outcomes indicate that Digitonin may selectively eradicate GC cells without harming normal gastric mucosa, highlighting its therapeutic promise. The anti-tumor efficacy of digitonin was further corroborated by clonogenic assays, which revealed inhibition rates of 31.0–35.5% in MKN1 and 32.5–46.5% in NUGC3 cells (Figure 1E and F). Wound healing assays (Figure 1G and H) and Transwell migration/invasion analyses demonstrated dose-dependent suppression of GC cell motility, with migration inhibition rates ranging from 34.2–67.0% (MKN1) and 31.0–71.9% (NUGC3), while invasion suppression rates reached 29.0–59.2% (MKN1) and 33.2–59.4% (NUGC3) (Figure 1I and J).
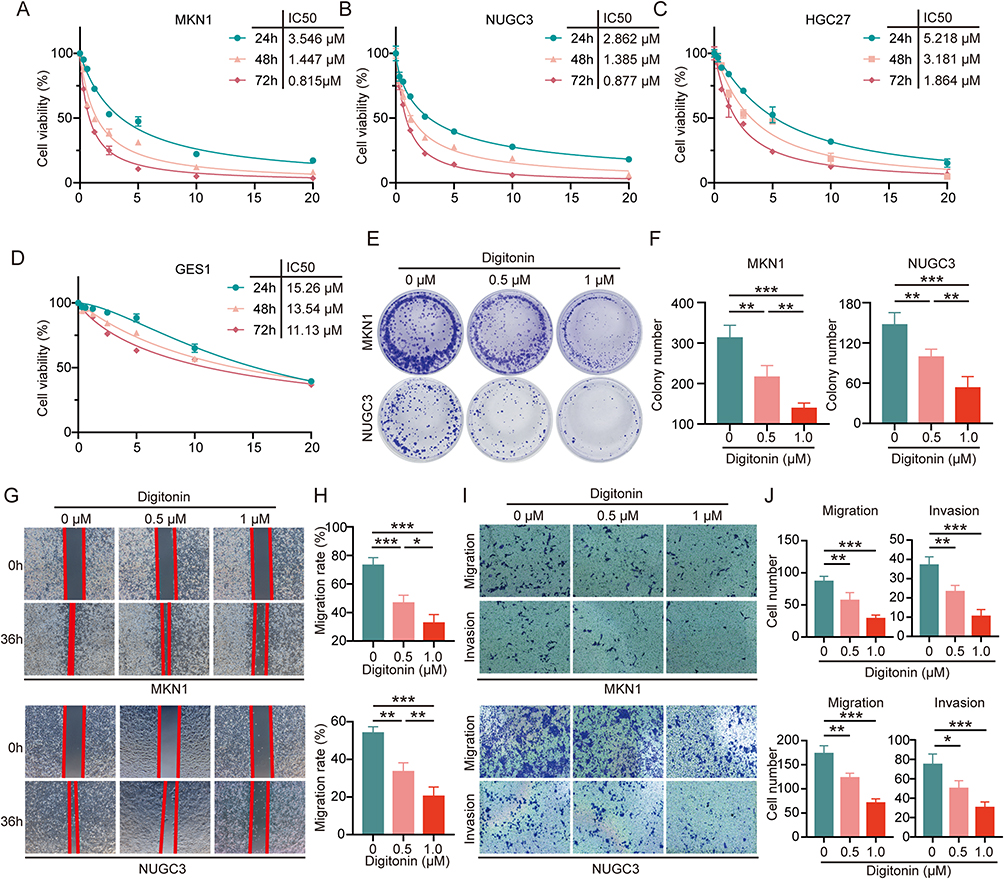 |
Figure 1 Digitonin inhibits the proliferation, migration, and invasion of GC cells in vitro. (A–D) CCK-8 assays were conducted on MKN1, NUGC3, HGC27, and GES-1 cell lines following treatment with varying concentrations of Digitonin (0–20 μM) for 24, 48, and 72 hours. (E and F) Clonogenic assays were performed on MKN1 and NUGC3 cell lines treated with Digitonin at concentrations of 0, 0.5 μM, and 1 μM, followed by statistical analysis of colony formation. (G and H) Wound healing assays were carried out on MKN1 and NUGC3 cell lines treated with Digitonin at 0, 0.5 μM, and 1 μM, with subsequent statistical evaluation of cell migration. (I and J) Transwell migration and invasion assays were conducted on MKN1 and NUGC3 cell lines treated with Digitonin at 0, 0.5 μM, and 1 μM, and statistical analyses were performed to assess cell motility and invasiveness. Data are presented as the mean ± SD (n=5) of at five independent experiments. * means p<0.05, ** means p<0.01, *** means p<0.001. |
Network Pharmacology Analysis Identifies Key Therapeutic Targets of Digitonin in GC
To identify key therapeutic targets of Digitonin in GC, we performed network pharmacology analysis. Potential GC targets were identified from the GeneCards (top 2000 targets), OMIM (56 targets), and DisGeNET (367 targets) databases, yielding a total of 2208 unique targets (Figure 2A). Digitonin-associated targets (58 targets) were obtained from the TCMSID database. Intersection analysis revealed 25 common genes (Figure 2B). KEGG pathway enrichment using the DAVID database showed significant involvement in cancer-related pathways, including HIF-1 signaling, Ras signaling, PI3K-Akt signaling, and MAPK signaling pathways (Figure 2C). Visualization of these 25 genes and the top 10 enriched KEGG pathways indicated that the “Pathway in cancer” contained the most genes (8 genes), and VEGFA was linked to the most pathways (9 pathways) (Figure 2D). Subsequent GO analysis revealed that these intersecting targets are involved in biological processes about tumor progression, such as regulation of transcription by RNA polymerase, regulation of cell population proliferation, and regulation of apoptotic processes (Figure 2E). Additionally, Metascape pathway analysis revealed that those were interacted to tumor-related processes, including morphogenesis of epithelium, photodynamic therapy-induced NF-kB survival signaling, and regulation of intracellular protein transport (Figure 2F).
 |
Figure 2 Network Pharmacology Analysis of Potential Targets of Digitonin in GC. (A) Venn diagram showing potential GC targets obtained from the GeneCards (top 2000), OMIM (56), and DisGeNET (360) databases. (B) Venn diagram illustrating the overlap between predicted GC targets and Digitonin targets retrieved from the TCMSID database. (C) KEGG pathway enrichment analysis of intersecting genes using the DAVID database. (D) Visualization of the association between intersecting targets and the top 5 KEGG pathways based on gene ratio. (E) GO term enrichment analysis of intersecting genes using the DAVID database. (F) Top 10 enriched pathways of the intersecting genes identified through Metascape analysis. |
Core Targets of Digitonin in GC Predicting by Cytoscape Network Analysis
To identify the core targets, further analysis of the intersecting genes were performed using Cytoscape software. Key genes were selected based on degree, betweenness, and closeness centrality measures, with thresholds set above the median values for each parameter (5.18, 22.73, and 0.024, respectively) (Figure 3A). This analysis revealed that ESR1, PTPN1, FGF2, IL2, ABCG2, HSP90AA1, CDK1, and NFKB1 are the core genes among the intersecting targets (Figure 3B). A subsequent visualization of these core targets, ranked by degree centrality, showed that HSP90AA1, ESR1, and NFKB1 had significantly higher degree values compared to the other targets (Figure 3C). These core targets were further subjected to enrichment analysis using the DAVID database. KEGG pathway analysis indicated significant enrichment in tumor-related pathways such as inflammatory bowel disease, chemical carcinogenesis-receptor activation, and PI3K-Akt signaling pathway (Figure 3D). GO enrichment analysis revealed that pathways related to transcription by RNA polymerase, regulation of apoptotic process, and regulation of DNA-templated transcription were also significantly enriched (Figure 3E).
 |
Figure 3 Identification of Core Targets Among Potential Digitonin Targets in GC. (A and B) Core targets were identified from the intersecting genes of Digitonin and GC using Cytoscape, based on median values of degree, betweenness, and closeness centrality measures. (C) Visualization of core genes according to degree centrality, with larger circles representing higher degree values. (D and E) KEGG pathway and GO enrichment analyses of the core genes were conducted using the DAVID database. |
Validation of Core Targets and Molecular Docking Analysis of Digitonin in GC
To validate the potential role of these core targets, particularly the three with high degree centrality, in Digitonin’s therapeutic effect against GC, we first analyzed their expression profiles using the TIMER2.0 public database. ESR1 expression did not increase in GC tissues compared to normal tissues, while HSP90AA1 and NFKB1 were significantly overexpressed in tumor tissues (Figure 4A). Further survival analysis using the Kaplan-Meier plotter showed that higher HSP90AA1 expression correlated with poorer prognosis (Figure 4B), whereas NFKB1 showed the opposite trend (Figure 4C). Additionally, molecular docking simulations revealed robust interactions between Digitonin and the target proteins HSP90AA1 and NFKB1, with binding energies of −7.593 and −6.92 kcal/mol, respectively (Figure 4D and E). Detailed analysis revealed that Digitonin forms four hydrogen bonds with three amino acid residues of HSP90AA1 (Figure 4F), and five hydrogen bonds with three amino acid residues of NFKB1 (Figure 4G), suggesting potential mechanisms through which Digitonin may exert its therapeutic effects on GC. Additionally, we performed molecular docking simulations with known HSP90 inhibitor (Alvespimycin) and NFκB inhibitor (Tryptanthrine). The results showed binding energies of −5.92 and −5.8 kcal/mol, respectively, which were not better than that of Digitonin (Figure 4H and I), supporting the potential of HSP90 and NFKB1 as targets for Digitonin.
 |
Figure 4 Expression Profiles, Prognostic Significance, and Molecular Docking Analysis of Digitonin with Core Targets in GC. (A) Expression levels of ESR1, HSP90AA1, and NFKB1 in tumor and adjacent normal tissues across various cancer types, as analyzed using the TIMER2.0 database. (B and C) Kaplan-Meier survival curves showing the correlation between high expression of HSP90AA1 and NFKB1 and overall survival in GC patients, generated using the Kaplan-Meier plotter. (D and E) Molecular docking models illustrating the binding interactions of Digitonin with HSP90AA1 and NFKB1. (F and G) Detailed views of the binding sites and hydrogen bond interactions between Digitonin and the amino acid residues of HSP90AA1 and NFKB1, including hydrogen bond lengths. (H and I) Molecular docking results of Alvespimycin with HSP90 and Tryptanthrine with NFκB, including specific binding hydrogen bonds. * means p<0.05, ** means p<0.01, *** means p<0.001. |
Digitonin Increased GC Cell Apoptosis by Modulating HSP90AA1 and Nuclear Translocation of NFKB1
HSP90AA1 and NFKB1 are considered oncogenes. Notably, the precursor form of NFKB1, p105, is proteolytically cleaved into p50, which dimerizes with another NF-κB subunit, p65 (RelA), to form the p50:p65 heterodimer, regulating the transcription of anti-apoptotic genes, thereby promoting oncogenic processes. Western blotting results showed that Digitonin significantly inhibited HSP90AA1 expression, but had no noticeable effect on the total levels of NFKB1 (Figure 5A and 5). Moreover, nuclear-cytoplasmic fractionation revealed a decrease in nuclear NFKB1 levels, with a concurrent increase in the cytoplasmic fraction (Figure 5C and D). Immunofluorescence analysis further confirmed that Digitonin treatment inhibited the nuclear translocation of NFKB1 (Figure 5E). Furthermore, Digitonin markedly induced apoptosis in GC cells (Figure 5F and G).
 |
Figure 5 Digitonin Induces Apoptosis in GC Cells by Modulating HSP90AA1 Expression and NFKB1 Nuclear Translocation. (A and B) Western blot analysis of HSP90AA1 and NFKB1 (p50) protein levels in MKN1 cells treated with Digitonin for 24 hours, followed by densitometric quantification. (C and D) Nuclear and cytoplasmic fractions were isolated from MKN1 cells treated with Digitonin for 24 hours. The expression of NFKB1 (p50) in the nuclear and cytoplasmic compartments was analyzed by Western blot and quantified. (E) Immunofluorescence staining of NFKB1 in MKN1 cells after 24 hours of Digitonin treatment, showing its subcellular localization. (F and G) Representative flow cytometry plots and quantitative analysis of apoptosis in MKN1 cells following 24 hours of Digitonin treatment. Data are presented as the mean ± SD (n=5) of at five independent experiments, “ns” means not significant, * means p<0.05, ** means p<0.01, *** means p<0.001. |
Digitonin Inhibits GC Growth In Vivo
To assess the therapeutic function of Digitonin in vivo, a subcutaneous xenograft model was established using MKN1 cells (Figure 6A). As expected, Digitonin markedly decreased tumor volume relative to the control group, with the high-dose Digitonin group demonstrating efficacy comparable to that of 5-FU (Figure 6B). Additionally, images and final tumor weights further confirmed the substantial anti-tumor effect of Digitonin, which was comparable to 5-FU (Figure 6C and D). Quantitative analysis revealed 40.0% and 73.8% tumor growth inhibition for low- and high-dose digitonin respectively, paralleling the 72.8% inhibition observed with 5-FU. Importantly, body weight remained stable in both the Digitonin-treated and control groups throughout the treatment period (Figure 6E). Furthermore, while ALT and AST levels were significantly elevated in the 5-FU treatment group, the Digitonin treatment group showed no significant change compared to the normal control group, indicating that Digitonin has lower drug toxicity compared to traditional chemotherapy (Figure 6F). Subsequent experiments confirmed that Digitonin treatment significantly downregulated HSP90AA1 expression, thereby inhibiting tumor growth in vivo (Figure 6G).
 |
Figure 6 Therapeutic Effects of Digitonin on MKN1 Subcutaneous Xenograft Model in Mice. (A) Schematic representation of the subcutaneous xenograft model using MKN1 cells and the treatment strategy. (B) Tumor volumes of subcutaneous xenografts in different treatment groups measured over time. (C and D) Representative images and final tumor weights of xenografts from each treatment group on the last day of treatment. (E) Body weights of mice in each treatment group, showing no significant difference, indicating low toxicity of Digitonin. (F) Serum ALT and AST levels were measured following Digitonin or 5-Fu administration to evaluate hepatorenal function. (G) Western blot showed HSP90AA1 expression levels in tumor tissues from control and Digitonin-treated groups. Data are presented as the mean ± SD (n=6) of at six independent experiments, “ns” means not significant, * means p<0.05, ** means p<0.01, *** means p<0.001. |
Discussion
Currently, the most established targeted therapies for GC include agents targeting HER2,20 VEGF,21 and PD-1/PD-L1 pathways.22 Antibody-drug conjugates (ADCs) represent a promising direction in targeted therapy. In a Phase 2 trial, Trastuzumab deruxtecan, targeting HER2, has shown significant efficacy in HER2-positive advanced GC.23 The DESTINY-Gastric01 trial demonstrated improved overall survival and response rates, leading to its approval for HER2-positive advanced GC.24 Beyond HER2, other targets like CLDN18.2 are being explored with ADCs like zolbetuximab.25 Despite these promising developments, the clinical application of ADCs in GC still faces challenges. One of the main issues is the heterogeneity of target expression within tumors, which can lead to variable efficacy and the development of resistance.2 Therefore, there is a pressing need for the newly therapeutics that are not only safe and effective but also exhibit low toxicity.
Current first-line therapies for advanced GC predominantly rely on platinum-based (eg, oxaliplatin) and fluoropyrimidine (eg, 5-FU) chemotherapies, often combined with taxanes or irinotecan.26 While these regimens demonstrate initial efficacy, >60% of patients develop multidrug resistance (MDR) within 12 months, frequently mediated by P-glycoprotein overexpression,27 alongside substantial toxicities including myelosuppression and peripheral neuropathy.28 Targeted therapies and immunotherapies remain limited by molecular stratification requirements and acquired resistance.29,30 In contrast, our findings demonstrate that Digitonin exhibits broad-spectrum anti-GC activity while maintaining minimal cytotoxicity in non-cancerous GES-1 cells. Furthermore, in vivo animal experiments confirm that Digitonin, compared to 5-FU, effectively inhibits tumor progression with lower host toxicity. This effect may be mediated through the regulation of multiple tumor targets involved in tumor promotion, and this polypharmacological action helps circumvent the limitations of single-target therapies. These attributes position Digitonin as a promising candidate for overcoming conventional therapy resistance while improving safety. However, its long-term impacts, including potential tumor recurrence post-treatment and delayed toxicity, warrant further investigation through chronic toxicity studies and relapse monitoring protocols.
This study investigates the anticancer effects of digitonin in GC cells, integrating network pharmacology and molecular docking approaches to delineate its regulatory effects on critical oncogenic targets, specifically HSP90AA1 and NFKB1. Heat shock proteins (HSPs) are highly conserved chaperones essential for proteostasis and stress adaptation, with emerging roles in malignant progression.31 Among these, HSP90AA1, a central node in the HSP90 chaperone system, has garnered significant attention as a therapeutic target due to its critical role in stabilizing oncogenic clients.32 Notably, recent studies identify extracellular HSP90AA1 (eHSP90α) as a secreted chaperone that facilitates cancer cell invasion through extracellular matrix remodeling and activation of matrix metalloproteinases.33 In addition, NFKB1 (p105/p50), a pivotal subunit of the NF-κB transcription factor family, exhibits context-dependent duality in tumorigenesis.34–37 NFKB1 homodimers primarily repress gene transcription, dampening inflammatory responses and abrogating anti-apoptotic signaling.36,38,39 In contrast, p50 heterodimers increase the expression of NFκB-regulated genes, promoting pro-inflammatory responses, anti-apoptotic pathways, and cellular proliferation in various cancers.35,40–42 This dual functionality makes NFKB1 a complex but valuable target in cancer therapy.43 This study demonstrates that Digitonin significantly inhibits the expression of HSP90AA1 and suppresses the nuclear translocation of NFKB1, thereby inducing apoptosis in tumor cells. These findings underscore the significance of targeting HSP90AA1 and NFKB1 pathways in the development of therapeutic strategies against GC, as previous studies have highlighted the involvement of these proteins in cancer progression and treatment resistance.33
The network pharmacology analysis conducted in this study reveals that the core targets of Digitonin are significantly involved in critical cancer-related pathways, including HIF-1 signaling, Ras signaling, and PI3K-Akt signaling pathways. These pathways are essential for cancer cell proliferation and survival, thus emphasizing the relevance of understanding how Digitonin modulates these networks. By elucidating the mechanisms through which Digitonin interacts with these pathways, we can identify potential biomarkers for prognosis and therapeutic targets for GC treatment. Future research may focus on the interplay between these signaling pathways to develop more effective combinatorial therapies, enhancing the efficacy of existing treatment modalities while minimizing adverse effects.
However, this study has some limitations. Firstly, network pharmacology relies on databases that may contain incomplete or inaccurate information, which can lead to erroneous results and the omission of important details. Although our in vitro and in vivo experiments confirmed that Digitonin affects GC growth by regulating HSP90AA1 and NFKB1, the absence of in vivo clinical validation limits our ability to fully assess the therapeutic potential of Digitonin in a physiological context, which is essential for translating preclinical findings into clinical practice. Therefore, future research should leverage the number of experimental repetitions advanced technologies, such as multi-omics data and clinical sample analysis, to further investigate the specific mechanisms of Digitonin in GC treatment and to establish a comprehensive understanding of its anticancer effects and clinical applicability.
Our experimental results further confirmed that Digitonin effectively downregulates HSP90AA1 expression and inhibits the nuclear translocation of NFKB1, resulting in increased apoptosis in GC cells. These findings highlight the potential of using network pharmacology in combination with traditional Chinese medicine to identify novel therapeutic targets and develop new anti-cancer agents. By integrating bioinformatics with experimental validation, this approach could advance GC therapy and unveil enhanced, low-toxicity treatment alternatives.
Conclusion
In this study, we showed that Digitonin has substantial anti-tumor effects in GC, both in experimental models and in live subjects. Through network pharmacology and docking analysis, HSP90AA1 and NFKB1 were identified as key proteins of Digitonin. Our findings indicate that Digitonin downregulates HSP90AA1 expression and inhibits the nuclear translocation of NFKB1, leading to increased apoptosis in GC cells. These findings indicate that Digitonin, targeting various oncogenic pathways, shows potential as a new therapeutic agent for GC. Furthermore, our study underscores the potential of integrating network pharmacology with traditional Chinese medicine to identify new therapeutic targets and develop effective multi-target anti-cancer therapies. Future studies should concentrate on clarifying the molecular mechanisms of Digitonin and assessing its clinical efficacy in managing GC.
Data Sharing Statement
Data will be made available upon reasonable request to the corresponding author.
Ethics Approval and Consent to Participate
All the animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the National Institutes of Health. It received approval from the Ethics and Laboratory Animal Management Committee of Zhejiang Chinese Medical University (IACUC-20211011-02). The human public data were approved by the Biomedical Ethics Committee of Ningbo No.2 hospital (no. PJ-NBEY-KY-2025-002-01).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81770535).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Laversanne M, Sung H. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca a Cancer J Clinicians. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Guan WL, He Y, Xu RH. Gastric cancer treatment: recent progress and future perspectives. J hematol oncol. 2023;16(1):57. doi:10.1186/s13045-023-01451-3
3. Zeng Y, Jin RU. Molecular pathogenesis, targeted therapies, and future perspectives for gastric cancer. Semi Cancer Biol. 2022;86(Pt 3):566–582. doi:10.1016/j.semcancer.2021.12.004
4. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396(10251):635–648. doi:10.1016/s0140-6736(20)31288-5
5. Reddavid R, Dagatti S, Franco C, et al. Molecularly Targeted Therapies for Gastric Cancer. State of the Art. Cancers. 2021;13. doi:10.3390/cancers13164094.
6. De Lucca AJ, Bland JM, Boue S, Vigo CB, Cleveland TE, Walsh TJ. Synergism of CAY-1 with amphotericin B and itraconazole. Chemotherapy. 2006;52(6):285–287. doi:10.1159/000095959
7. Kim SM, Lee SY, Cho JS. Combination of ginsenoside Rg3 with docetaxel enhances the susceptibility of prostate cancer cells via inhibition of NF-kappaB. Eur J Pharmacol. 2010;631(1–3):1–9. doi:10.1016/j.ejphar.2009.12.018
8. Wang W, Rayburn ER, Hao M. Experimental therapy of prostate cancer with novel natural product anti-cancer ginsenosides. Prostate. 2008;68(8):809–819. doi:10.1002/pros.20742
9. Herrmann F, Wink M. Synergistic interactions of saponins and monoterpenes in HeLa cells, Cos7 cells and in erythrocytes. Phytomedicine. 2011;18(13):1191–1196. doi:10.1016/j.phymed.2011.08.070
10. Wink M. Evolutionary advantage and molecular modes of action of multi-component mixtures used in phytomedicine. Curr Drug Metab. 2008;9(10):996–1009. doi:10.2174/138920008786927794
11. Cai J, Liu M, Wang Z, Ju Y. Apoptosis induced by dioscin in Hela cells. Biol Pharm Bull. 2002;25(2):193–196. doi:10.1248/bpb.25.193
12. Haridas V, Higuchi M, Jayatilake GS. Avicins: triterpenoid saponins from Acacia victoriae (Bentham) induce apoptosis by mitochondrial perturbation. Proc Natl Acad Sci USA. 2001;98(10):5821–5826. doi:10.1073/pnas.101619098
13. Yang HT, Chien MY, Chiang JH, Lin PC. Literature-based translation from synthetic lethality screening into therapeutics targets: CD82 is a novel target for KRAS mutation in colon cancer. Comput Struct Biotechnol J. 2022;20:5287–5295. doi:10.1016/j.csbj.2022.09.025
14. Eid SY, El-Readi MZ, Wink M. Synergism of three-drug combinations of sanguinarine and other plant secondary metabolites with digitonin and doxorubicin in multi-drug resistant cancer cells. Phytomedicine. 2012;19(14):1288–1297. doi:10.1016/j.phymed.2012.08.010
15. Eid SY, El-Readi MZ, Wink M. Digitonin synergistically enhances the cytotoxicity of plant secondary metabolites in cancer cells. Phytomedicine. 2012;19(14):1307–1314. doi:10.1016/j.phymed.2012.09.002
16. Eid SY, El-Readi MZ, Eldin EE, Fatani SH, Wink M. Influence of combinations of digitonin with selected phenolics, terpenoids, and alkaloids on the expression and activity of P-glycoprotein in leukaemia and colon cancer cells. Phytomedicine. 2013;21(1):47–61. doi:10.1016/j.phymed.2013.07.019
17. Zhao Z, Zhu Y, Long F, Ma Y, Tang Q, Wang T. Exploring the potential of Huangqin Tang in breast cancer treatment using network pharmacological analysis and experimental verification. BMC Complement Med Therap. 2024;24(1):221. doi:10.1186/s12906-024-04523-0
18. Guo P, Cai C, Wu X. An Insight Into the Molecular Mechanism of Berberine Towards Multiple Cancer Types Through Systems Pharmacology. Front Pharmacol. 2019;10:857. doi:10.3389/fphar.2019.00857
19. Guo JY, Wang YJ, Li SQ, Wu YP. Molecular targets of metformin against ovarian cancer based on network pharmacology. Chem Biol Drug Des. 2023;102(1):88–100. doi:10.1111/cbdd.14234
20. Bang Y-J, Van Cutsem E, Feyereislova A. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a Phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. doi:10.1016/s0140-6736(10)61121-x
21. Wilke H, Muro K, Van Cutsem E. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15(11):1224–1235. doi:10.1016/s1470-2045(14)70420-6
22. Fukuoka S, Hara H, Takahashi N. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: an Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J Clinl Oncol. 2020;38(18):2053–2061. doi:10.1200/jco.19.03296
23. Shitara K, Bang Y-J, Iwasa S. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. New Engl J Med. 2020;382(25):2419–2430. doi:10.1056/NEJMoa2004413
24. Shitara K, Bang Y-J, Iwasa S. Trastuzumab deruxtecan in HER2-positive advanced gastric cancer: exploratory biomarker analysis of the randomized, phase 2 DESTINY-Gastric01 trial. Nat Med. 2024;30(7):1933–1942. doi:10.1038/s41591-024-02992-x
25. Nakayama I, Qi C, Chen Y, Nakamura Y, Shen L, Shitara K. Claudin 18.2 as a novel therapeutic target. Nat Rev Clin Oncol. 2024;21(5):354–369. doi:10.1038/s41571-024-00874-2
26. Wang F-H, Zhang X-T, Tang L. The Chinese Society of Clinical Oncology (CSCO): clinical guidelines for the diagnosis and treatment of gastric cancer, 2023. Cancer Commun. 2024;44(1):127–172. doi:10.1002/cac2.12516
27. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. doi:10.1038/nrc706
28. Al-Batran S-E, Van Cutsem E, Oh SC. Quality-of-life and performance status results from the Phase III RAINBOW study of ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated gastric or gastroesophageal junction adenocarcinoma. Annals Oncol. 2016;27(4):673–679. doi:10.1093/annonc/mdv625
29. Janjigian YY, Kawazoe A, Bai Y. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet. 2023;402(10418):2197–2208. doi:10.1016/s0140-6736(23)02033-0
30. Janjigian YY, Kawazoe A, Yañez P. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature. 2021;600(7890):727–730. doi:10.1038/s41586-021-04161-3
31. Xiao X, Wang W, Li Y. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J Experiment Clin Cancer Res. 2018;37(1):201. doi:10.1186/s13046-018-0880-6
32. Wang Z, Fan L, Xu H. HSP90AA1 is an unfavorable prognostic factor for hepatocellular carcinoma and contributes to tumorigenesis and chemotherapy resistance. Transl Oncol. 2024;50:102148. doi:10.1016/j.tranon.2024.102148
33. Eustace BK, Sakurai T, Stewart JK. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. 2004;6(6):507–514. doi:10.1038/ncb1131
34. Sun F, Qu Z, Xiao Y. NF-κB1 p105 suppresses lung tumorigenesis through the Tpl2 kinase but independently of its NF-κB function. Oncogene. 2016;35(18):2299–2310. doi:10.1038/onc.2015.299
35. Porta C, Ippolito A, Consonni FM. Protumor Steering of Cancer Inflammation by p50 NF-κB Enhances Colorectal Cancer Progression. Cancer Immunol Res. 2018;6(5):578–593. doi:10.1158/2326-6066.Cir-17-0036
36. Wilson CL, Jurk D, Fullard N, et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat Commun. 2015;6(1):6818. doi:10.1038/ncomms7818
37. Huang T, Kang W, Zhang B, et al. miR-508-3p concordantly silences NFKB1 and RELA to inactivate canonical NF-κB signaling in gastric carcinogenesis. mol Cancer. 2016;15(1):9. doi:10.1186/s12943-016-0493-7
38. Elsharkawy AM, Oakley F, Lin F, Packham G, Mann DA, Mann J. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol. 2010;53(3):519–527. doi:10.1016/j.jhep.2010.03.025
39. Schmitt AM, Crawley C, Kang S, et al. p50 (NF-κB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol Cell. 2011;44(5):785–796. doi:10.1016/j.molcel.2011.09.026
40. Mathas S, Jöhrens K, Joos S. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood. 2005;106(13):4287–4293. doi:10.1182/blood-2004-09-3620
41. Li J, Jia H, Xie L. Association of constitutive nuclear factor-kappaB activation with aggressive aspects and poor prognosis in cervical cancer. Int J Gynecoll Cancer. 2009;19(8):1421–1426. doi:10.1111/IGC.0b013e3181b70445
42. Saccani A, Schioppa T, Porta C, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66(23):11432–11440. doi:10.1158/0008-5472.Can-06-1867
43. Concetti J, Wilson CL. NFKB1 and Cancer: friend or Foe? Cells. 2018;7(9):133. doi:10.3390/cells7090133
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.