")
Back to Journals » Infection and Drug Resistance » Volume 18
Whole-Genome Analysis of Paenibacillus campinasensis from Blood of a Hand-Foot-Mouth Patient
Authors Yu X , Lin J, Jiang W, Zhu T, Lin X, Liu X, Zeng Y
Received 18 October 2024
Accepted for publication 24 April 2025
Published 24 May 2025 Volume 2025:18 Pages 2663—2672
DOI https://doi.org/10.2147/IDR.S501740
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sandip Patil
Xiaoling Yu,1 Jun Lin,2 Wenqian Jiang,2 Tianzhu Zhu,2 Xiao Lin,3 Xiaolong Liu,4 Yongyi Zeng5
1Department of Pharmacy, Mengchao Hepatobiliary Hospital of Fujian Medical University, Fuzhou, People’s Republic of China; 2Institute of Applied Genomics, Fuzhou University, Fuzhou, People’s Republic of China; 3Department of Critical Care Medicine, the First Affiliated Hospital of Fujian Medical University, Fuzhou, People’s Republic of China; 4The United Innovation of Mengchao Hepatobiliary Technology Key Laboratory of Fujian Province, Mengchao Hepatobiliary Hospital of Fujian Medical University, Fuzhou, People’s Republic of China; 5Department of Hepatobiliary Surgery, Mengchao Hepatobiliary Hospital of Fujian Medical University, Fuzhou, People’s Republic of China
Correspondence: Xiaolong Liu, The United Innovation of Mengchao Hepatobiliary Technology Key Laboratory of Fujian Province, Mengchao Hepatobiliary Hospital of Fujian Medical University, No. 312 Xihong Road, Gulou District, Fuzhou, 350025, Fujian Province, Email [email protected] Yongyi Zeng, Department of Hepatobiliary Surgery, Mengchao Hepatobiliary Hospital of Fujian Medical University, No. 66 Jintang Road, Cangshan District, Fuzhou, 350001, Fujian Province, Email [email protected]
Purpose: This study aims to investigate the antibiotic resistance and virulence factors of Paenibacillus campinasensis, an understudied opportunistic pathogen, through whole-genome sequencing and resistance genes. The findings will contribute to the development of effective clinical strategies for infection control.
Patients and Methods: A 1-year and 3-month-old boy presenting with fever and rash was admitted to the hospital and clinically diagnosed with hand-foot-mouth disease. A blood culture was obtained, and the pathogen was preliminarily identified as belonging to the genus Paenibacillus using MALDI-TOF-MS. Subsequent 16S rDNA PCR product Sanger sequencing confirmed the isolate as Paenibacillus campinasensis. Whole-genome sequencing and bioinformatics analysis were performed to evaluate the antibiotic resistance and virulence factors of the strain.
Results: Whole-genome sequencing and bioinformatics analysis identified 18 resistance genes in the Paenibacillus campinasensis strain, spanning multiple antibiotic categories. These findings suggest the potential for multidrug resistance in this bacterium, despite its isolation from blood without associated clinical infection in the patient.
Conclusion: The identification of resistance genes in Paenibacillus campinasensis underscores the challenges in clinical treatment and emphasizes the critical role of whole-genome analysis in pathogen detection and characterization. These findings are vital for informing effective clinical strategies to address infections, particularly given the potential for antibiotic resistance to hinder treatment outcomes, even in the absence of direct disease causation by the bacterium.
Keywords: antibiotic resistance, whole-genome sequencing, multidrug resistance, bioinformatics analysis, virulence factors
Introduction
Paenibacillus campinasensis (P. campinasensis) is a Gram-variable bacillus, first isolated from soil in Brazil by Yoon et al in 1998.1 Since its discovery, it has been generally utilized in the industrial production of β-cyclodextrin glucanosyltransferase, xylanase, and cellulase.1–3 There is only one report of its isolation from the prostatic fluid of a patient with chronic prostatitis, with no literature on clinical infections caused by P. campinasensis.4
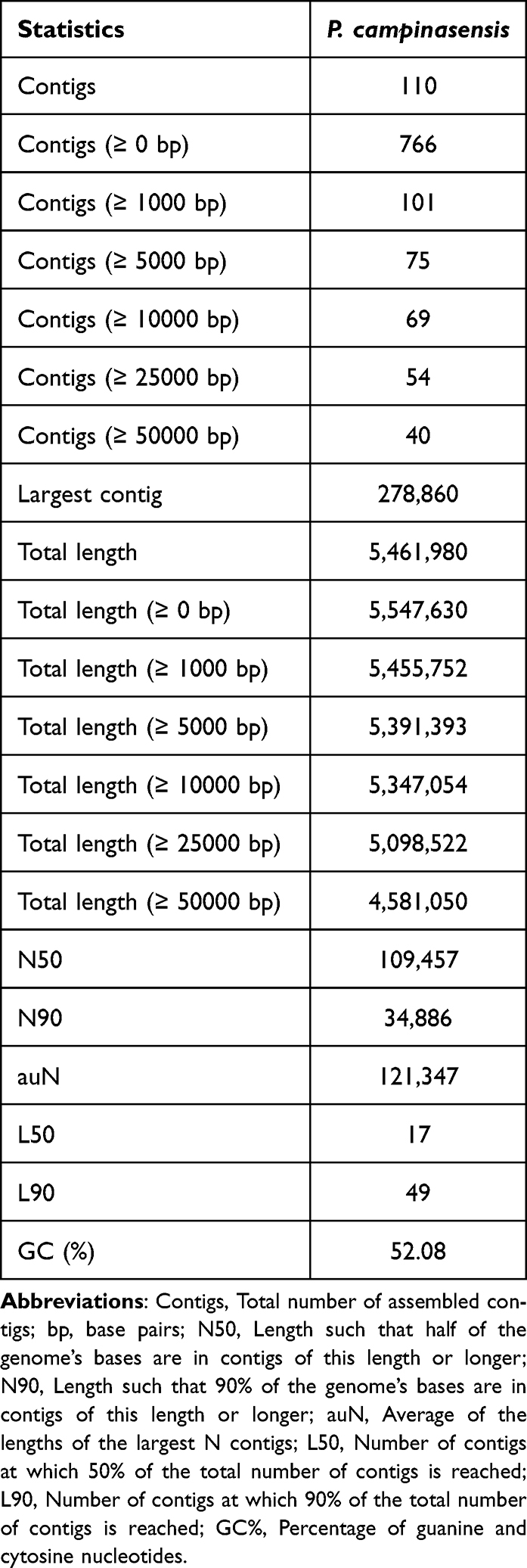 |
Table 1 P. Campinasensis Genome Assembly Contig and Scaffold Statistics |
A 1-year and 3-month-old male patient presented to our hospital with a chief complaint of “fever with rash for 1 day”. The patient’s examination results were as follows: weight 10 kg; body temperature 36.8°C; scattered, medium-sized, red maculopapular rashes on both hands, elbows, knees, feet, and buttocks without petechiae; multiple granulated herpes on the oral cavity; heart rate 130 beats/min; respiratory rate 28 breaths/min; blood pressure 117/70 mm Hg; enterovirus universal type tested positive. Initial complete blood count showed the following: white blood cell counts 12.77×10^9/L [reference range: 3.5–9.5×10^9/L]; lymphocyte percentage 64.8% [reference range: 20–50%]; neutrophil percentage 28.6% [reference range: 40–75%]; interleukin-6: 47.8 pg/mL [reference range: 0–7 pg/mL]. Routine biochemical test results showed the following: alkaline phosphatase 283 U/L [reference range: 35–100 U/L]; lactate dehydrogenase 409 U/L [reference range: 120–250 U/L]. Abdominal ultrasound showed hepatomegaly and coarse liver echo. After admission, the patient was treated with methylprednisolone to counteract inflammatory cytokine responses, omeprazole to protect the gastric mucosa, pediatric amino acid support therapy, Jinlian liquid for oral spray to clear heat and detoxify, and Bifidobacterium bifidum and Lactobacillus bulgaricus granules to regulate intestinal flora. The rash and oral granulated herpes subsided, and the patient’s condition improved. Clinically, there was no specific anti-infection treatment for P. campinasensis, and the patient was discharged with the doctor’s consent.
Materials and Methods
Bacterial Identification and Genome Sequencing
Blood culture results indicated microbial growth, which was preliminarily identified as Pantoea spp. using MALDI-TOF-MS (VITEK MS, BioMerieux SA, BioMerieux Inc., France). Species-level identification was achieved through Sanger sequencing (ABI 3730xl DNA Sequencer) following bacterial genome extraction with a one-step genomic extraction kit (Weiyin, China) and 16S rDNA PCR amplification (universal primers: 27F: 5′ AGAGTTTGATCCTGGCTCAG 3′, 1492: 5′ TACGGYTACCTTGTTACGACTT 3′). NCBI Blast analysis confirmed the isolate as P. campinasensis (16S rRNA gene sequence data are provided in Table S1), not Pantoea spp. A next-generation sequencing (NGS) library was constructed using the Illumina NextEra XT DNA Library Preparation Kit, and metagenomic next-generation sequencing was performed on the Illumina NextSeq platform.
Bioinformatics Analysis of High-Throughput Sequencing Data
Following the acquisition of preliminary high-throughput sequencing data, a rigorous quality assessment of the raw sequencing data was performed using bioinformatics tools such as seqkit, fastqc, and multiqc.5,6 Subsequently, trim_galore software was employed to further refine sequence quality. Clean sequences obtained after quality control were aligned to P. campinasensis reference genome using bwa software to validate sequencing accuracy and consistency.7 Genome assembly was conducted using SOAPDenovo2 (version 2.40) with optimized parameters: “-K 63” (kmer size), “-p 64” (activation of 64 CPU cores), “-d 0” (retention of low-frequency kmers), “-D 1” (elimination of low-coverage edges), “-M 1” (merging intensity of similar sequences), and “-L 65” (shortest contig length for scaffolding).8 Additional configuration parameters included “max_rd_len=150” (maximum read length), “avg_ins=350” (average insert fragment length), “reverse_seq=0” (no reverse-complementary operation), “asm_flags=3” (assembly markers), “rd_len_cutoff=150” (read length cutoff), and “rank=1” (data priority). These parameters collectively ensured successful genome assembly. For comprehensive analysis of antibiotic resistance and virulence factors of P. campinasensis, gene annotation was performed using ABRicate 1.0.1 with the following parameters: “--minid 50” (minimum 50% sequence similarity), “--mincov 50” (minimum 50% coverage of target genes), and “--threads 8” (8 BLAST+ threads for stable parallel processing). Annotation accuracy was further enhanced by integrating multiple databases, including NCBI AMRFinderPlus, CARD, ARG-ANNOT, Resfinder, MEGARES, EcOH, PlasmidFinder, Ecoli_VF, and VFDB.9–16
Gene annotation was further performed using Prokka 1.14.6. Among its key “Matching” parameters, “--evalue” was set to the default value of “1e - 09”. This extremely low e-value was used to meticulously filter out highly similar and reliable sequences, ensuring the precision of the annotation. The “--coverage” parameter was set to the default “80”, stipulating that the coverage of the aligned protein sequence over the target protein must be at least 80%, guaranteeing the comprehensiveness and credibility of the annotation results. The “--cpus 8” parameter specified the use of 8 CPU cores for computation, striking a balance between computing resources and time expenditures to accelerate the annotation process and provide efficient and high-quality data support for subsequent in-depth gene function analyses.
To comprehensively uncover the antibiotic resistance and virulence factors of P. campinasensis, we employed the ABRicate software to conduct a comparative analysis of the assembled scaffolds, and the results were compared with nine authoritative databases, namely NCBI AMRFinderPlus, CARD, ARG-ANNOT, Resfinder, MEGARES, EcOH, PlasmidFinder, Ecoli_VF, and VFDB. Different databases vary in data sources, coverage scopes, and annotation standards, which may lead to similarities and differences in the detection results. For instance, there might be a high overlap between NCBI AMRFinderPlus and Resfinder in the inclusion of common β-lactam resistance genes, which will strengthen our confidence in the detection results of such resistance genes. On the other hand, MEGARES may detect unique resistance genes that are not covered by other databases. Through this analysis, we can more accurately evaluate the reliability of the results, thereby enhancing our confidence in the research findings.
Results
Figure 1A presents the coverage of sequences after quality control by Trim_galore software when aligned with the reference sequence of Paenibacillus campinasensis (GCF_002272015.1) using BWA software across various genomes (only the top 30 genomes in terms of length are shown in the figure). This figure is presented in the form of a bar chart. The red part visually represents the full length of each genome, while the green part accurately indicates the length of the covered region. The text above each bar represents the average sequencing depth. From the data in the figure, it can be seen that most genes not only exhibit extremely high coverage but also possess a considerably high sequencing depth.
 |
Figure 1 Quality Check of the Sequences After Quality Control. (A) Mean Coverage of Genome Mapping; (B) Base Quality Distribution After Quality Control. |
Figure 1B shows the base quality distribution. Through a detailed analysis of this figure, it can be clearly observed that both the Q20 and Q30 quality indicators of the obtained sequences significantly exceed 90%. Taken together, the information reflected in Figures 1A and B fully demonstrates that the sequencing data obtained in this study, in terms of both quantity and quality, have reached the ideal standards for scientific research. This has laid an extremely solid foundation for the subsequent in-depth bioinformatics analysis work, ensuring that follow-up research can conduct precise and reliable analyses and investigations based on high-quality data resources.
The quality-controlled sequences were aligned to the P. campinasensis reference genome using BWA software. Alignment results demonstrated an 88.20% match rate (15,053,371/17,067,368), indicating high sequences purity and reliability. Based on these data, de novo assembly was performed using SOAPDenovo2 software to identify potential key gene differences between this strain and the reference genome. This approach aimed to uncover potential biological characteristics and functional variations unique to this bacterial strain.
The genome assembly results were evaluated using QUAST software, as summarized in Table 1.17 The assembly produced a total of 766 scaffolds, with lengths ranging from 100 bp to 278,860 bp, an average length of approximately 7,242.3 bp, and an N50 of 109.4 kbp.
Based on the QUAST assessment results, the current genome assembly has shown positive outcomes, particularly with the maximum contig length reaching 278,860 base pairs (bp), which indicates the effective formation of longer continuous sequences during the assembly process. As can be seen from the Nx length distribution graph of sample MC2017006996 (Figure 2), the N50 value exceeded 100,000 bp, suggesting that most of the genomic information is integrated into longer contigs. This is not only conducive to subsequent gene annotation and functional analysis but also demonstrates the high continuity of the assembly results. The total length is close to the total length after removing contigs of zero length, indicating that the assembly results cover most of the genomic sequences, providing a solid foundation for the integrity of the genome. In addition, the GC content is within a reasonable range, which helps to maintain the stability of the genome and provides favorable conditions for subsequent research. In summary, the assembly process has successfully reconstructed the genomic structure of P. campinasensis, providing a more complete sequence framework for subsequent drug resistance analysis.
 |
Figure 2 Nx Length Distribution Curve. |
To identify potential functional regions associated with drug resistance that may be contained in the assembled scaffolds, Prokka was employed to annotate and predict the functional regions of 766 scaffolds with a total of 5,547,630 bp of base pairs.18 The results presented in Figure 3, include the annotation of over 5,000 protein-coding regions, i among which are proteins and systems associated with drug resistance. These annotations primarily focus on the mechanisms of antibiotic resistance in bacteria. They include: 1) Multidrug resistance proteins: Multidrug resistance ABC transporter ATP-binding/permease protein BmrA, Multidrug resistance protein 3, Multidrug resistance protein MdtA, Multidrug resistance protein MdtG, Multidrug resistance protein MdtK, Multidrug resistance protein NorM; 2) Antibiotic-specific resistance: Tetracycline resistance protein, class C, Daunorubicin/doxorubicin resistance ABC transporter permease protein DrrB, Daunorubicin/doxorubicin resistance ATP-binding protein DrrA; 3) Regulatory proteins for drug resistance: Transcriptional regulatory protein CusR, Transcriptional regulatory protein CymR, Transcriptional regulatory protein QacR.
 |
Figure 3 Prokka Annotation of Functional Regions. |
These proteins and systems play a key role in the formation of bacterial drug resistance. They can help bacteria resist the effects of antibiotics through various mechanisms, such as pumping out antibiotics, altering the target structure of antibiotics, producing enzymes to destroy antibiotics, or changing the cell wall structure to reduce the entry of antibiotics.
As mentioned earlier, to assess the drug resistance of P. campinasensis, we conducted a comprehensive genomic analysis. On this basis, we compared the assembled scaffolds with multiple drug resistance gene databases to identify potential drug resistance genes. Through this process, we identified a total of 18 drug resistance genes, as shown in Table 2. These genes are involved in various categories of antibiotics, including macrolides, rifampicin, streptomycin, lincosamides, and tetracyclines, indicating that the bacterial strain may have multiple-drug resistance. For example, the coverage and identity percentages of the VatI gene are 99.05% and 75.92%, respectively, which suggests a high correlation with known streptomycin resistance. In addition, the rphB gene is not only resistant to rifampicin but is also marked as resistant to lincosamides in the megares database, further confirming the strain’s multiple-drug resistance characteristics. The high coverage and identity percentages of these drug resistance genes indicate that the identified drug resistance gene sequences have a high degree of similarity to the reference sequences in the databases. The diversity of drug resistance mechanisms is also significant, including the inactivation of antibiotics (such as rphC and rphD genes), the efflux of antibiotics (such as the TaeA gene), and the modification of antibiotic target sites (such as the LlmA_23S_ribosomal_RNA_methyltransferase gene). The existence of these mechanisms suggests that P. campinasensis may acquire drug resistance through various pathways, which poses a challenge to clinical treatment and may require the use of a broader range of antibiotics or combination therapies to effectively treat infections.
 |
Table 2 ABRicate Drug Resistance Database Annotation Results |
P. campinasensis, as a microorganism with a Gram-variable staining phenotype, may exhibit resistance to antibiotics traditionally targeted against both Gram-positive and Gram-negative bacteria. This characteristic not only makes this bacterial strain a focus of attention but also suggests that it may develop resistance to a broad spectrum of antibiotics through various mechanisms. Therefore, an in-depth study of its resistance mechanisms is crucial for developing effective treatment strategies.
In the clinical setting, a wide array of antibiotics is used to treat infections caused by both Gram-negative and Gram-positive bacteria. Table 3 includes some common classes and specific drugs:
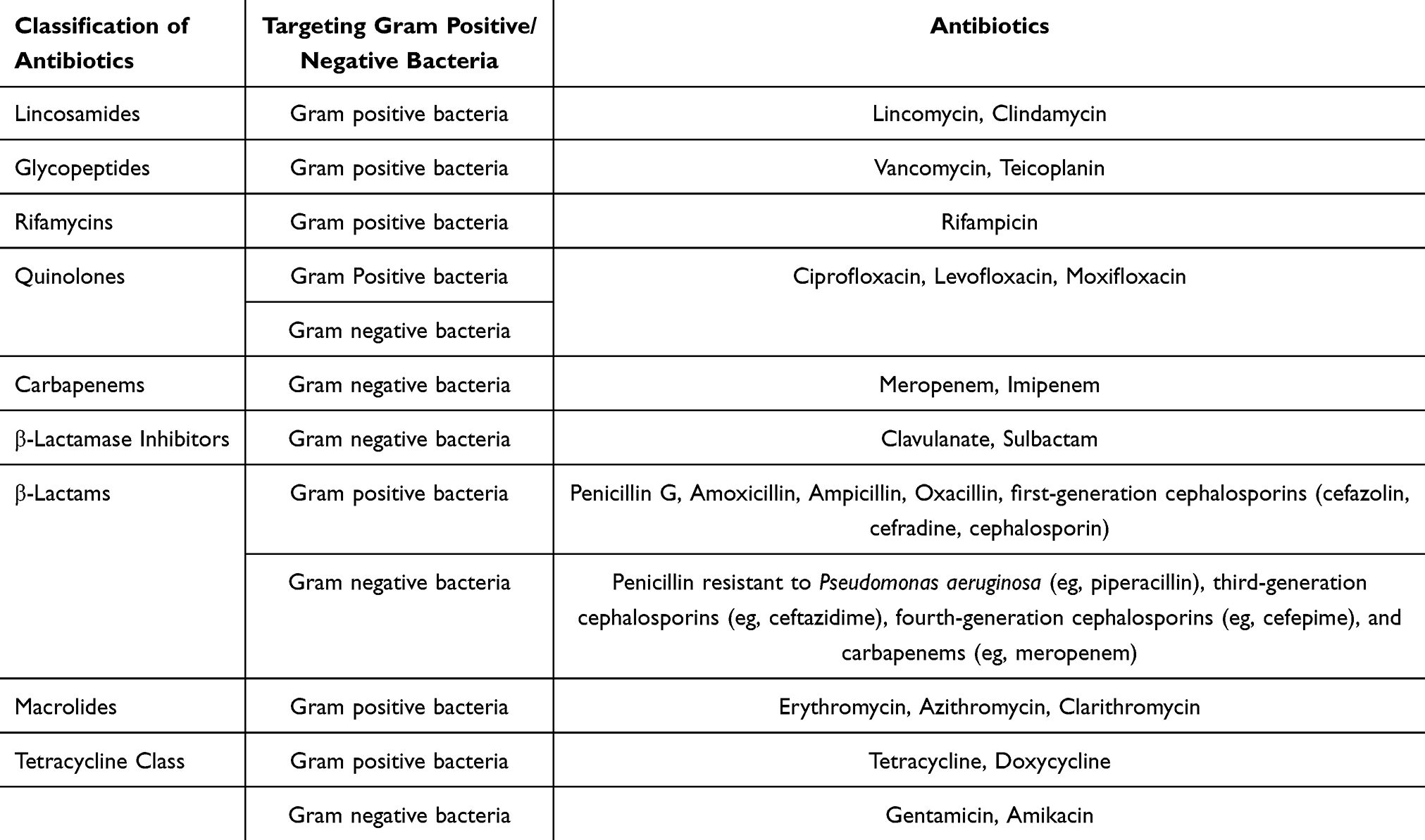 |
Table 3 Antibiotics Used in Clinical Practice |
Through the results of Table 2, we have identified the resistance genes of P. campinasensis to the following antibiotics (the particulars are elaborated upon in Table 4):
 |
Table 4 Types of Antibiotics and Genes Conferring Resistance in P. Campinasensis |
The Gram-variable staining characteristic of P. campinasensis provides a biological basis for the broad spectrum of its resistance genes, which may lead to cross-resistance to various classes of antibiotics. The identification of these resistance genes is significant for understanding the resistance mechanisms of this strain, predicting its potential response to new antibiotics, and providing targeted treatment options for clinicians facing P. campinasensis infections. This helps to optimize treatment plans, improve therapeutic outcomes, reduce unnecessary drug exposure, and slow the development of antibiotic resistance.
Discussion
This study introduced a case of P. campinasensis derived from blood culture of a child with hand-foot-mouth disease. Although P. campinasensis did not cause clinical infection in the patient, our detailed investigation through whole-genome sequencing and bioinformatics analysis revealed a complex profile of antibiotic resistance genes. Specifically, we identified 18 resistance genes involving multiple antibiotic classes. This is the first study to analyze the whole genome of P. campinasensis and evaluate its drug resistance. The presence of numerous resistance genes within this isolate, despite the lack of active infection, poses a potential risk for the development of infections that are difficult to treat. Our findings highlight the importance of surveillance for multidrug resistance (MDR) in clinically relevant isolates, even when they do not cause immediate infections. This approach is crucial for early detection and prevention of MDR spread, which is a growing global health concern. Through genomic analysis, this study reveals that the strain has a complex profile of antibiotic resistance genes, indicating its potential to develop multidrug resistance. This finding not only emphasizes the necessity for continuous monitoring and research on the genetic determinants of bacterial resistance, especially in the face of the increasingly severe global situation of multidrug-resistant pathogens, but also highlights the application value of whole-genome sequencing and bioinformatics tools in analyzing bacterial genetic composition. This is significant for developing effective clinical treatment strategies and addressing the spread of antibiotic resistance.
Further in-depth research on the drug resistance of P. campinasensis is of great significance for understanding its resistance patterns to current antibiotics and predicting its potential resistance gene spectrum. This knowledge has a direct guiding role in preventing potential resistance issues in the future and formulating public health safety and disease control strategies. Against the backdrop of growing global concern over antibiotic resistance, in-depth research on the drug resistance of this strain will provide a solid scientific basis for the rational use of antibiotics, the development of new antibiotics, and the exploration of potentially effective new compounds or combination therapy regimens.
Conclusion
In summary, the study of drug resistance in P. campinasensis is not only of significant clinical guidance for treatment but also has far-reaching scientific value in the formulation of antibiotic management policies and the field of drug development. By identifying the resistance genes of this strain, its resistance characteristics can be more accurately grasped, thereby guiding clinical treatment, enhancing therapeutic outcomes, reducing unnecessary drug use, and effectively responding to the global challenge of antibiotic resistance.
Future research should focus on the following directions: Systematic monitoring of resistance patterns in both environmental and clinical isolates is essential to identify emerging resistance mechanisms and their potential spread. This surveillance will enable early detection of novel resistance genes and inform targeted interventions. Further studies should explore the role of horizontal gene transfer in the dissemination of resistance genes, particularly in clinical settings. Understanding the mechanisms and vectors of gene transfer will provide critical insights for developing strategies to mitigate resistance spread. Research should also focus on elucidating the environmental factors that drive the emergence and persistence of resistance genes. This includes investigating the impact of anthropogenic activities, such as antibiotic use in agriculture and wastewater management, on resistance dynamics.
Data Sharing Statement
The sequencing raw data is stored in the SRA database of NCBI, accession number PRJNA1106201.
Ethics Approval and Informed Consent
This study was reviewed and approved by the Medical Ethics Committee of Mengchao Hepatobiliary Hospital of Fujian Medical University with the approval number: 2024_049_01, dated November 10, 2023. Written informed consent has been provided by the patient’s mother to have the case details published.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation of Fujian Province (2021J01238), Key Clinical Specialty Discipline Construction Program of Fuzhou, Fujian, P.R.C (20230101), Fuzhou Health science and technology plan soft science research project, Fujian, P.R.C (2022-S-wr4), Young and middle-aged talent research project of Fuzhou, Fujian, P.R.C (2022-S-rc5), Fuzhou University Testing Fund of precious apparatus (2024T021), Natural fund project of Fujian Province, China (2024J011223).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Yoon JH, Yim DK, Lee JS, et al. Paenibacillus campinasensis sp. nov. a cyclodextrin-producing bacterium isolated in Brazil. Int J Syst Bacteriol. 1998;48(3):833–837. doi:10.1099/00207713-48-3-833
2. Ko C-H, Tsai C-H, Lin P-H, et al. Characterization and pulp refining activity of a Paenibacillus campinasensis cellulase expressed in Escherichia coli. Biores Technol. 2010;101(20):7882–7888. doi:10.1016/j.biortech.2010.05.043
3. Wang L, Wang Y, Chang S, et al. Identification and characterization of a thermostable GH11 xylanase from Paenibacillus campinasensis NTU-11 and the distinct roles of its carbohydrate-binding domain and linker sequence. Colloids Surfaces B. 2022;209:112167. doi:10.1016/j.colsurfb.2021.112167
4. Yan X, Wang Y, Yue Q, et al. Isolation and identification of Paenibacillus campinasensis sp. J Pathogen Biol. 2013;8(2):120–122.
5. Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11(10):e0163962. doi:10.1371/journal.pone.0163962
6. Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32(19):3047–3048. doi:10.1093/bioinformatics/btw354
7. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013. doi:10.48550/arXiv.1303.3997
8. Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):18. doi:10.1186/2047-217X-1-18
9. Feldgarden M, Brover V, Haft DH, et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother. 2019;63(11). doi:10.1128/AAC.00483-19
10. Jia B, Raphenya AR, Alcock B, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–D573. doi:10.1093/nar/gkw1004
11. Gupta SK, Padmanabhan BR, Diene SM, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 2014;58(1):212–220. doi:10.1128/AAC.01310-13
12. Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi:10.1093/jac/dks261
13. Doster E, Lakin SM, Dean CJ, et al. MEGARes 2.0: a database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2020;48(D1):D561–D569. doi:10.1093/nar/gkz1010
14. Ingle DJ, Valcanis M, Kuzevski A, et al. In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microb Genom. 2016;2(7):e000064. doi:10.1099/mgen.0.000064
15. Carattoli A, Zankari E, Garcia-Fernandez A, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–3903. doi:10.1128/AAC.02412-14
16. Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis--10 years on. Nucleic Acids Res. 2016;44(D1):D694–7. doi:10.1093/nar/gkv1239
17. Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics. 2018;34(13):i142–i150. doi:10.1093/bioinformatics/bty266
18. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.