")
Back to Journals » International Journal of Nanomedicine » Volume 20
A Review on QbD-Driven Optimization of Lipid Nanoparticles for Oral Drug Delivery: From Framework to Formulation
Authors Hidayat AF , Wardhana YW , Suwendar S, Mohammed AFA, Mahmoud SA, Elamin KM , Wathoni N
Received 13 April 2025
Accepted for publication 20 June 2025
Published 3 July 2025 Volume 2025:20 Pages 8611—8651
DOI https://doi.org/10.2147/IJN.S534137
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Misra
Aulia Fikri Hidayat,1,2,* Yoga Windhu Wardhana,3,* Suwendar Suwendar,2,* Ahmed Fouad Abdelwahab Mohammed,4,* Safwat A Mahmoud,5,* Khaled M Elamin,6,* Nasrul Wathoni3,*
1Doctoral Program of Pharmacy, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, Indonesia; 2Department of Pharmacy, Faculty of Mathematics and Natural Sciences, Universitas Islam Bandung, Bandung, Indonesia; 3Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, Indonesia; 4Department of Pharmaceutics, Faculty of Pharmacy, Minia University, Minia, Egypt; 5Center for Scientific Research and Entrepreneurship, Northern Border University, Arar, 73213, Saudi Arabia; 6Graduate School of Pharmaceutical Sciences, Kumamoto University, Kumamoto, Japan
*These authors contributed equally to this work
Correspondence: Nasrul Wathoni, Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia, Tel +62-22-842-888-888, Email [email protected]
Abstract: Oral administration is the most preferred route for drug delivery due to its convenience, non-invasiveness, and patient compliance. However, it is challenged by gastrointestinal barriers, enzymatic degradation, and first-pass metabolism, which reduce drug bioavailability. Lipid nanoparticles (LNPs), including solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs), offer a promising strategy to overcome these limitations by enhancing drug stability, permeability, and absorption. The Quality by Design (QbD) framework provides a systematic approach for LNP development to ensure consistent product quality. By promoting process understanding and control, QbD not only supports scientific formulation development but also enhances industrial scalability by reducing the experimental workload, shortening the development time, and lowering the production costs. This review highlights key QbD elements such as the quality target product profile (QTPP), critical quality attributes (CQAs), critical material attributes (CMAs), critical process parameters (CPPs), and design of experiments (DoE) and their roles in guiding formulation and process optimization. The effects of various CMAs and CPPs on the CQAs such as particle size, polydispersity index, encapsulation efficiency, zeta potential, and drug release are discussed. Furthermore, the in vitro, in vivo, and ex vivo performances of optimized LNPs were explored in detail. Overall, QbD offers a robust platform for the rational design and scalable production of high-quality lipid-based drug delivery systems for oral administration.
Keywords: quality by design, optimization, solid lipid nanoparticles, nanostructured lipid carriers, oral administration
Introduction
Nanotechnology offers broader opportunities for the development of novel drug delivery systems to overcome the limitations of traditional pharmaceutical formulations. Conventional drugs suffer from drawbacks such as poor solubility and low permeability, which subsequently result in poor bioavailability and therapeutic efficiency.1 These issues are particularly pronounced in oral drug delivery, owing to environmental variability, enzymatic degradation, and first-pass metabolism.2 Nanotechnology-based drug delivery systems exploit their size and specific structure to circumvent these hurdles. Various nanocarriers, including micelles, dendrimers, polymer-based nanosystems, and lipid-based nanosystems, have been developed to enhance the effectiveness of drug delivery via various routes.3–5 Lipid-based nanosystems, which are often categorized as vesicular systems, such as liposomes, niosomes, ethosomes, transferosomes, and matrix systems (or lipid nanoparticles, LNPs), such as solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs), have attracted considerable interest owing to their potential as drug delivery systems.6–9 In oral drug delivery, LNPs can protect the drug from degradation in the acidic environment of the stomach and enzymatic hydrolysis of the intestines.10,11 They also proved to be effective in enhancing permeability across epithelial membranes, facilitating lymphatic transport, and bypassing first-pass metabolism, thereby improving systemic bioavailability.12–14
Despite their potential as novel drug delivery systems, a comprehensive understanding in LNPs production remains lacking. The influences of input materials and process parameters on product properties and therapeutic performance have not been clearly explained, which can be attributed to inconsistencies such as batch-to-batch variations in particle size, zeta potential, encapsulation efficiency, and drug release profiles during formulation. Furthermore, the traditional experimental procedure of changing one-factor-at-a-time (OFAT) is deemed inefficient.15 For instance, modifying the lipid type without adjusting surfactant concentration may not improve drug loading, and important interactions between formulation variables may be overlooked. Correspondingly, it was realized that adding more tests did not enhance the product quality. The trial-and-error nature of this method is also resource-intensive, as researchers often need to restart the entire experiment when target quality attributes are not achieved. This approach poses challenges not only in terms of cost, but also in meeting predefined product quality requirements.16,17
The quality by design (QbD) approach offers a more systematic design for production, reducing exhaustive experimental procedures to more efficient ones. The QbD strategy for pharmaceutical formulations has been implemented since the publication of several guidelines by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH).18–23 The foundation of QbD is based on scientific design and manufacturing. Its core value is that the quality should be incorporated into the product. In addition to pharmaceutical knowledge, a mathematical-statistical understanding is important when applying QbD elements in pharmaceutical production. As part of QbD, optimization via design of experiment (DoE) is crucial for defining the mathematical model of the relationship between independent and dependent variables. DoE can also identify the statistical significance and optimal conditions to achieve the desired drug quality.24
In this review, the challenges of oral administration and the corresponding role of LNPs are discussed. Subsequently, we explored the implementation of QbD principles by identifying the quality profile in relation to the selection of materials and preparation methods based on risk assessment to understand the path for improvement in SLN and NLC production for oral drug delivery. Additionally, this article summarizes the findings from recent studies regarding the impact of input variables on output responses based on DoE results, where the optimized products were evaluated in vitro, in vivo, and ex vivo for various purposes.
Challenges of Oral Drug Delivery
Oral delivery is generally considered a more favorable drug administration route for patients and physicians owing to its effortlessness, noninvasive nature, and high patient acceptability.25 Additionally, orally administered drugs may specifically target certain regions, thereby localizing therapy in gastrointestinal (GI) diseases, such as gastroesophageal reflux disease, inflammatory bowel disease, GI cancer, and colorectal cancer.26–29 Despite these advantages, oral drug delivery still faces challenges associated with the inherent properties of drugs and the complexity of the GI system during their traversal.
The GI tract is particularly intriguing because of the variability in its environment. Greatly different pH values are encountered by the drugs, from acidic conditions in the stomach (pH 1 − 2.5), duodenum (pH 6.1), intestines (pH 7.1 − 7.5), and higher pH conditions in the colon (pH 7 − 8).30 Furthermore, the presence of various GI enzymes is particularly challenging for protein-based drugs. They may be hydrolyzed and degraded by pepsin in the stomach or by other proteolytic enzymes in the small intestine.31 In contrast, the enzymatic activity of pancreatic lipase may promote lipolysis in the GI tract and enhance the solubilization of lipophilic drugs or lipid-based formulations.32 GI enzymes also play pivotal roles in the first-pass metabolism. This presystemic metabolism is mediated mainly by cytochrome P450 enzymes such as CYP3A4, which are predominantly found in the liver and small intestine.33 The large surface area and low blood flow in the small intestine might prolong CYP3A4 exposure toward drugs, enabling more extensive metabolism and thus decreasing the oral bioavailability of the drugs.34 This intestinal first-pass effect is markedly enhanced by drugs that are substrates of CYP3A4, such as felodipine, nifedipine, atorvastatin, and simvastatin.35
The small intestine is considered the primary site of oral drug absorption, owing to its extensive surface area and various transport modes. The microvilli in the small intestine are lined with goblet cells, which facilitate glycoprotein secretion and form a mucosal layer.36 This layer is primarily composed of mucin, an oligosaccharide-rich glycosylated protein that provides an overall negative charge to mucus, thus facilitating electrostatic interactions with positively charged substances.37,38 Intestinal mucus is also involved in the formation of an unstirred water layer (UWL) between the intestinal bulk fluid phase and the epithelial brush border. The presence of UWL can be detrimental to lipophilic substances because of hindered access to the epithelium, limiting their passage to the systemic circulation.39
To reach the systemic circulation, drugs must be able to cross the intestinal epithelium, which primarily comprises enterocytes, along with a smaller number of other cells, such as goblet cells, microfold cells (M cells), and Paneth cells.40 Epithelium crossing involves several different transport mechanisms owing to its particular structure and constituents. Passive diffusion is the most common pathway, which relies on the movement of drugs along their concentration gradient, from a higher amount on the apical side to a lower amount on the basolateral side of the membrane. This mechanism may occur via the paracellular or transcellular routes. In the paracellular route, drugs traverse the enterocytes through intercellular tight junctions, allowing the movement of smaller hydrophilic molecules. In contrast, lipophilic molecules can naturally diffuse through the cell membrane transcellularly, owing to their similar affinities.41 Furthermore, transcellular absorption may also occur via endocytic mechanisms, such as phagocytosis by immune cells or M cells, macropinocytosis, clathrin-mediated or clathrin-independent endocytosis, and caveolae-mediated or caveolae-independent endocytosis.42
Unlike passive diffusion, active transport allows molecules to move against the concentration gradient. This transporter-mediated movement offers specific pathways during drug traversal and requires a certain amount of energy derived from adenosine triphosphate (ATP). Among the ATP-dependent transporters, the role of P-glycoprotein (P-gp) has been particularly highlighted in oral drug administration. P-gp is an efflux pump that acts as a defense mechanism by actively pushing xenobiotics out of enterocytes.36 However, this mechanism can be disadvantageous for orally administered drugs because it reduces the intracellular concentration of drugs, thereby limiting drug absorption into systemic circulation and reducing oral bioavailability. This limitation is particularly pronounced for the absorption of drugs that are P-gp substrates.43
Role of Lipid Nanoparticles in Oral Drug Delivery
Drug incorporation into LNPs, such as solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs), is a feasible strategy for overcoming the variability of GI barriers and the complexity of transport modes during oral administration (Figure 1). As a nanoscale system, the structure of LNPs intrinsically generates a larger surface area. This results in increased interaction between the systems and biological membranes, facilitating enhanced absorption into blood circulation.44 The components of LNPs, which mainly comprise lipids and surfactants as well as additional excipients, provide specific advantages for oral drug traversal.
 |
Figure 1 Drug transport mechanisms across intestine. Created in BioRender. Suliman, (K) (2025) https://BioRender.com/vmkadsf. |
Protection From Degradation
The solid lipid components in LNPs, particularly long-chain fatty acids, can slow the degradation process by digestive enzymes, resulting in higher drug stability in the GI environment.40 This allows the drug to reach its target active form, thereby enhancing its therapeutic effectiveness.45 A study conducted by Veni and Gupta showed that the drug release of linagliptin formulated into SLN using stearic acid as a solid lipid at pH 6.8 was higher than that at pH 1.2. This signifies the ability of stearic acid to protect linagliptin from the gastric environment, thereby ensuring intestinal release of the drug.46
Using cetyl palmitate as a solid lipid component in the LNP formulation can also be advantageous for minimizing drug degradation owing to its low susceptibility to lipase hydrolysis, leading to prolonged drug retention in the formulation.47 El-Dakroury et al reported that the cumulative release of fexofenadine HCl in an acidic medium was lower when formulated in SLN using cetyl palmitate compared with the pure drug.48 Similarly, the release of doxorubicin from NLC containing the same solid lipid was lower in simulated gastric fluid than in simulated intestinal fluid.49
Inhibition of Cytochrome P450
First-pass metabolism, mainly mediated by cytochrome P450 enzymes, such as CYP3A4, is a major drawback of effective oral administration. Several LNP constituents may act as modulators of CYP3A4 in bypassing the first-pass metabolism, and subsequently increasing drug absorption.50 Unsaturated fatty acids, such as oleic acid, can inhibit CYP isoforms including CYP3A4. This may be due to the ability of fatty acids to disrupt the microsomal membrane and prevent drug binding to the enzyme active site.51 Several reports have shown that formulating NLC using oleic acid as a liquid lipid can improve the oral bioavailability of telmisartan and fexofenadine HCl.52,53 In a study by Sharma et al, the use of oleic acid in NLC markedly increased the plasma concentration and bioavailability of atorvastatin, which is a CYP3A4 substrate, compared to the marketed drug.54
Surfactants also play a pivotal role in the inhibition of CYP3A4. Non-ionic surfactants, such as Cremophor® EL and Cremophor® RH-40 have inhibitory effects on CYP3A4, with IC50 values ranging between 0.40 to 0.80 mM. Cremophores can alter drug absorption owing to agent-produced membrane fluidization, causing perturbations toward the CYP3A4 microenvironment, thus decreasing enzyme function.51 An in vivo pharmacokinetic study demonstrated increased bioavailability of silybin in SLN formulated using Cremophor® RH40 as a surfactant.55 In another study, it was observed that using Cremophor® EL in the SLN of fenofibrate, a CYP3A4 substrate, resulted in higher plasma concentrations than those of the pure drug.56
Inhibition of P-Glycoprotein
P-glycoprotein (P-gp) is an ATP-binding cassette transporter predominantly found in the apical layer of epithelial cells. Its primary function as a xenobiotic efflux pump can be detrimental for oral delivery due to restricted drug transport across the basolateral layer, thereby limiting the amount of drug in the systemic circulation.57 Designing LNPs with appropriately selected constituents that act as P-gp inhibitors is an effective strategy to improve drug transport across the intestinal barrier. In general, the commonly proposed P-gp inhibitory mechanism involves obstruction of drug-binding sites, disruption of ATP hydrolysis, and alteration of cell membrane integrity.58 Lipid-based excipients, such as glyceryl monooleate and hard fat, as well as surfactants, such as Cremophor®, poloxamers, and polysorbates, are among the components that can be utilized in LNP formulations for these purposes.59 Tween® 80 (polysorbate 80) has been specifically recognized for its synergistic role as an inhibitor of P-gp and CYP3A4.60 Beloqui et al showed that the transport of saquinavir, a known P-gp substrate, across Caco-2 cells was enhanced when incorporated into an NLC system prepared with polysorbate 80.61 Furthermore, an in vitro permeability study demonstrated higher apical-basolateral transport of tilmicosin-loaded NLC prepared using poloxamer 188 and polysorbate 80 as surfactants.62 The use of Gelucire® 44/14 and polysorbate 80 as liquid lipids and surfactants in NLC also significantly increased the plasma concentration of iloperidone, confirming the potential of both excipients as P-gp inhibitors.63
Enhancement of Lymphatic Pathway
The enhancement of oral bioavailability offered by LNPs can also be facilitated through lymphatic transport, allowing lipophilic drugs to bypass first-pass metabolism. Lipids in LNPs may promote the formation of chylomicrons, which are large lipoproteins that are partly responsible for transcellular drug absorption.36 In the GI tract, triglycerides from lipids are broken down by lipase into monoglycerides and free fatty acids, which are then absorbed by enterocytes and incorporated into chylomicrons. These drug-loaded chylomicrons subsequently enter the lymphatic capillaries and avoid hepatic metabolism to finally reach the systemic circulation.45 Molecules with Log P > 5 are naturally transported via the lymphatic pathway. However, drugs with lower Log P values, such as atazanavir (Log P = 4.1), can also utilize this route when encapsulated within the NLC system. Using the chylomicron flow block model, Gurumukhi and Bari revealed the ability of NLC to circumvent first-pass metabolism, resulting in a higher plasma concentration of atazanavir.64
Lymphatic transport can also be facilitated transcellularly through uptake by M cells, which are specific epithelial cells essential for the intestinal immune system. These cells are primarily found in the gut-associated lymphoid tissue (GALT) or Peyer’s patches.40 Functionalization of LNPs with targeting ligands such as lectins enables specific binding to receptors on the M cell surface, thereby enhancing absorption and subsequent transport into the lymphatic system. A study by Hädrich et al demonstrated increased phagocytosis of quercetin NLC when its surface was functionalized with wheat germ agglutinin.65
Enhancement of Mucoadhesion
LNPs can also contribute to enhanced mucoadhesion in oral drug delivery via electrostatic, covalent, hydrogen, and van der Waals interactions.66 Intestinal epithelial cells are protected by a hydrophilic negatively charged mucus layer that serves as a barrier against foreign particles. The functionalization of LNP using positively charged polymers, such as chitosan, is considered a feasible strategy to enhance mucoadhesion via electrostatic interactions, subsequently prolonging the drug residence time in the GI tract, enabling controlled drug release, and improving oral bioavailability. A study by Pyo et al showed higher plasma concentrations of chitosan-coated fenofibrate NLC than of uncoated fenofibrate NLC. The authors suggested that chitosan contributes to enhanced mucoadhesion while also acting as a tight junction modulator in intestinal enterocytes, thus facilitating drug transport into the systemic circulation.67 In another study, a chitosan-functionalized SLN of thymoquinone showed a higher mucoadhesive efficiency than that of free thymoquinone. This may be due to electrostatic interactions between the cationic chitosan-functionalized SLN and anionic mucin molecules. In addition, the hydrophilic properties of chitosan further intensified mucoadhesion.68
Mucoadhesion can also be enhanced through covalent bonding between mucosal cysteine residues and thiomer (thiolated polymer) molecules. For instance, it was found that the use of thiolated polyoxyethylene oleyl ether surfactant in aprepitant-loaded NLC displayed prolonged adhesion to goat intestinal mucosa compared to unmodified surfactant-based aprepitant NLC and aprepitant suspensions. Furthermore, the modified NLC formulation also exhibited increased plasma concentration and relative oral bioavailability compared to the drug suspension and unmodified NLC.69
Quality by Design Framework
The development of LNPs involves complex interactions between formulation components and process parameters, which often result in challenges such as variability in particle size distribution, low encapsulation efficiency, poor drug loading, low zeta potential, and inconsistent drug release behavior. These attributes are critical to the performance of LNPs but are difficult to optimize using traditional OFAT approaches because of the absence of interaction analysis and the need for numerous trial-and-error iterations. In this context, the QbD framework serves as a powerful and structured approach for systematically exploring the formulation space. By employing design of experiments (DoE), QbD enables the identification and control of critical material attributes (CMAs) and critical process parameters (CPPs) that influence the critical quality attributes (CQAs). Beyond its conceptual strengths, QbD also offers practical advantages such as shortened development time, reduced experimental workload, and enhanced precision in targeting desired product characteristics. This is particularly valuable for complex systems like SLNs and NLCs, where small variations in formulation or processing can significantly impact particle size, encapsulation efficiency, and release profile, which subsequently translates into therapeutic performances. Consequently, QbD not only improves formulation robustness and scalability but also supports regulatory alignment and cost-effective LNP development.70,71
Quality Target Product Profile
Quality target product profile (QTPP) is a foundational component of the QbD framework, outlining the desired profile of a drug product to provide optimal safety and efficacy.70 In pharmaceutical product development, the QTPP provides a prospective summary of the final drug product, including dosage form, delivery system, route of administration, dosage strength, container closure system, drug release, pharmacokinetic properties, purity, sterility, and stability.18 In the context of oral drug delivery, the identification of QTPP related to the enhancement of systemic and/or lymphatic absorption is particularly crucial.72 For example, in a QbD-based study of atazanavir-loaded NLC, the QTPP stated that the defined pharmacokinetic parameters should be higher than the reference to provide higher drug concentrations, ensuring higher lymphatic uptake of the drug.64
In the identification of the QTPP, researchers should consider the regulatory requirements for bioequivalence and patient adherence, ensuring that the product matches the therapeutic performance of the reference products.73 The QTPP not only guides formulation and manufacturing strategies but also provides a benchmark for assessing critical quality attributes (CQAs) throughout the development process, aiming for a high-quality pharmaceutical product that meets both safety standards and therapeutic goals.
Critical Quality Attributes
As defined in the ICH Q8 (R2) document, CQA is another important element of QbD that represents the physical, chemical, biological, or microbiological properties of a drug product, which must remain within a defined range, limit, or distribution to ensure the desired quality.18 Quality attributes can be either critical or noncritical. When failure to achieve a specified range results in no efficacy or potential harm to the patient, an attribute should be considered critical.73 Along with the QTPP, CQAs play a vital role in guiding product and process development, ensuring that the safety and efficacy standards are met. However, CQAs differ from QTPP in scope and function within the QbD framework. QTPP outlines the overall desired characteristics and quality of the final drug product, such as the expected release profile and therapeutic effect, whereas CQAs may include specific parameters, such as particle size and encapsulation efficiency, which need to be tightly controlled to meet the target profile.74 Furthermore, unlike the more fixed profile of the QTPP, CQAs serve as adjustable responses to changes in the formulation attributes or process parameters. Thus, CQAs play a critical role in bridging the quality objectives outlined in the QTPP with the practical aspects of formulation and process development.71
In the production of lipid-based nanocarriers, common CQAs include particle size, polydispersity index, encapsulation efficiency, drug-loading capacity, cumulative drug release, and zeta potential.75,76 For orally administered drugs, the control of uniformly small particles is substantial because nanosized particles (<1000 nm) provide a greater surface contact area, subsequently increasing the intestinal absorption.77 Furthermore, nano-sized particles can be transported both paracellularly and transcellularly (via endocytosis by enterocytes or via M cell uptake).25,78 However, a larger particle size may be useful for extended drug release. LNPs larger than 150 nm are more likely to be taken up by phagocytes, which act as reservoirs and accumulate inside the liver or spleen over an extended period before being gradually released into systemic circulation.79,80
The selection of zeta potential as a CQA also affects the performance of the final product. Zeta potential describes the surface charge of a colloidal particle, which is measured as the electrical potential at a layer relative to a certain point in the bulk medium. A higher absolute value of the zeta potential (≥ ±30 mV) indicates stronger repulsive electrostatic interactions between particles, thus preventing aggregation and ensuring the stability of the dispersion system.81 Moreover, from the perspective of drug delivery, the surface charge of nanoparticles is partially responsible for stronger membrane binding and cellular uptake enhancement.82,83 Nisini et al found that positively charged liposomes could interact with the negatively charged mucosal surfaces of tumor cells, facilitating liposome endocytosis by antigen-presenting cells, thus enhancing cell-mediated immune responses.84 The dependence of cytotoxicity on the zeta potential was also demonstrated in a study by Shao et al, where positively charged nanoparticles resulted in higher cytotoxicity toward L929 cells.85
Another property that may be considered a CQA in orally administered LNPs is the percentage of unpleasant taste, which describes the palatability of the drug product. In a study of diacerein-loaded SLN, it was found that an optimum amount of lipid was suitable for producing a palatable preparation.86 Several studies have also revealed the ability of lipids to control the release of bitter drugs in saliva, effectively sustaining the concentration of drugs that reach bitter taste receptors.87,88
Critical Material Attributes
Critical material attributes (CMAs) mainly identify the state of the input materials, such as the drug substances and excipients employed during production. These attributes encompass a wide range of material properties within an acceptable range, which can influence the quality profile of the final product.89 Determining the interrelationship between CMAs and CQAs is fundamental during the QbD process, where material attributes are systematically identified, screened, and controlled based on their impact on quality.90 In the development of SLNs and NLCs for the oral route, determination of CMAs is particularly crucial in relation to their objectives, both to protect the drugs from the GI environment and to deliver them into the systemic circulation.
Generally, the type and amount of lipids are the primary considerations in LNPs production. Changes in the type of lipid, drug-to-lipid ratio, solid-to-liquid lipid ratio, or total lipid concentration can affect the particle size, drug encapsulation efficiency, and release profile of nanoparticles.62,91 For example, the use of solid lipids in different polymorphs can influence the phase transition temperature, which affects lipid crystallinity and the likelihood of drug expulsion from the nanoparticle matrix.92 In NLCs, the optimum amount of liquid lipids may provide greater encapsulation efficiency because of the lower melting point of the system, which subsequently enhances the dissolving capacity of the matrix.93 Furthermore, solid and liquid lipid compositions also affect the type of NLCs, which are classified as imperfect, amorphous, or multiple NLCs. In imperfect NLCs, a lower amount of liquid lipid is blended with solid crystalline lipids, such as glycerides, reducing crystallinity and promoting the formation of an unstructured matrix during cooling. This facilitated a higher drug loading inside the matrix voids.94 In the amorphous type, specific non-crystalline solid lipids, such as medium-chain triglycerides, hydroxystearate, or isopropyl myristate, form an amorphous core along with liquid lipids. The disordered nature of the matrix can minimize drug leakage during storage.95 In multiple NLCs, liquid lipids such as medium-chain and long-chain triglycerides or oleic acid are employed in higher amounts, which enables phase separation and oil compartment formation within the solid lipid. This compartment provides a suitable environment for solubilizing lipophilic drugs, and subsequently promotes sustained or controlled drug release during oral administration.96
Surfactants also play an important role as CMAs in LNPs formulation. The type and concentration of the surfactant can modify the surface properties of the nanoparticles, thereby influencing drug loading, stability, particle size distribution, and pharmacokinetic profile.97 For instance, cationic surfactants such as hexadecyltrimethylammonium bromide (CTAB) can improve mucoadhesion by forming electrostatic interactions with negatively charged endocytosis-inducing biological membranes, thus enhancing the cellular uptake of nanoparticles.98,99 Both the lipids and surfactants selected in the SLN and NLC formulations should be generally recognized as safe based on their biocompatibility, biodegradability, and non-toxicity (Table 1).
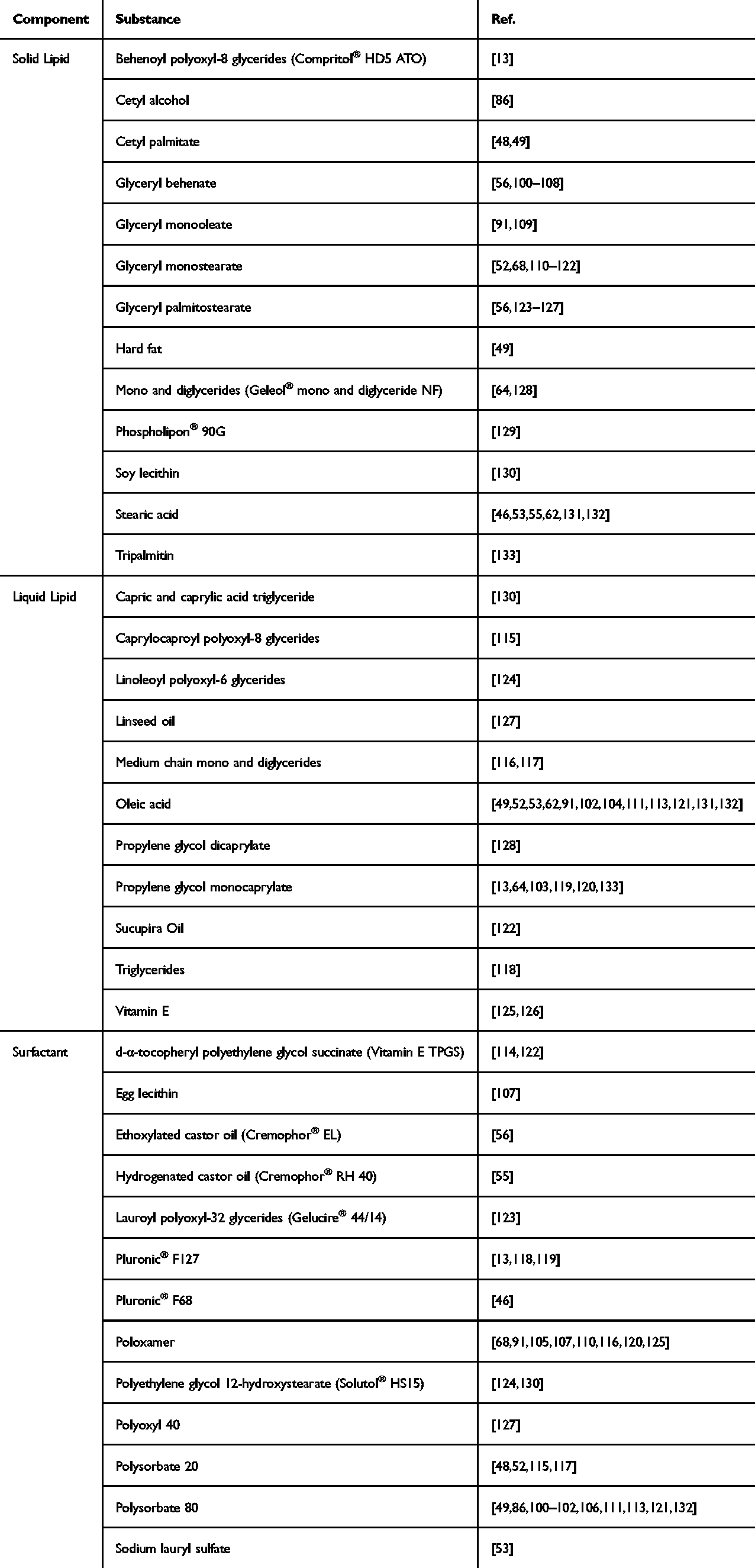 |
Table 1 Common Excipients in LNPs for Oral Drug Delivery |
In surface-modified LNPs, the coating materials can also be considered CMAs. A study by Veni et al indicated that increasing the amount of eudragit, a pH-sensitive polymer, delayed the drug release from the SLN matrix. Eudragit-coated SLN remain stable in acidic gastric environments, whereas the drug is gradually released upon arrival in alkaline intestinal environments.46 The addition of surface charge modifiers to LNPs may also be beneficial. El-say et al reported that a higher zeta potential was obtained by increasing the concentration of stearylamine, a positive charge-inducing agent, thus improving the stability of LNP.123
Critical Process Parameters
Critical process parameters (CPPs) refer to specific input operating or process state variables that are controlled and monitored during production.89 These parameters can impact CQAs, and highly impactful factors should be prioritized. The criticality of a process parameter lies in its ability to satisfy the desired product quality. In LNPs production, the identification of potential CPPs is directly related to the selection of the preparation method (Figure 2). Based on preliminary information, the operating range of a specific parameter can be established to obtain the optimal conditions for the preferred CQA results.70 Several preparation methods, such as melt emulsification-ultrasonication, high-pressure homogenization (HPH), and high-shear homogenization (HSH), have been widely employed for the QbD-driven development of LNPs.100,110,111 Other methods such as hot-melt extrusion, phase inversion temperature, solvent diffusion, solvent evaporation, and spray drying have also been reported.91,130,134,135 However, there have been fewer QbD studies conducted using these methods.
 |
Figure 2 Critical process parameters in various LNP preparation methods. |
Each preparation technique offers a different mechanism along with its advantages and disadvantages. For example, in HPH, input materials are accelerated by high pressure (100 − 2000 bar) through a micron-size gap, where the resulting cavitation force and shear stress can break down the particle to the nanometer size.136 However, the high-energy nature of HPH potentially leads to a suboptimal polydispersity index (PDI) owing to uneven particle disruption.64 It is worth noting that process variations in HPH involving hot and cold conditions facilitate distinctive LNP characteristics. In hot HPH, the drug-lipid melt is combined with a surfactant above the lipid melting point. The mixture was emulsified. The hot emulsion was subsequently homogenized for several cycles at a specific pressure and cooled to room temperature to obtain the SLNs or NLCs. Conversely, in cold HPH, the drug-lipid melt is first solidified using dry ice or liquid nitrogen. The solid was then crushed to obtain micron-sized particles, which were dispersed in a cold surfactant solution, followed by high-pressure homogenization.137 Relatively smaller particles can be obtained using the hot process because of the decreased viscosity at higher temperatures. Nevertheless, high temperatures may also promote rapid degradation of the LNP system;136 thus, certain conditions should be optimized to control the quality of the final product. During the HPH process, the number of homogenization cycles also contributes to the particle size and PDI and is commonly considered a CPP in the HPH technique.138
High-shear homogenization and ultrasonication-based methods are other approaches involving high energy. Both techniques offer a similar mechanism. Prior to nanosizing, the molten lipid was prepared at 5 − 10 °C above its melting point and dispersed in a surfactant solution under stirring at the same temperature. The resulting emulsion was homogenized or sonicated to reduce the droplet size to the nanometer scale.139 The homogenization speed and cavitation-generating ultrasound amplitude are responsible for particle breakdown and thus may be considered as CPPs in their respective methods.140 Concurrently, the duration of sonication and homogenization should be optimized to obtain the desired LNP characteristics without overheating the sample.141
In the spray-drying method, LNPs can be converted into dry powders that offer better physicochemical stability than the dispersion form. Atomization of the aqueous LNP dispersion may occur because of the high inlet temperature of the spray dryer.142 The feed flow rate also promotes physical quality alteration of the LNPs. Mozaffar et al reported that, at a higher temperature (180 °C) and flow rate (15 mL/min), the particles were more evenly distributed and free of aggregates.135
The hot-melt extrusion (HME) technique is a relatively less explored method for LNPs preparation, despite its scalability and environmentally friendly characteristics.143 During HME, the main ingredients are pumped and mixed inside an extruder barrel at 10 − 15 °C above the solid lipid melting temperature. Excipients can be subsequently added to certain feeding zones during extrusion. The resultant hot pre-emulsion was then subjected to either sonication or high-pressure homogenization to reduce the particle size.56,134,144 In this method, varying the extrusion barrel temperature and screw speed evidently influenced the particle size and encapsulation efficiency of the LNPs, and thus could be selected as CPPs during production.
In the phase-inversion temperature (PIT) method, a certain type of emulsion is transformed into its reversed type by continuously changing the mixture temperature.136 This technique primarily exploits the temperature-dependent properties of the hydrophilic-lipophilic balance (HLB) of surfactants. A mixture of drugs, lipids, and surfactants was prepared prior to the phase inversion. The emulsion was then subjected to several cycles of heating and cooling, followed by rapid cooling using cold water (0 °C). This treatment breaks down the emulsion system, resulting in the formation of stable LNPs.130 The temperatures for phase inversion and the heating/cooling rate may influence the resulting nanoparticles; thus, they can be considered as CPPs in the PIT method.145
To select CPPs for each preparation technique, it is necessary to recognize the tunable operating parameters and process state variables that influence the CQAs of the product. Subsequently, the established potential operating space could be further employed during continuous manufacturing of LNPs.
Risk Assessment
In the QbD framework, risk assessment is a vital yet distinct component that supports decision-making by identifying and prioritizing material attributes and process parameters that may impact CQAs.18 While QbD focuses on defining and achieving the QTPP through the systematic control of formulation and process variables, risk assessment serves to anticipate potential sources of variability or failure that could compromise product quality, safety, or cost efficiency.
Several risk assessment tools can be used to guide this process. For example, the Ishikawa (fishbone) diagram provides a broad overview of the contributing factors from each category, describing the cause-and-effect relationships between the variables (Figure 3).112,124 The risk estimation matrix (REM) ranks variables based on their qualitative impact on CQAs.101 In contrast, quantitative tools like failure mode and effects analysis (FMEA) assign numerical values to severity, occurrence, and detectability of potential failures to generate a risk priority number (RPN).90 This allows researchers to prioritize experimental efforts based on criticality. Thus, while QbD is primarily concerned with achieving product quality through robust design, risk assessment complements it by systematically evaluating failure points and hazards, ultimately enhancing the efficiency and reliability of the development process.
 |
Figure 3 Example of an Ishikawa diagram in LNP optimization. |
Design of Experiments
During the QbD course of action, the interactions between independent variables (CMAs and/or CPPs) and dependent variables (CQAs) were formalized through the design of experiments (DoE). In the optimization step, these interactions are typically represented by a polynomial equation that captures single-factor effects, two-factor interactions, and quadratic relationships.146 The generated polynomial equation describes the interplay between factors, providing recommended paths to follow upscaled production. Various aspects are considered when selecting DoE models, such as the purpose of the investigation, factors and responses to be investigated (number, levels, qualitative or quantitative), resources (materials, time, and budget), prior knowledge, and historical data.147 Based on the objective of this study, DoE is generally categorized into screening and optimization designs.148 To concise this article, the DoE models that are commonly used in the production of LNPs are briefly reviewed. More in-depth information regarding the technical details of DoE models has been discussed extensively in several other studies.149–151
Screening Designs
A screening design is considered an initial approach to isolate potentially more significant factors from the numerous possible factors influencing responses. Although methodically different, the function of the screening design is similar to that of risk assessment.152 In general, a screening step is used only to determine the important variables experimentally observed in subsequent optimization designs. Several models, such as two-level full factorial, fractional factorial, Plackett-Burman, and Taguchi, are the most frequently employed screening designs in pharmaceutical formulations.153
Factorial design is one of the most comprehensive DoE models which allows multiple factors to be screened simultaneously. The full factorial design (FFD) examines all possible combinations of factor levels, ensuring a comprehensive analysis of the main effects and interactions. A two-level FFD is commonly employed during the screening step, with levels denoted as high (+1) and low (−1) (Figure 4a). The total number of experimental runs is n = 2k, where 2 and k represent the number of levels and factors, respectively.70 This design allows the evaluation of both the main effects and the interaction effects between the variables. However, as the number of factors increases, the required number of experiments increases exponentially, making it impractical for a large number of factors. To address this, fractional factorial design (FrFD) offers a more efficient alternative by selecting only a subset (fraction) of the full factorial runs while still capturing the significant effects (Figure 4b). Instead of performing all the 2k experiments, a fraction, such as half (2k−1) or a quarter (2k−2) of the total runs, was conducted.151 For example, in a four-factor scenario, instead of performing all 16 runs for a full 24 design, an FrFD with 24–1 = 8 runs may be sufficient for the initial screening.
 |
Figure 4 Schematic illustration of Full Factorial Design, FFD (A); Fractional Factorial Design, FrFD (B); Box-Behnken Design, BBD (C); Central Composite Circumscribed Design, CCCD (D); Central Composite Inscribed Design, CCID (E); Central Composite Face-centered Design, CCFD (F). Created in BioRender. Suliman, (K) (2025) https://BioRender.com/10a19ff. |
The Plackett-Burman design (PBD) is a highly efficient screening design that focuses solely on identifying the most critical factors among a large number of factors while maintaining a minimum number of experiments. Unlike factorial designs, PBD focuses exclusively on estimating the main effects and assumes that interaction effects are negligible.150 The total number of runs in a PBD follows a multiplication of four greater than the number of factors. For instance, the design may require only twelve runs when there are seven or eight factors to screen.154,155 This makes PBD particularly useful for preliminary screening, where the goal is to quickly eliminate insignificant factors before moving to a more detailed optimization phase. However, because interaction effects are not accounted for, PBD is best suited for cases in which factor interactions are either minimal or of no primary interest.71
The Taguchi design is a specialized factorial design that incorporates orthogonal arrays to systematically reduce the number of experiments while ensuring robust results.156 However, in contrast to traditional factorial designs that focus on the main effects and interactions, the Taguchi design emphasizes the improvement of process stability using a signal-to-noise (S/N) ratio to quantify the stability and performance of a system under varying conditions. In the experimental setting, the signal (S) represents the desired output quality, whereas the noise (N) represents undesired response variability due to uncontrolled factors or external disturbances.157 Depending on the desired outcome of the experiment, the S/N ratio can be classified as larger-the-better (LTB), smaller-the-better (STB), or nominal-the-best (NTB).158 For example, in a study on erythrocyte-coated NLC, coating factors affecting particle size and PDI were screened based on the STB S/N ratio, successfully yielding ultrasmall NLC with potential for glioblastoma therapy.159 The orthogonal arrays in the Taguchi design allowed for a wide range of experimental possibilities. A two-level design is commonly used to screen for multiple factors in fewer runs. For instance, Pant et al conducted a 7-factor, 2-level experiment with only 8 runs (L8) using the Taguchi design, instead of 128 (27), to identify the critical factors in the production of raloxifene-loaded NLC.129 To effectively perform screening using the Taguchi design, it is essential to carefully select experiments that maintain a statistical balance and provide an unbiased estimation of the effect of each factor on the responses.
Optimization Designs
While FFD are often used for screening, they can also be applied in optimization by incorporating three or more levels per factor. A three-level FFD (3k) introduces an additional intermediate level (coded as 0), allowing for a more precise estimation of the quadratic effects.147 This is particularly useful when researchers suspect that the relationships between factors and responses are nonlinear. However, similar to two-level FFD, the total number of runs increases exponentially, making this approach efficient only when working with a small number of factors. Notably, a two-level FFD may be applied in an optimization that employs fewer factors.160 Mendes et al constructed two types of designs to develop NLC containing atorvastatin calcium: 32 (nine runs) and 22 (four runs) FFD. An observation of the influence of surfactant concentration on the particle size revealed similar interactions in both designs.159
Central composite design (CCD) is one of the most extensively used response surface methodology (RSM) designs for optimization because it is effective for modeling curvature and optimizing nonlinear processes. The CCD consists of three main components: a full factorial or fractional factorial cube (2k), axial (star) points (2k), and center points. The total number of runs required is determined by the formula 2k + 2k + cp, where k is the number of factors, and cp is the number of center points. Thus, for a 3-factor optimization, a minimum of 15 experiments were required. The axial points (coded as +α and − α) may extend the design space beyond the factorial region, allowing observations at extreme values.141 Based on the selection of α values, CCD can be categorized as: a) circumscribed (CCCD): the axial points extend beyond the factorial space (Figure 4d); b) inscribed (CCID): the axial points remain within the factorial region (Figure 4e); and c) face-centered (CCFD): the axial points are positioned on the faces of the factorial cube (Figure 4f).161 The α value in the CCD varies between 1 and 2k/4, with the latter usually selected to maintain the design rotatability. For example, Ayed et al used a rotatable CCD to optimize two factors, the lipid and surfactant amounts, over 13 runs (including five center points) in the production of quetiapine fumarate-loaded NLC, with an α value of 1.414 (22/4).131 A similar design was employed in a 3-factor, 20-run optimization of NLC containing ifosfamide (including six center points), with an α value of 1.682 (23/4).91 It is also noteworthy that the α value in CCD, particularly in face-centered design, can be set at the same level as the low (−1) and high (+1) values of the factorial space, omitting observations at extreme points, as demonstrated in the study of diacerein-loaded SLN performed by Al-Remawi et al.86
The Box-Behnken design (BBD) is another widely used optimization design, particularly when the relationship between the factors and responses is expected to be nonlinear. Unlike CCD, which includes axial points that may extend beyond the factorial space, BBD distributes the experimental runs evenly at the midpoint of the factor pairs, eliminating the need to test for extremely high and low values (Figure 4c).146 CCD, particularly the circumscribed design, includes axial points that extend beyond the factorial region, which can be valuable for exploring a broader response surface but may introduce impractical conditions. On the other hand, face-centered CCD maintains experimental points within the factorial space but requires more runs because of more levels on account of axial points (+α and −α).153 The number of runs in a BBD is given by the formula 2k (k−1) + cp, where k represents the number of factors, and cp represents the number of center points. This means that to optimize three factors, a minimum of 13 experiments are required, whereas CCD requires at least 15 runs. This makes BBD a more efficient choice for optimization in fewer runs, while still maintaining a robust quadratic model. In the development of LNPs, BBD can be used as a follow-up to the screening stage. For instance, in a study of lurasidone HCl-loaded SLN fabricated by high-pressure homogenization, Patel et al screened seven factors using the Plackett-Burman design. Highly critical factors, namely lipid concentration, homogenization pressure, and homogenization cycle, were subsequently optimized to obtain the preferred particle size and entrapment efficiency using a 15-run BBD (including three center points).112 Nevertheless, numerous studies have reported the direct implementation of BBD for SLN or NLC optimization using 1 − 5 center points, resulting in 13 − 17 experiments.102,110,113,125
Design Space
In developing LNPs, identifying an optimal design space is essential to ensure a well-balanced formulation that meets predefined quality attributes because each factor can influence a response differently, sometimes even in a contradictory manner. For example, increasing the sonication time may reduce the particle size, but it can also lead to an undesirably low entrapment efficiency.68,103,104,124 Therefore, it is important to describe an optimal space that simultaneously achieves a balanced response for multiple factors. The selection of the most optimized formula is guided by the established DoE results, which help map the relationships between the critical factors and their respective responses. This process involves defining a design space that serves as a multidimensional region where the combination of input variables ensures the achievement of optimal responses.74 One of the most effective tools in this process is contour plot overlay, which enables the simultaneous evaluation of multiple responses by superimposing their individual contour plots.162 This graphical approach helps to identify an intersection where all quality attributes meet predefined requirements, thereby defining the most suitable formulation space. In addition, a desirability function is commonly used to simultaneously optimize multiple responses. This method transforms each interaction into a desirability scale ranging from 0 (least desirable) to 1 (most desirable), allowing the calculation of a composite desirability index that reflects the overall optimization outcome.163 By assigning specific weights to different responses based on their importance, this function aids in resolving conflicting optimization criteria, ensuring that the selected formulation maintains a balance among all critical parameters.
Optimization of Lipid Nanoparticles for Oral Drug Delivery
In QbD-based studies on the development of LNPs for oral administration, various independent variables (CMAs and/or CPPs) have been examined to elucidate their effects on the dependent variables (CQAs). The formation of LNPs relies on physicochemical principles such as lipid melting and recrystallization, emulsification, and colloidal stabilization. Upon cooling, the dispersed lipid phase solidifies into nanoparticles, while surfactants reduce the interfacial tension and provide steric or electrostatic stabilization to maintain colloidal stability.137 The nature and concentration of lipids, their crystallinity, and the compatibility with surfactants play key roles in controlling particle characteristics and drug incorporation. Process parameters, such as homogenization speed, pressure, and sonication time, directly influence the nucleation and growth of particles. Thus, the relationship between the independent variables and LNP formation mechanisms underpins their influence on CQAs. The most commonly evaluated CQAs during the optimization stage included particle size, polydispersity index, entrapment efficiency, drug loading, zeta potential, and drug release, as presented in Table 2.
 |
Table 2 Design of Experiments in the Optimization of LNPs for Oral Drug Delivery |
Influences of Independent Variables on Particle Size
Particle size is considered to be one of the principal CQAs in LNP development. SLNs and NLCs with smaller particle sizes inherently have larger surface areas, facilitating higher drug dissolution and absorption. A smaller particle size is also preferable because of the varied transport mechanisms during oral administration.25 Several variables significantly affect the size of the LNPs. In most cases, a higher amount of solid lipids leads to an increase in particle size. At higher solid lipid concentrations, the increased viscosity may resist oil droplet breakdown, resulting in a larger particle size.114 For example, Diwan et al demonstrated that, with other variables held constant, increasing the solid lipid amount from 50 mg to 300 mg led to a particle size increase from 279.2 nm to 837.6 nm during production of SLN containing cilnidipine.105 A similar trend is also observed in NLC optimization when the total lipid content or solid-to-liquid lipid (S/L) ratio is considered a CMA, particularly when the solid lipid concentration exceeds that of the liquid lipid, as evident in various reports.53,115,116,128 However, contradictory interactions have been observed in several studies. For instance, in the study of paliperidone-loaded NLC by Rehman et al, a three-fold increase in total lipid concentration (at an S/L ratio of 70:30) resulted in a particle size reduction from 487.9 nm to 332.6 nm.117 This finding aligns with the results of Pant et al in the development of raloxifene-loaded NLC, in which smaller particles were obtained at higher solid lipid levels.129
The use of higher liquid lipid concentrations has only been investigated in a few studies. Predominantly, an increased amount of liquid lipids is associated with a reduction in the particle size. For instance, in the optimization of eplerenone-loaded NLC, Abd-Elhakeem et al demonstrated that increasing the liquid-to-solid lipid (L/S) ratio from 1:1 to 2:1 markedly reduced the particle size.118 This reduction can be attributed to the low viscosity of the liquid lipid, which facilitates the rapid movement of surfactant molecules, effectively preventing aggregation, and promoting the formation of smaller droplets.164
Surfactants are another key factor that influences the particle size. A higher surfactant concentration primarily leads to particle size reduction by enhancing the emulsifying capacity, which prevents droplet agglomeration and concurrently stabilizes the dispersion system of the LNPs.91,106,116 This trend has been consistently reported in numerous studies on SLN and NLC optimization. For example, while maintaining the other variables constant, increasing the surfactant amount from 1% to 3% effectively decreased the particle size from 49 nm to 20 nm, as reported by El-say et al123 However, contrasting results have been reported in several studies. This can be attributed to the accumulation of surfactant molecules on the nanoparticles, which enveloped the surface excessively, thereby increasing their size.119,131
The preparation method also has a direct impact on the LNP particle size, with CPPs generally selected from adjustable parameters specific to each method. Typically, the application of higher energy, represented by the HPH pressure, homogenization speed, or ultrasound amplitude, leads to a reduction in particle size. A similar effect was observed when pre-LNP primary emulsion was subjected to a high-energy process for an extended period during production. For instance, the HPH pressure accelerates the input materials at high energy, where the resulting cavitation force and shear stress effectively break down particles into smaller sizes.111 Additionally, an increased number of HPH cycles can further reduce the particle size owing to the prolonged exposure to high pressures. As an example, during the preparation of ezetimibe-loaded NLC, Agrawal et al illustrated that increasing the number of HPH cycles from 5 to 7 at 700 bars significantly reduced the particle size from 614.4 nm to 262.0 nm.120 In the HME method, increased barrel temperature also reportedly decreases SLN particle size, possibly due to the complete melting of lipids and drugs, which effectively lowers the viscosity of the drug-lipid system.56
Influences of Independent Variables on Polydispersity Index
The polydispersity index (PDI) is another commonly selected CQA, which is usually measured along with particle size. This describes the particle size distribution, which indicates the dispersion characteristics of LNPs. A monodisperse system with lower PDI is preferable to ensure homogeneous drug delivery and release. Generally, CMAs influence PDI in a manner similar to that of the particle size. Increased solid lipid content may increase LNP viscosity, obstruct uniform particle breakdown, and ultimately lead to a higher PDI.103 Formulating NLCs with higher liquid lipid and surfactant concentrations can reduce overall viscosity and surface tension, respectively, subsequently reversing this effect, as demonstrated in several studies.68,109,118,130
The influence of CPPs on PDI is not as straightforward, as contrasting interactions have been found in multiple reports. On several occasions, prolonged high-energy applications can be beneficial because extended exposure to cavitation energy promotes a low-PDI system with uniformly distributed smaller particles.103,124,126 However, beyond the optimal point, excessive high-energy exposure may generate heat, disrupting the surfactant layer and diminishing its stabilizing capacity, ultimately resulting in a more polydisperse LNP system.165 This phenomenon has been observed in several studies that have utilized ultrasonication or the HPH method.64,104,113,127
Influences of Independent Variables on Entrapment Efficiency and Drug Loading
In LNP formulations, entrapment efficiency (EE) and drug loading (DL) determine the extent of drug incorporation within the lipid matrix, ensuring effective delivery to target sites. For the most part, a higher amount of lipids results in a higher EE, owing to the greater space available inside the matrix. This effect was particularly pronounced when drug compatibility with a specific lipid was considered. For example, El Assasy et al demonstrated a considerable increase in EE in amisulpride-loaded NLC when the solid lipid was changed from tripalmitin to Gelucire® 43/1. This may be due to the greater compatibility of amisulpride with Gelucire® 43/1 than with tripalmitin. Furthermore, in the same study, it was also observed that increasing the lipid-to-drug ratio from 7:1 to 13:1 resulted in an improvement in EE from 49.50% to 69.06%.133 A higher lipid concentration may also increase DL, which can be attributed to the reduction in drug expulsion from the lipid matrix. However, it is notable that the DL of LNP with high lipid content may still be low when the drug makes up only a small fraction of the total mass, as reported in several studies.104,105,130
Surfactants have also been found to significantly affect EE in several ways. Gilani et al illustrated that surfactant concentration positively influenced EE, as a higher surfactant amount may prevent drug leakage by facilitating layer formation in the aqueous phase.114 In contrast, in a study on sulforaphane-loaded NLC, Soni et al argued that excessively high surfactant levels could promote drug partitioning into the external aqueous phase by increasing drug solubility, ultimately leading to greater drug expulsion and resulting in low EE. A similar interaction was also observed for DL in the same study.125
The influence of CPPs on EE generally followed a converging trend, where a higher applied energy tended to lower EE. For example, in ultrasonication-based preparation, a higher ultrasound amplitude or prolonged sonication time may expose LNPs to greater acoustic cavitation energy, potentially disrupting lipid matrix integrity and reducing their ability to retain the drug, thereby decreasing EE.13,68 However, contradictory interactions, where an increased EE was achieved with higher energy levels, have also been observed on several occasions. Agrawal et al revealed that high pressure can alter the lipid matrix, creating more space within its imperfect structure and thus enhancing drug incorporation.120 It has also been reported that during the production of fenofibrate-loaded SLN using the HME method, increasing the screw speed can enhance EE by generating a higher shear force inside the barrel, which promotes the formation of a homogeneous emulsion comprising the drug, lipid, and surfactant.56
Influences of Independent Variables on Zeta Potential
From a structural standpoint, a zeta potential (ZP) value above +30 mV or below −30 mV signifies stronger electrostatic repulsion between particles, which helps prevent aggregation and maintains the stability of the LNP dispersion system. Additionally, in oral drug delivery, the surface charge of the LNP may facilitate charge-dependent interactions across GI barriers.37,166 The excipients used in the LNP influence the zeta potential in various ways. In the optimization of quetiapine fumarate-loaded NLC, Ayed et al used a combination of poloxamer 188 and soybean lecithin as surfactants, resulting in a zeta potential ranging from −36.2 mV to −32.5 mV. The observed anionic surface charge was likely attributed to the phospholipid component of lecithin. Additionally, the study indicated that increasing the surfactant concentration led to a more negative ZP value.131 Dudhipala et al demonstrated a similar interaction in the preparation of zaleplon-loaded SLN using poloxamer 188 and egg lecithin. Their polynomial equation analysis further revealed that poloxamer 188 had an insignificant impact on the ZP.107
In functionalized LNPs, the use of charge-modifying excipients evidently influences ZP. During optimization of carvedilol-loaded SLN, El say et al demonstrated that increasing the concentration of stearylamine, a positive-charge modifier, from 4% to 6% markedly elevating the ZP from +8.4 mV to +25.2 mV.123 El-Dakroury et al used chitosan with various molecular weights as a coating agent for fexofenadine-loaded SLN with the objective of improving the mucoadhesion property of the nanoparticles. These findings indicate that changing the chitosan type from low to high molecular weight increases the positive value of ZP to some extent, which may be due to the higher density of the positively charged amino groups in chitosan.48,167
Influences of Independent Variables on Drug Release
For oral administration of LNPs, drug release studies are typically conducted either directly as a CQA during optimization or evaluated separately after the optimized formulation has been established. It was found that a specific variable may exhibit different interactions with drug release. Several reports have indicated that a higher solid lipid concentration results in a lower amount of released drug, which can be attributed to the increased viscosity and lipid core thickness, thus hindering drug expulsion from the matrix.115,123 Contrasting results have been observed in other studies, in which the addition of solid lipids increased the cumulative drug release percentage. This effect may be attributed to prolonged drug retention within the matrix, allowing for gradual release, which ultimately results in a higher cumulative release at a certain time period.109,129
Surfactants also variably influence the LNP drug release. A higher surfactant concentration can improve drug release by enhancing emulsification, reducing particle size, and increasing surface area, which collectively facilitates drug release. However, an excessive amount of surfactant may accumulate on the particle surface, leading to an increase in size and ultimately slowing the drug release, as reported in several studies.108,130
A direct correlation between CPPs and drug release has been explored less frequently, as most studies that consider drug release as a CQA have primarily focused on optimizing CMAs. In the optimization of dapagliflozin-loaded NLC, Zafar demonstrated that an increased number of HPH cycles positively influenced drug release, likely due to reduced particle size, which ultimately increased the surface contact area with the release medium.115 Similarly, in a study on SLN containing rosuvastatin calcium, it was observed that a shorter time was required for 80% drug release when stirring speed was increased, which may also be attributed to reduced particle size.101
It is also noteworthy that unlike other dependent variables such as particle size, PDI, EE, and ZP, where the desirable values are well-defined (particle size and PDI should be low, ZP should exceed ±30 mV, and EE should be high), the preferred drug release profile can vary among investigators. For instance, in the development of NLC containing olmesartan medoxomil, Beg et al demonstrated the positive impact of surfactant concentration on the quantity of drug released within 4 h (Q4h), where the minimum value was considered optimal because of the goal of achieving sustained release.132 A similar surfactant-drug release interaction was also observed in a study of abiraterone acetate-loaded SLN by Konatham et al. However, they were optimized for a maximum value owing to their selection of a 24-hour release period (Q24h), as it is more favorable for obtaining a higher cumulative release.109 Hence, when considering drug release as a CQA, the optimization direction can vary depending on the assigned constraints, despite the same factor-response interaction.
Performance of Optimized Lipid Nanoparticles for Oral Drug Delivery
The ultimate objective of QbD-driven LNP formulation is to develop a high-performance product tailored to the intended use of the drug. Thus, it is essential to investigate the performance of LNPs optimized through DoE to ensure that they satisfy a predefined target profile. This performance is intrinsically governed by CQAs, such as particle size, polydispersity index, zeta potential, encapsulation efficiency, drug loading, and release profile, each of which is influenced by specific CMAs and CPPs identified during the QbD process. The performance of optimized LNPs for oral drug delivery is typically evaluated using in vitro, in vivo, or ex vivo methods across various aspects, such as drug release kinetics, permeability, pharmacokinetics, and therapeutic efficacy (Table 2).
Drug Release and Kinetics
Optimized LNPs should exhibit a controlled and sustained drug release to ensure prolonged therapeutic action. In vitro drug release studies, often conducted using dialysis membrane techniques, help to assess the release profile under simulated physiological conditions. Low- and high-pH settings are commonly implemented in specific media to evaluate release profiles in both gastric and intestinal environments. For example, an acyclovir-loaded SLN drug release study was conducted in simulated gastric fluid (pH 1.2) and simulated intestinal fluid (pH 6.8) for 24 h. The results indicated that the release rates in both media were lower than that of free acyclovir, suggesting a sustained release pattern for 24 h. It is also displayed that the release plots in both media were superimposed, implying that acyclovir release from the optimized SLN was not pH-dependent.106
It is also common to evaluate drug release under different pH conditions as a continuation, rather than as a separate measurement, where the release profile is depicted as a single continuous plot. This is typically conducted by first assessing the drug release in a low-pH medium for 2 h, followed by a transition to a high-pH medium for the remaining 10 or 22 h, assuming that the gastric transit time is shorter than the intestinal transit time.107 A biphasic release pattern is frequently observed, characterized by a rapid initial release in an acidic medium, followed by sustained release under higher pH conditions. The initial burst release occurred due to the presence of the adsorbed drug on the LNP surface, which was readily released upon contact with the medium, whereas the subsequent sustained release was governed by the gradual diffusion of the encapsulated drug from the lipid matrix.13,112,121,133 For instance, in a drug release study of pioglitazone from SLNs, Shaveta et al demonstrated that more than 30% of the drug was released within the first 2 h, whereas a sustained release of up to 89.56% was achieved after 24 h. Furthermore, the study revealed that the cumulative release of pioglitazone from the optimized SLN was significantly higher than that of the pure drug under the same conditions, which reached only 43.12%.100
Several mathematical models, including zero-order, first-order, Higuchi, Hixson-Crowell, and Korsmeyer-Peppas models, are commonly used to analyze drug release kinetics. The coefficient of determination (R²) for each model was subsequently examined to determine the best fit, allowing for a more precise interpretation of drug release behavior. Vieira et al demonstrated that the release kinetics of sucupira oil from the NLC matrix were best described by the first-order model (R² = 0.9829), compared to the zero-order (R² = 0.7722), Higuchi (R² = 0.9268), and Korsmeyer-Peppas (R² = 0.8403) models. This indicates that the release process is primarily governed by the concentration gradient of sucupira oil over time.122 Conversely, zero-order release kinetics were observed in an Eudragit-coated SLN containing linagliptin, demonstrating that drug release occurred at a constant rate, independent of the remaining drug concentration. This may be attributed to the pH-responsive Eudragit coating, which prevents premature drug release in the stomach and dissolves in the intestine, allowing for controlled water penetration and diffusion. The lipid matrix further regulates the drug release, prevents burst effects, and ensures a controlled drug profile.46 In another study, release kinetics were best described by the Higuchi model, which is primarily governed by diffusion through a lipid matrix.55,111,116,125 The Hixson-Crowell model best fits highly uniform LNPs, where drug release is driven by dissolution following a gradual reduction in particle size, as demonstrated in a study of carvedilol-loaded SLN.123
Several studies have also reported that the Korsmeyer-Peppas model is the most suitable for describing release kinetics, which is predominantly regulated by a combination of multiple mechanisms such as swelling, diffusion, and lipid matrix erosion.52,68,124 Additionally, the exponent value (n) calculated from the Korsmeyer-Peppas equation provides further insight into the drug release mechanisms. These are generally classified as Fickian diffusion (n < 0.45), non-Fickian diffusion or anomalous transport (0.45 < n < 0.85), and case II transport (n = 0.85).115,130 For example, the n values for quetiapine fumarate (QTF) released from NLC in fasted-state simulated gastric fluid (FaSSGF) and fasted-state simulated intestinal fluid (FaSSIF) were 0.508 and 0.624, respectively. The lower n value in FaSSGF, which is closer to Fickian diffusion, suggests that QTF release in the gastric environment may be primarily governed by simple diffusion from the outer shell of the NLC, with a partial contribution from erosion. Conversely, the higher n value in FaSSIF indicates that intestinal drug release is likely driven by a combination of non-Fickian diffusion and erosion mechanisms.131 A study by Beg et al illustrated similar findings to some extent, demonstrating that the release mechanisms of rosuvastatin calcium from SLN range from Fickian to non-Fickian diffusion. The matrix-type structure of SLN plays a crucial role in the regulation of drug diffusion. Furthermore, the release kinetics exhibited a direct relationship with solid lipid concentration, which negatively affected the diffusional drug release mechanism. A higher lipid content favored the predominance of non-Fickian diffusion, suggesting that an increase in lipid levels contributed to a greater influence of erosion-based release mechanisms.101
Drug Permeability and Intestinal Absorption
Effective oral drug delivery requires sufficient permeability of intestinal epithelium. Optimized LNP formulations may enhance drug permeability by modulating P-gp efflux or by utilizing lipid-mediated transport mechanisms. Several methods, such as ex vivo gut permeation models and in vitro cell line studies, are frequently used to evaluate drug permeability and transport across the intestines.
In gut permeation studies, rat or goat intestines are frequently used as models, employing either everted or non-everted techniques. The everted method involves turning the intestine inside out. The mucosal side faces outward, whereas the serosal side faces inside, allowing direct access to the absorptive surface. Meanwhile, the non-everted method maintains the intestine in its natural orientation, where the mucosal surface remains inside.168 In an everted gut sac study of ritonavir-loaded NLC, it was revealed that the apparent permeability coefficient (Papp) of the optimized formulation was markedly higher than that of the pure drug suspension.13 In another study, a non-everted gut sac method was used to compare the permeability of an optimized NLC-containing efavirenz with that of the free drug. The sample was placed on the mucosal side and aliquots were withdrawn from the serosal compartment at predetermined time intervals. The results showed a two-fold increase in both the cumulative permeated drug and Papp of the NLC, confirming the potential of lipid-based nanocarriers to enhance intestinal permeability.130
In vitro cellular permeability studies offer a deeper understanding of the drug transport across the intestinal barrier. Caco-2 cells are commonly used as models in these studies because of their morphological and functional similarities to the human intestinal epithelium, making them reliable for assessing drug permeability and absorption.122 In a permeability study using the Caco-2 cell line, an optimized SLN formulation reportedly increased the Papp of lurasidone HCl six-fold compared to that of the pure drug. Further analysis was performed to elucidate the transport mechanism, using nystatin and chlorpromazine as inhibitors of caveolae-mediated endocytosis and clathrin-mediated endocytosis, respectively. These findings suggest that both transport pathways play a significant role in drug uptake across Caco-2 cells.112 In a transport study of rosuvastatin calcium-loaded SLN, filipin and sucrose were utilized as inhibitors of caveolae-mediated and clathrin-mediated endocytosis, respectively. The reduction in drug uptake was significant only in the presence of sucrose, suggesting that the transport of rosuvastatin calcium across Caco-2 cells is likely clathrin-mediated.101
Besides Caco-2 cells, other cell lines, such as Madin-Darby canine kidney (MDCK) cells, can also serve as epithelial models. MDCK cells are particularly useful for studying paracellular transport owing to their distinct tight junction properties. In a cellular permeability study of tilmicosin-loaded NLC using the MDCK cell model, the optimized formulation exhibited a bidirectional transport mechanism, enhancing cellular uptake while simultaneously inhibiting efflux transport. Additionally, this study confirmed that drug uptake is primarily mediated through caveolae-dependent endocytosis, highlighting the role of LNPs in facilitating transcellular absorption.62
Pharmacokinetic Profile and Bioavailability
One of the primary advantages of LNPs is their ability to improve the pharmacokinetic profiles of poorly soluble drugs. Parameters such as the maximum plasma concentration (Cmax), time to reach maximum concentration (Tmax), area under the curve (AUC), and relative bioavailability are critical indicators of systemic drug exposure. Comparative in vivo pharmacokinetic studies between the optimized LNP formulation and pure drug suspensions or marketed drugs will help to validate the effectiveness of the experimental design. For instance, the optimized formula of NLC-based capsules containing amisulpride demonstrated a significantly higher Cmax and AUC than the reference drug Amipride®. This extended to a 2.52-fold increase in relative bioavailability, highlighting the potential of NLC formulations to enhance the oral absorption and overall systemic availability of amisulpride.133 It is also notable that other pharmacokinetic parameters, such as the elimination rate constant (Kel), can provide further insight into the fate of the drug prior to entering the systemic circulation. Several studies have reported a decrease in Kel for encapsulated drugs compared to free drugs, suggesting prolonged exposure, slower elimination, and enhanced absorption. This also demonstrates the ability of LNPs to facilitate lymphatic uptake, allowing the drug to bypass the first-pass metabolism.102,116,169
The role of LNP formulations in facilitating lymphatic transport has often been investigated using the chylomicron flow block model. In this approach, cycloheximide is administered to inhibit chylomicron formation, allowing researchers to differentiate between the lymphatic and non-lymphatic drug absorption pathways. The effect of lymphatic transport on systemic drug exposure was assessed using the pharmacokinetic profile of each formulation. Rangaraj et al investigated the lymphatic uptake capability of ibrutinib-loaded NLC. The resulting Cmax and AUC of the NLC without cycloheximide were each 2.75-fold and 3.57-fold higher compared to NLC that administered along with cycloheximide.119 Cycloheximide inhibits chylomicron secretion, blocking systemic entry through the lymphatic pathway and ultimately leading to lower plasma concentrations. Several reports of drug-loaded LNPs have consistently shown similar results, confirming their potential to enhance lymphatic transport.64,124,170
Therapeutic Efficacy
Following the QbD-based preparation, optimized LNPs should demonstrate improved therapeutic outcomes. Based on the predefined QTPP and inherent characteristics of the loaded drug, further in vitro or in vivo studies are commonly conducted to assess the efficacy of the formulation in achieving the desired pharmacological effects.
Inflammatory Disorders Treatment
Based on in vivo studies in rats, orally administered bergenin and nabumetone encapsulated in NLC displayed a significant reduction in carrageenan-induced paw edema compared to the pure drug suspension, revealing the superiority of lipid-based nanoformulations as anti-inflammatory agents. Furthermore, the optimized bergenin-loaded NLC exhibited a more prolonged effect than the standard drug indomethacin, which was attributed to higher absorption and longer circulation time.102,113
The anti-inflammatory potential of drug-loaded LNPs may further elucidate their efficacy for specific disorders. In rheumatoid arthritis, pro-inflammatory cytokines such as TNF-α and IL-1β promote the formation of osteoclasts, which are specific cells responsible for bone destruction.171 The downregulation of these cytokines is a key target in arthritis therapy as it helps mitigate inflammation and preserve joint integrity. An in vivo study on apigenin-loaded SLN demonstrated that the optimized formulation significantly reduced TNF-α and IL-1β levels compared to pure apigenin. This orally administered formulation enhances systemic absorption and distribution to tissues and bones, leading to higher drug concentrations in the synovial joint, highlighting the potential of SLN as an anti-arthritic agent.114 A similar pattern was observed in a study on SLN containing fexofenadine HCl for ulcerative colitis therapy. Oral administration of this formulation evidently decreased TNF-α and IL-6 levels in rat colon tissue by 70.79% and 72.99%, respectively. Moreover, optimized SLN also downregulated phosphatidylinositol-3 kinase (PI3K) and protein kinase B (Akt), both of which play crucial roles in the inflammatory signaling pathways implicated in the pathogenesis of ulcerative colitis.48
Lipid-Lowering Treatment
Optimized LNPs have also been shown to enhance the therapeutic potential against hyperlipidemia. In separate studies, ezetimibe-loaded NLC and rosuvastatin calcium-loaded SLN reportedly reduced total cholesterol (TC), triglycerides (TG), and low-density lipoprotein (LDL) levels and increased high-density lipoprotein (HDL) levels in rats treated with a high-fat diet (HFD).101,120 Similar improvements in the lipid profile have been observed in studies investigating the antidiabetic potential of dapagliflozin-loaded and pioglitazone-loaded SLNs. In these studies, streptozotocin was administered along with HFD to induce diabetes. It was revealed that, in addition to lowering TC, TG, and LDL levels while increasing HDL, the optimized SLN formulations also reduced SGOT, SGPT, ALP, and blood glucose levels, confirming the potential benefits of LNPs in liver function regulation.100,108
Cancer Treatment
The use of LNPs for oral administration in cancer treatment has attracted considerable interest because of their prolonged circulation and ability to achieve site-specific targeting. Moraes et al developed an NLC for the oral delivery of doxorubicin and assessed its potential in breast cancer treatment. The optimized DOX-NLCs, functionalized with PEG and folic acid, demonstrated enhanced cytotoxicity against MDA-MB-231 cells and exhibited rapid cellular internalization with over 80% uptake within 30 min, highlighting its superior cellular uptake efficiency.49 Similar performance was observed in lycopene-loaded NLC, where cell death was higher than that of the free drug. This may be attributed to higher lycopene endocytosis and subsequent accumulation within cells.126 Another breast cancer model, MCF-7 cells, was employed to elucidate the potential of NLC to deliver exemestane and thymoquinone (EXE-THY). The EXE-THY-loaded NLC simultaneously demonstrated higher cellular uptake and higher cytotoxicity against MCF-7 cells, as illustrated by the significantly lower IC50 compared to each drug and their combination. The sustained release of EXE-THY from the NLC is particularly beneficial for extending the drug exposure toward cancer cells.103
The anticancer potential of the optimized LNPs has also been evaluated in a prostate cancer model. Sabale et al formulated NLC containing flutamide, a nonsteroidal anti-androgen commonly used in prostate cancer treatment. However, unlike the conventional approach in which the performance of the LNP formulation is compared with that of the free drug, this study aimed to investigate whether nanonization affects the therapeutic activity of flutamide. The results showed that PC3 cell viability upon treatment with flutamide-loaded NLC was comparable to that of free flutamide, but remarkably lower than that of blank NLC, confirming that nanonization did not compromise the anti-cancer activity of the drug. The authors suggested that incorporating flutamide into the NLC matrix is especially advantageous for facilitating chylomicron formation, which enhances lymphatic uptake and ultimately enables a more efficient oral delivery for prostate cancer therapy.127
HIV Treatment
A major challenge in HIV treatment is the persistence of viral reservoirs, which allow the virus to evade conventional antiretroviral therapy. Poor oral bioavailability further complicates effective drug delivery because many antiretroviral drugs have low solubility, rapid metabolism, and limited penetration into these reservoirs.172 The use of LNP formulations for antiretroviral drugs has been increasingly explored to address these challenges. In a QbD-based study, Muheem et al developed a dual drug-loaded NLC containing etravirine and darunavir ethanolate functionalized with d-α-tocopheryl polyethylene glycol succinate (TPGS). An in vitro study in TZM-bl cell lines showed that the optimized NLC formulation inhibited HIV-1 infection by 50% at a drug concentration 58 times lower than that of the pure drug suspensions. Furthermore, the results confirmed chylomicron formation and P-gp efflux inhibition by TPGS in the NLC formulation, highlighting its potential as an oral antiretroviral therapy.124
Schizophrenia Treatment
Owing to the chronic nature of psychotic disorders, long-term treatment is often necessary to manage the symptoms and prevent relapse.173 Developing an effective oral formulation for psychotic therapies is crucial to enhance patient compliance and reduce dosing frequency, thereby minimizing the risk of adverse effects, including drug-induced Parkinsonism, tardive dyskinesia, and metabolic disturbances.174 Aligning with this objective, Patel et al developed an SLN formulation containing lurasidone HCl to evaluate its efficacy in the treatment of schizophrenia in MK-801-induced rats. The optimized SLN significantly reduced the escape latency during the pole-climbing test, indicating improved cognitive function. Furthermore, the same formulation demonstrated a reduction in extrapyramidal side effects after 7−21 days of catalepsy testing, further highlighting its potential to effectively manage schizophrenia, while minimizing motor-related adverse effects.112
Challenges and Future Perspectives
Despite the advantages of QbD in the development of LNPs for oral drug delivery, several challenges persist, particularly the formulation complexity, scalability, and regulatory acceptance. One of the primary difficulties lies in the nonlinear relationship between formulation variables, including CMAs, CPPs, and CQAs, which often makes optimization unpredictable Although DoE helps to systematically analyze these interactions, the experimental workload and associated costs remain significant. To address this, future advancements can focus on the integration of artificial intelligence (AI) into QbD modeling. As part of AI technology, machine learning tools, such as artificial neural networks (ANN), can analyze the complex relationships between the properties of drugs, excipients, and process parameters. This leads to an accurate prediction of the formulation performance in pharmacokinetics/pharmacodynamics (PK/PD) studies, enabling a more efficient approach in the optimization step during pharmaceutical manufacturing.175,176
Another key challenge is the scalability of the optimized formulations. Many LNP formulations that perform well at laboratory scale fail to retain their properties during large-scale manufacturing. Conventional preparation techniques, such as HPH, HSH, and ultrasonication, may not always be feasible for industrial-scale production, owing to batch-to-batch variations and process inefficiencies. Continuous manufacturing and Process Analytical Technology (PAT) should be emphasized to ensure real-time monitoring of particle size, zeta potential, and encapsulation efficiency, reducing batch inconsistencies while improving process efficiency.144,177,178
From a regulatory perspective, nanoformulations face evolving challenges owing to the lack of standardized evaluation criteria for safety, bioavailability, and quality control. Although ICH guidelines outline the general principles for QbD-driven pharmaceutical development, a globally harmonized framework for lipid-based nanomedicines is still lacking. This regulatory uncertainty can delay clinical translation and commercialization.179 The establishment of regulatory standards across different regions to streamline approval processes can ensure safety compliance and ascertain bioequivalence assessment protocols for LNP-based drug delivery systems.180
The future of QbD-driven LNP development will also be shaped by personalized medical approaches. The current formulations are designed for broad patient populations; however, individual metabolic variations can significantly affect drug absorption, distribution, and therapeutic efficacy. Integrating pharmacogenomics and precision medicine principles into LNP design can lead to tailored drug delivery systems that optimize therapeutic outcomes for individual patients.181 Furthermore, nanoinformatics and high-throughput screening techniques can be employed to refine LNP formulations based on patient-specific factors such as genetic makeup, microbiome composition, and disease pathology.182
Conclusion
The QbD framework has emerged as a powerful approach for the systematic development and optimization of LNPs for oral-drug delivery. Given the challenges of oral administration, including gastric degradation, enzymatic metabolism, limited intestinal permeability, and first-pass metabolism, SLNs and NLCs have demonstrated significant potential for improving drug bioavailability and therapeutic outcomes. Unlike traditional one-factor-at-a-time (OFAT) methods, QbD enables a structured approach through DoE, allowing CMAs, CPPs, and CQAs to be identified, modeled, and optimized. This facilitates fine control over key properties, such as particle size, drug encapsulation, zeta potential, and release behavior, supporting the development of reproducible and high-performance formulations.
Despite these advancements, challenges remain in the clinical translation, regulatory harmonization, and industrial scalability of oral LNPs. Future directions include the integration of artificial intelligence in QbD-based optimization, the adoption of continuous manufacturing techniques, and alignment of regulatory standards across regions. Moreover, the application of QbD in the context of personalized medicine offers new possibilities for individualized oral therapies. Overall, the integration of QbD principles in LNP optimization not only enhances drug performance but also ensures a more reliable, scalable, and compliant approach to the development of oral drug delivery systems. Continuous innovations in this field have immense potential to improve patient outcomes and expand the therapeutic landscape of nanomedicine.
Acknowledgments
The authors are grateful to the Rector of Universitas Padjadjaran for covering the APC. The authors would also like to extend their appreciation to Northern Border University, Arar, KSA for supporting this work through the project number “NBU-CRP-2025-2292”.
Funding
This work was funded by Indonesia Endowment Fund for Education Agency (LPDP RI) through doctoral scholarship program.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Afzal O, Altamimi ASA, Nadeem MS, et al. Nanoparticles in drug delivery: from history to therapeutic applications. Nanomaterials. 2022;12(24). doi:10.3390/nano12244494
2. Patil-Gadhe A, Pokharkar V. Montelukast-loaded nanostructured lipid carriers: part i Oral bioavailability improvement. Eur J Pharm Biopharm. 2014;88(1):160–168. doi:10.1016/j.ejpb.2014.05.019
3. Yellepeddi VK, Ghandehari H. Pharmacokinetics of oral therapeutics delivered by dendrimer-based carriers. Expert Opin Drug Deliv. 2019;16(10):1051–1061. doi:10.1080/17425247.2019.1656607
4. De Frates K, Markiewicz T, Gallo P, et al. Protein polymer-based nanoparticles: fabrication and medical applications. Int J Mol Sci. 2018;19(6). doi:10.3390/ijms19061717
5. Limongi T, Susa F, Marini M, et al. Lipid-based nanovesicular drug delivery systems. Nanomaterials. 2021;11(12). doi:10.3390/nano11123391
6. Chen ZL, Huang M, Wang XR, et al. Transferrin-modified liposome promotes α-mangostin to penetrate the blood-brain barrier. Nanomedicine. 2016;12(2):421–430. doi:10.1016/j.nano.2015.10.021
7. Ascenso A, Raposo S, Batista C, et al. Development, characterization, and skin delivery studies of related ultradeformable vesicles: transfersomes, ethosomes, and transethosomes. Int J Nanomed. 2015;10:5837–5851. doi:10.2147/IJN.S86186
8. Aparajay P, Dev A. Development and evaluation of eberconazole-loaded niosomes. MDPI AG. 2022;28. doi:10.3390/ecsoc-25-11664
9. Nasirizadeh S, Malaekeh-Nikouei B. Solid lipid nanoparticles and nanostructured lipid carriers in oral cancer drug delivery. J Drug Deliv Sci Technol. 2020;55. doi:10.1016/j.jddst.2019.101458
10. Li HL, Bin ZX, Ma YK, Zhai GX, Li LB, Lou HX. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J Control Release. 2009;133(3):238–244. doi:10.1016/j.jconrel.2008.10.002
11. Poonia N, Kharb R, Lather V, Pandita D. Nanostructured lipid carriers: versatile oral delivery vehicle. Future Sci OA. 2016;2(3). doi:10.4155/fsoa-2016-0030
12. Talegaonkar S, Bhattacharyya A. Potential of lipid nanoparticles (SLNs and NLCs) in enhancing oral bioavailability of drugs with poor intestinal permeability. AAPS Pharm Sci Tech. 2019;20(3). doi:10.1208/s12249-019-1337-8
13. Gurumukhi VC, Bari SB. Development of ritonavir-loaded nanostructured lipid carriers employing quality by design (QbD) as a tool: characterizations, permeability, and bioavailability studies. Drug Deliv Transl Res. 2022;12(7):1753–1773. doi:10.1007/s13346-021-01083-5
14. Patel P, Pailla SR, Rangaraj N, Cheruvu HS, Sampathi S, Dodoala S. Quality by design approach for developing lipid-based nanoformulations of gliclazide to improve oral bioavailability and anti-diabetic activity. AAPS Pharm Sci Tech. 2019;20(2). doi:10.1208/s12249-018-1214-x
15. Jacyna J, Kordalewska M, Markuszewski MJ. Design of Experiments in metabolomics-related studies: an overview. J Pharm Biomed Anal. 2019;164:598–606. doi:10.1016/j.jpba.2018.11.027
16. Rathod M, Suthar D, Patel H, Shelat P, Parejiya P. Microemulsion based nasal spray: a systemic approach for non-CNS drug, its optimization, characterization and statistical modelling using QbD principles. J Drug Deliv Sci Technol. 2019;49:286–300. doi:10.1016/j.jddst.2018.11.017
17. Taleuzzaman M, Sartaj A, Kumar Gupta D, Gilani SJ, Mirza MA. Phytosomal gel of Manjistha extract (MJE) formulated and optimized with central composite design of Quality by Design (QbD). J Dispers Sci Technol. 2023;44(2):236–244. doi:10.1080/01932691.2021.1942036
18. International Conference on Harmonisation. ICH Guideline Q8 (R2) on Pharmaceutical Development.; 2009. Available from: https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf.
19. International Conference on Harmonisation. ICH Guideline Q9 (R1) on Quality Risk Management; 2023. Available from: https://database.ich.org/sites/default/files/ICH_Q9%28R1%29_Guideline_Step4_2023_0126_0.pdf.
20. International Conference on Harmonisation (2008). ICH Guideline Q10 on Pharmaceutical Quality System.; 2008. Available from: https://database.ich.org/sites/default/files/Q10%20Guideline.pdf.
21. International Conference on Harmonisation. ICH Guideline Q11 on Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities); 2012. Available from: https://database.ich.org/sites/default/files/Q11%20Guideline.pdf.
22. International Conference on Harmonisation. ICH Guideline Q12 on Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management.; 2019. Available from: https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119.pdf.
23. International Conference on Harmonisation. ICH Guideline Q13 on Continuous Manufacturing of Drug Substances and Drug Products; 2022. Available from: https://database.ich.org/sites/default/files/ICH_Q13_Step4_Guideline_2022_1116.pdf.
24. Sousa AS, Serra J, Estevens C, Costa R, Ribeiro AJ. A quality by design approach in oral extended release drug delivery systems: where we are and where we are going? J Pharm Investig. 2023;53(2):269–306. doi:10.1007/s40005-022-00603-w
25. Alqahtani MS, Kazi M, Alsenaidy MA, Ahmad MZ. Advances in Oral Drug Delivery. Front Pharmacol. 2021;12. doi:10.3389/fphar.2021.618411
26. Enwereuzo OO, Akakuru OC, Uwaoma RC, Elemike EE, Akakuru OU. Self-assembled membrane-polymer nanoparticles of top-notch tissue tolerance for the treatment of gastroesophageal reflux disease. J Nanostruct Chem. 2021;11(4):707–719. doi:10.1007/s40097-021-00394-w
27. Sharma M, Sharma S, Wadhwa J. Improved uptake and therapeutic intervention of curcumin via designing binary lipid nanoparticulate formulation for oral delivery in inflammatory bowel disorder. Artif Cells Nanomed Biotechnol. 2019;47(1):45–55. doi:10.1080/21691401.2018.1543191
28. Daşkın D, Erdoğar N, Iskit AB, Bilensoy E. Oral docetaxel delivery with cationic polymeric core-shell nanocapsules: in vitro and in vivo evaluation. J Drug Deliv Sci Technol. 2023;80. doi:10.1016/j.jddst.2023.104163
29. Ahmed SS, Baba MZ, Wahedi U, et al. Oral delivery of solid lipid nanoparticles surface decorated with hyaluronic acid and bovine serum albumin: a novel approach to treat colon cancer through active targeting. Int J Biol Macromol. 2024:279. doi:10.1016/j.ijbiomac.2024.135487.
30. Yamamura R, Inoue KY, Nishino K, Yamasaki S. Intestinal and fecal pH in human health. Front Microbiomes. 2023;2. doi:10.3389/frmbi.2023.1192316
31. Zhu Q, Chen Z, Paul PK, Lu Y, Wu W, Qi J. Oral delivery of proteins and peptides: challenges, status quo and future perspectives. Acta Pharm Sin B. 2021;11(8):2416–2448. doi:10.1016/j.apsb.2021.04.001
32. Carrière F. Impact of gastrointestinal lipolysis on oral lipid-based formulations and bioavailability of lipophilic drugs. Biochimie. 2016;125:297–305. doi:10.1016/j.biochi.2015.11.016
33. Zhang R, Dong K, Wang Z, Miao R, Lu W, Wu X. Nanoparticulate drug delivery strategies to address intestinal cytochrome p450 cyp3a4 metabolism towards personalized medicine. Pharmaceutics. 2021;13(8). doi:10.3390/pharmaceutics13081261
34. Alqahtani S, Bukhari I, Albassam A, Alenazi M. An update on the potential role of intestinal first-pass metabolism for the prediction of drug–drug interactions: the role of PBPK modeling. Expert Opin Drug Metab Toxicol. 2018;14(6):625–634. doi:10.1080/17425255.2018.1482277
35. Kato M. Intestinal first-pass metabolism of CYP3A4 substrates. Drug Metab Pharmacokinet. 2008;23(2):87–94. doi:10.2133/dmpk.23.87
36. Azman M, Sabri AH, Anjani QK, Mustaffa MF, Hamid KA. Intestinal absorption study: challenges and absorption enhancement strategies in improving oral drug delivery. Pharmaceuticals. 2022;15(8). doi:10.3390/ph15080975
37. Lock JY, Carlson TL, Carrier RL. Mucus models to evaluate the diffusion of drugs and particles. Adv Drug Deliv Rev. 2018;124:34–49. doi:10.1016/j.addr.2017.11.001
38. Boegh M, Nielsen HM. Mucus as a barrier to drug delivery - Understanding and mimicking the barrier properties. Basic Clin Pharmacol Toxicol. 2015;116(3):179–186. doi:10.1111/bcpt.12342
39. Ebert A, Hannesschlaeger C, Goss KU, Pohl P. Passive permeability of planar lipid bilayers to organic anions. Biophys J. 2018;115(10):1931–1941. doi:10.1016/j.bpj.2018.09.025
40. Plaza-Oliver M, Santander-Ortega MJ, Lozano MV. Current approaches in lipid-based nanocarriers for oral drug delivery. Drug Deliv Transl Res. 2021;11(2):471–497. doi:10.1007/s13346-021-00908-7
41. Padhye T, Maravajjala KS, Swetha KL, Sharma S, Roy A. A comprehensive review of the strategies to improve oral drug absorption with special emphasis on the cellular and molecular mechanisms. J Drug Deliv Sci Technol. 2021;61. doi:10.1016/j.jddst.2020.102178
42. Beloqui A, Des Rieux A, Préat V. Mechanisms of transport of polymeric and lipidic nanoparticles across the intestinal barrier. Adv Drug Deliv Rev. 2016;106:242–255. doi:10.1016/j.addr.2016.04.014
43. Elmeliegy M, Vourvahis M, Guo C, Wang DD. Effect of P-glycoprotein (P-gp) Inducers on exposure of P-gp substrates: review of clinical drug–drug interaction studies. Clin Pharmacokinet. 2020;59(6):699–714. doi:10.1007/s40262-020-00867-1
44. Jiwankar M, Sabale V. Potential of nanostructured lipid carriers in oral delivery of the poorly soluble drugs. J Nanopart Res. 2023;25(9). doi:10.1007/s11051-023-05840-0
45. Mahor AK, Singh PP, Gupta R, et al. Nanostructured lipid carriers for improved delivery of therapeutics via the oral route. J Nanotechnol. 2023;2023. doi:10.1155/2023/4687959
46. Veni DK, Gupta NV. Development and evaluation of Eudragit coated environmental sensitive solid lipid nanoparticles using central composite design module for enhancement of oral bioavailability of linagliptin. Int J Polym Mater Polym Biomater. 2020;69(7):407–418. doi:10.1080/00914037.2019.1570513
47. Zöller K, To D, Knoll P, Bernkop-Schnürch A. Digestion of lipid excipients and lipid-based nanocarriers by pancreatic lipase and pancreatin. Eur J Pharm Biopharm. 2022;176:32–42. doi:10.1016/j.ejpb.2022.05.003
48. El-Dakroury WA, Zewail MB, Asaad GF, et al. Fexofenadine-loaded chitosan coated solid lipid nanoparticles (SLNs): a potential oral therapy for ulcerative colitis. Eur J Pharm Biopharm. 2024;196. doi:10.1016/j.ejpb.2024.114205
49. Moraes S, Marinho A, Lima S, et al. Targeted nanostructured lipid carriers for doxorubicin oral delivery. Int J Pharm. 2021:592. doi:10.1016/j.ijpharm.2020.120029.
50. Preeti SS, Saharan R, Narwal S,et al. Exploring LIPIDs for their potential to improves bioavailability of lipophilic drugs candidates: a review. Saudi Pharm J. 2023;31(12). doi:10.1016/j.jsps.2023.101870
51. Patel R, Barker J, Elshaer A. Pharmaceutical excipients and drug metabolism: a mini-review. Int J Mol Sci. 2020;21(21):1–21. doi:10.3390/ijms21218224
52. Thapa C, Ahad A, Aqil M, Imam SS, Sultana Y. Formulation and optimization of nanostructured lipid carriers to enhance oral bioavailability of telmisartan using Box–Behnken design. J Drug Deliv Sci Technol. 2018;44:431–439. doi:10.1016/j.jddst.2018.02.003
53. Abdelhameed AH, Abdelhafez WA, Saleh KI, Mohamed MS. Formulation, optimization, and in-vivo evaluation of nanostructured lipid carriers loaded with Fexofenadine HCL for oral delivery. J Drug Deliv Sci Technol. 2022;74. doi:10.1016/j.jddst.2022.103607
54. Sharma K, Hallan SS, Lal B, Bhardwaj A, Mishra N. Development and characterization of floating spheroids of atorvastatin calcium loaded NLC for enhancement of oral bioavailability. Artif Cells Nanomed Biotechnol. 2016;44(6):1448–1456. doi:10.3109/21691401.2015.1041637
55. Nazem Z, Firoozian F, Khodabandelou S, Mohammadi M, Mahboobian MM. Systematic optimization of solid lipid nanoparticles of silybin for improved oral drug delivery by box-behnken design: in vitro and in vivo evaluations. J Pharm Innov. 2023;18(2):472–484. doi:10.1007/s12247-022-09637-x
56. Patil H, Feng X, Ye X, Majumdar S, Repka MA. Continuous production of fenofibrate solid lipid nanoparticles by hot-melt extrusion technology: a systematic study based on a quality by design approach. AAPS J. 2015;17(1):194–205. doi:10.1208/s12248-014-9674-8
57. Chaturvedi S, Verma A, Saharan VA. Lipid drug carriers for cancer therapeutics: an insight into lymphatic targeting, P-gp, CYP3A4 modulation and bioavailability enhancement. Adv Pharm Bull. 2020;10(4):524–541. doi:10.34172/apb.2020.064
58. Varma MVS, Ashokraj Y, Dey CS, Panchagnula R. P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol Res. 2003;48(4):347–359. doi:10.1016/S1043-6618(03)00158-0
59. Nguyen TTL, Duong VA, Maeng HJ. Pharmaceutical formulations with p-glycoprotein inhibitory effect as promising approaches for enhancing oral drug absorption and bioavailability. Pharmaceutics. 2021;13(7). doi:10.3390/pharmaceutics13071103
60. Tompkins L, Lynch C, Haidar S, Polli J, Wang H. Effects of commonly used excipients on the expression of CYP3A4 in colon and liver cells. Pharm Res. 2010;27(8):1703–1712. doi:10.1007/s11095-010-0170-2
61. Beloqui A, Solinís MÁ, Gascón AR, Pozo-Rodríguez A D, Des Rieux A, Préat V. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Control Release. 2013;166(2):115–123. doi:10.1016/j.jconrel.2012.12.021
62. Wen J, Gao X, Zhang Q, et al. Optimization of tilmicosin-loaded nanostructured lipid carriers using orthogonal design for overcoming oral administration obstacle. Pharmaceutics. 2021;13(3):1–17. doi:10.3390/pharmaceutics13030303
63. Mandpe L, Pokharkar V. Quality by design approach to understand the process of optimization of iloperidone nanostructured lipid carriers for oral bioavailability enhancement. Pharm Dev Technol. 2015;20(3):320–329. doi:10.3109/10837450.2013.867445
64. Gurumukhi VC, Bari SB. Quality by design (QbD)–based fabrication of atazanavir-loaded nanostructured lipid carriers for lymph targeting: bioavailability enhancement using chylomicron flow block model and toxicity studies. Drug Deliv Transl Res. 2022;12(5):1230–1252. doi:10.1007/s13346-021-01014-4
65. Hädrich G, Vaz GR, Bidone J, et al. Development of a novel lipid-based nanosystem functionalized with wga for enhanced intracellular drug delivery. Pharmaceutics. 2022;14(10). doi:10.3390/pharmaceutics14102022
66. Cheng YH, Fung MP, Chen YQ, Chiu YC. Development of mucoadhesive methacrylic anhydride-modified hydroxypropyl methylcellulose hydrogels for topical ocular drug delivery. J Drug Deliv Sci Technol. 2024;93. doi:10.1016/j.jddst.2024.105450
67. Pyo YC, Tran P, Kim DH, Park JS. Chitosan-coated nanostructured lipid carriers of fenofibrate with enhanced oral bioavailability and efficacy. Colloids Surf B Biointerfaces. 2020;196. doi:10.1016/j.colsurfb.2020.111331.
68. Rahat I, Rizwanullah M, Gilani SJ, et al. Thymoquinone loaded chitosan - Solid lipid nanoparticles: formulation optimization to oral bioavailability study. J Drug Deliv Sci Technol. 2021:64. doi:10.1016/j.jddst.2021.102565.
69. Hashmi AR, Eltayib EM, Qaisar MN, et al. Mucoadhesive aprepitant-loaded nanostructured lipid carriers containing sulfhydryl surfactant for enhanced oral drug bioavailability. J Drug Deliv Sci Technol. 2024:98. doi:10.1016/j.jddst.2024.105904.
70. Buya AB, Mahlangu P, Witika BA. From lab to industrial development of lipid nanocarriers using quality by design approach. Int J Pharm X. 2024;8. doi:10.1016/j.ijpx.2024.100266
71. Camacho Vieira C, Peltonen L, Karttunen AP, Ribeiro AJ. Is it advantageous to use quality by design (QbD) to develop nanoparticle-based dosage forms for parenteral drug administration? Int J Pharm. 2024;657. doi:10.1016/j.ijpharm.2024.124163
72. Murthy A, Rao Ravi P, Kathuria H, Malekar S. Oral bioavailability enhancement of raloxifene with nanostructured lipid carriers. Nanomaterials. 2020;10. doi:10.3390/nano10051085
73. Yu LX, Amidon G, Khan MA, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–783. doi:10.1208/s12248-014-9598-3
74. Mohseni-Motlagh SF, Dolatabadi R, Baniassadi M, Baghani M. Application of the quality by design concept (qbd) in the development of hydrogel-based drug delivery systems. Polymers. 2023;15(22). doi:10.3390/polym15224407
75. Atre P, Rizvi SAA. A brief overview of quality by design approach for developing pharmaceutical liposomes as nano-sized parenteral drug delivery systems. RSC Pharmaceutics. 2024. doi:10.1039/d4pm00201f
76. Merlo-Mas J, Tomsen-Melero J, Corchero JL, et al. Application of Quality by Design to the robust preparation of a liposomal GLA formulation by DELOS-susp method. J Supercritical Fluids. 2021:173. doi:10.1016/j.supflu.2021.105204.
77. Banerjee A, Qi J, Gogoi R, Wong J, Mitragotri S. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J Control Release. 2016;238:176–185. doi:10.1016/j.jconrel.2016.07.051
78. Salah E, Abouelfetouh MM, Pan Y, Chen D, Xie S. Solid lipid nanoparticles for enhanced oral absorption: a review. Colloids Surf B Biointerfaces. 2020;196. doi:10.1016/j.colsurfb.2020.111305.
79. Ju Y, Guo H, Edman M, Hamm-Alvarez SF. Application of advances in endocytosis and membrane trafficking to drug delivery. Adv Drug Deliv Rev. 2020;157:118–141. doi:10.1016/j.addr.2020.07.026
80. Danaei M, Dehghankhold M, Ataei S, et al. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics. 2018;10(2). doi:10.3390/pharmaceutics10020057
81. Fagionato Masiero J, Barbosa EJ, de Oliveira Macedo L, et al. Vegetable oils in pharmaceutical and cosmetic lipid-based nanocarriers preparations. Ind Crops Prod. 2021:170. doi:10.1016/j.indcrop.2021.113838.
82. Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. doi:10.1016/j.jconrel.2010.01.036
83. Németh Z, Csóka I, Semnani Jazani R, et al. Quality by design-driven zeta potential optimisation study of liposomes with charge imparting membrane additives. Pharmaceutics. 2022;14(9). doi:10.3390/pharmaceutics14091798
84. Nisini R, Poerio N, Mariotti S, De Santis F, Fraziano M. The multirole of liposomes in therapy and prevention of infectious diseases. Front Immunol. 2018;9(FEB). doi:10.3389/fimmu.2018.00155
85. Shao XR, Wei XQ, Song X, et al. Independent effect of polymeric nanoparticle zeta potential/surface charge, on their cytotoxicity and affinity to cells. Cell Prolif. 2015;48(4):465–474. doi:10.1111/cpr.12192
86. Al-Remawi M, Elsayed A, Saleem A, Saleem M. Development of solid lipid nanoparticles of diacerein in a stable oral liquid dosage form using a central composite design. J Dispers Sci Technol. 2024;45(9):1880–1893. doi:10.1080/01932691.2023.2234483
87. Dandagi PM, Rath SP, Gadad AP, Mastiholimath VS. Taste masked quinine sulphate loaded solid lipid nanoparticles for flexible pediatric dosing. INDIAN J PHARM EDUC RES. 2014;48(4):93–99. doi:10.5530/ijper.48.4s.12
88. Banerjee S, Joshi U, Singh A, Saharan VA. Lipids for Taste masking and Taste assessment in pharmaceutical formulations. Chem Phys Lipids. 2021;235. doi:10.1016/j.chemphyslip.2020.105031
89. Lionberger RA, Lee LM, Lee SL, Raw A, Yu LX. Quality by design: concepts for ANDAs. AAPS J. 2008;10(2):268–276. doi:10.1208/s12248-008-9026-7
90. Zhang L, Mao S. Application of quality by design in the current drug development. Asian J Pharm Sci. 2017;12(1):1–8. doi:10.1016/j.ajps.2016.07.006
91. Velmurugan R, Selvamuthukumar S. Development and optimization of ifosfamide nanostructured lipid carriers for oral delivery using response surface methodology. Appl Nanosci. 2016;6(2):159–173. doi:10.1007/s13204-015-0434-6
92. Mazuryk J, Deptuła T, Polchi A, et al. Rapamycin-loaded solid lipid nanoparticles: morphology and impact of the drug loading on the phase transition between lipid polymorphs. Colloids Surf a Physicochem Eng Asp. 2016;502:54–65. doi:10.1016/j.colsurfa.2016.05.017
93. Dudhipala N, Janga KY, Gorre T. Comparative study of nisoldipine-loaded nanostructured lipid carriers and solid lipid nanoparticles for oral delivery: preparation, characterization, permeation and pharmacokinetic evaluation. Artif Cells Nanomed Biotechnol. 2018;46(sup2):616–625. doi:10.1080/21691401.2018.1465068
94. Chauhan I, Yasir M, Verma M, Singh AP. Nanostructured lipid carriers: a groundbreaking approach for transdermal drug delivery. Adv Pharm Bull. 2020;10(2):150–165. doi:10.34172/apb.2020.021
95. Gaba B, Fazil M, Ali A, Baboota S, Sahni JK, Ali J. Nanostructured lipid (NLCs) carriers as a bioavailability enhancement tool for oral administration. Drug Deliv. 2015;22(6):691–700. doi:10.3109/10717544.2014.898110
96. Wathoni N, Suhandi C, Elamin KM, et al. Advancements and challenges of nanostructured lipid carriers for wound healing applications. Int J Nanomed. 2024;19:8091–8113. doi:10.2147/IJN.S478964
97. Bnyan R, Khan I, Ehtezazi T, et al. Surfactant effects on lipid-based vesicles properties. J Pharm Sci. 2018;107(5):1237–1246. doi:10.1016/j.xphs.2018.01.005
98. Spleis H, Sandmeier M, Claus V, Bernkop-Schnürch A. Surface design of nanocarriers: key to more efficient oral drug delivery systems. Adv Colloid Interface Sci. 2023;313. doi:10.1016/j.cis.2023.102848
99. yan YX, xia LY, Li M, Zhang L, xia FL, Zhang N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–345. doi:10.1016/J.CANLET.2012.07.002
100. Shaveta S, Singh J, Afzal M, et al. Development of solid lipid nanoparticle as carrier of pioglitazone for amplification of oral efficacy: formulation design optimization, in-vitro characterization and in-vivo biological evaluation. J Drug Deliv Sci Technol. 2020:57. doi:10.1016/j.jddst.2020.101674.
101. Beg S, Jain S, Kushwah V, et al. Novel surface-engineered solid lipid nanoparticles of rosuvastatin calcium for low-density lipoprotein-receptor targeting: a quality by design-driven perspective. Nanomedicine. 2017;12(4):333–356. doi:10.2217/nnm-2016-0336
102. Zafar A, Yasir M, Panda DS, Singh L. Bergenin nano-lipid carrier to improve the oral delivery: development, optimization, in vitro and in vivo evaluation. J Drug Deliv Sci Technol. 2024;96. doi:10.1016/j.jddst.2024.105655
103. Gupta P, Sharma S, Neupane YR, et al. Co-delivery of exemestane and thymoquinone via nanostructured lipid carriers for efficient breast cancer therapy. J Drug Deliv Sci Technol. 2023:88. doi:10.1016/j.jddst.2023.104892.
104. Alhalmi A, Amin S, Beg S, Al-Salahi R, Mir SR, Kohli K. Formulation and optimization of naringin loaded nanostructured lipid carriers using Box-Behnken based design: in vitro and ex vivo evaluation. J Drug Deliv Sci Technol. 2022;74. doi:10.1016/j.jddst.2022.103590
105. Diwan R, Ravi PR, Pathare NS, Aggarwal V. Pharmacodynamic, pharmacokinetic and physical characterization of cilnidipine loaded solid lipid nanoparticles for oral delivery optimized using the principles of design of experiments. Colloids Surf B Biointerfaces. 2020;193. doi:10.1016/j.colsurfb.2020.111073
106. Hassan H, Adam SK, Alias E, Affandi MMRMM, Shamsuddin AF, Basir R. Central composite design for formulation and optimization of solid lipid nanoparticles to enhance oral bioavailability of Acyclovir. Molecules. 2021;26(18). doi:10.3390/molecules26185432
107. Dudhipala N, Janga KY. Lipid nanoparticles of zaleplon for improved oral delivery by Box–Behnken design: optimization, in vitro and in vivo evaluation. Drug Dev Ind Pharm. 2017;43(7):1205–1214. doi:10.1080/03639045.2017.1304957
108. Unnisa A, Chettupalli AK, Al HT, et al. Development of dapagliflozin solid lipid nanoparticles as a novel carrier for oral delivery: statistical design, optimization, in-vitro and in-vivo characterization, and evaluation. Pharmaceuticals. 2022;15(5). doi:10.3390/ph15050568
109. Konatham S, Patangay S. Abiraterone acetate-loaded solid lipid nanoparticles for improved oral bioavailability: design of experiments-based formulation optimization, in vitro, ex vivo, and in vivo characterization. Int J Appl Pharm. 2023;15(2):131–139. doi:10.22159/ijap.2023v15i2.46710
110. Hosny KM. Alendronate sodium as enteric coated solid lipid nanoparticles; preparation, optimization, and in vivo evaluation to enhance its oral bioavailability. PLoS One. 2016;11(5). doi:10.1371/journal.pone.0154926
111. Patil K, Gujarathi N, Sharma C, Ojha S, Goyal S, Agrawal Y. Quality-by-design-driven nanostructured lipid scaffold of apixaban: optimization, characterization, and pharmacokinetic evaluation. Pharmaceutics. 2024;16(7). doi:10.3390/pharmaceutics16070910
112. Patel MH, Mundada VP, Sawant KK. Fabrication of solid lipid nanoparticles of lurasidone HCl for oral delivery: optimization, in vitro characterization, cell line studies and in vivo efficacy in schizophrenia. Drug Dev Ind Pharm. 2019;45(8):1242–1257. doi:10.1080/03639045.2019.1593434
113. Kawish SM, Ahmed S, Gull A, et al. Development of nabumetone loaded lipid nano-scaffold for the effective oral delivery; optimization, characterization, drug release and pharmacodynamic study. J Mol Liq. 2017;231:514–522. doi:10.1016/j.molliq.2017.01.107
114. Gilani SJ, Bin‐jumah MN, Imam SS, Alshehri S, Jahangir MA, Zafar A. Formulation and optimization of nano lipid based oral delivery systems for arthritis. Coatings. 2021;11(5). doi:10.3390/coatings11050548
115. Zafar A. Development of oral lipid based nano-formulation of dapagliflozin: optimization, in vitro characterization and ex vivo intestinal permeation study. J Oleo Sci. 2020;69(11):1389–1401. doi:10.5650/jos.ess20162
116. Agrawal YO, Patil KD, More KR, et al. Amelioration of bioavailability through formulating and optimizing Azilsartan Entrapped nanostructured lipid carriers and its pharmacokinetic assessment. J Drug Deliv Sci Technol. 2022:77. doi:10.1016/j.jddst.2022.103894.
117. Rehman S, Nabi B, Baboota S, Ali J. Tailoring lipid nanoconstructs for the oral delivery of paliperidone: formulation, optimization and in vitro evaluation. Chem Phys Lipids. 2021;234. doi:10.1016/j.chemphyslip.2020.105005
118. Abd-Elhakeem E, El-Nabarawi M, Shamma R. Lipid-based nano-formulation platform for eplerenone oral delivery as a potential treatment of chronic central serous chorioretinopathy: in-vitro optimization and ex-vivo assessment. Drug Deliv. 2021;28(1):642–654. doi:10.1080/10717544.2021.1902023
119. Rangaraj N, Pailla SR, Shah S, Prajapati S, Sampathi S. QbD aided development of ibrutinib-loaded nanostructured lipid carriers aimed for lymphatic targeting: evaluation using chylomicron flow blocking approach. Drug Deliv Transl Res. 2020;10(5):1476–1494. doi:10.1007/s13346-020-00803-7
120. Agrawal YO, Mahajan UB, Agnihotri VV, et al. Ezetimibe-loaded nanostructured lipid carrier based formulation ameliorates hyperlipidaemia in an experimental model of high fat diet. Molecules. 2021;26(5). doi:10.3390/molecules26051485
121. Agrawal Y, Patil K, Mahajan H, et al. In vitro and in vivo characterization of Entacapone-loaded nanostructured lipid carriers developed by quality-by-design approach. Drug Deliv. 2022;29(1):1112–1121. doi:10.1080/10717544.2022.2058651
122. Vieira R, Severino P, Nalone LA, et al. Sucupira oil-loaded nanostructured lipid carriers (NLC): lipid screening, factorial design, release profile, and cytotoxicity. Molecules. 2020;25(3). doi:10.3390/molecules25030685
123. El-Say KM, Hosny KM. Optimization of carvedilol solid lipid nanoparticles: an approach to control the release and enhance the oral bioavailability on rabbits. PLoS One. 2018;13(8). doi:10.1371/journal.pone.0203405
124. Muheem A, Waseem M, Sartaj A, et al. D-α-tocopheryl polyethylene glycol succinate-decorated dual drug-loaded lipidic nanocarriers: a strategic approach for targeting lymphatic uptake and p-gp efflux modulation to enhance oral bioavailability in HIV-1 viral reservoirs. J Drug Deliv Sci Technol. 2024:98. doi:10.1016/j.jddst.2024.105831.
125. Soni K, Rizwanullah M, Kohli K. Development and optimization of sulforaphane-loaded nanostructured lipid carriers by the Box-Behnken design for improved oral efficacy against cancer: in vitro, ex vivo and in vivo assessments. Artif Cells Nanomed Biotechnol. 2018;46(sup1):15–31. doi:10.1080/21691401.2017.1408124
126. Singh A, Neupane YR, Panda BP, Kohli K. Lipid Based nanoformulation of lycopene improves oral delivery: formulation optimization, ex vivo assessment and its efficacy against breast cancer. J Microencapsul. 2017;34(4):416–429. doi:10.1080/02652048.2017.1340355
127. Sabale V, Nikam M, Sabale P. Formulation, optimization and in vitro studies of flutamide-loaded nanostructured lipid carrier based oral drug delivery for enhanced anticancer activity. J Pharm Innov. 2024;19(5). doi:10.1007/s12247-024-09859-1
128. Gurumukhi VC, Bari SB. Fabrication of efavirenz loaded nano-formulation using quality by design (QbD) based approach: exploring characterizations and in vivo safety. J Drug Deliv Sci Technol. 2020;56. doi:10.1016/j.jddst.2020.101545
129. Pant A, Sharma G, Saini S, et al. QbD-driven development of phospholipid-embedded lipidic nanocarriers of raloxifene: extensive in vitro and in vivo evaluation studies. Drug Deliv Transl Res. 2024;14(3):730–756. doi:10.1007/s13346-023-01427-3
130. Varshosaz J, Taymouri S, Jahanian-Najafabadi A, Alizadeh A. Efavirenz oral delivery via lipid nanocapsules: formulation, optimisation, and ex-vivo gut permeation study. IET Nanobiotechnol. 2018;12(6):795–806. doi:10.1049/iet-nbt.2018.0006
131. Ben Hadj Ayed O, Lassoued MA, Sfar S. Quality-by-design approach development, characterization, and in vitro release mechanism elucidation of nanostructured lipid carriers for quetiapine fumarate oral delivery. J Pharm Innov. 2022;17(3):840–855. doi:10.1007/s12247-021-09567-0
132. Beg S, Saini S, Bandopadhyay S, Katare OP, Singh B. QbD-driven development and evaluation of nanostructured lipid carriers (NLCs) of Olmesartan medoxomil employing multivariate statistical techniques. Drug Dev Ind Pharm. 2018;44(3):407–420. doi:10.1080/03639045.2017.1395459
133. El Assasy AEHI, Younes NF, Makhlouf AIA. Enhanced oral absorption of amisulpride via a nanostructured lipid carrier-based capsules: development, optimization applying the desirability function approach and in vivo pharmacokinetic study. AAPS Pharm Sci Tech. 2019;20(2). doi:10.1208/s12249-018-1283-x
134. Shadambikar G, Marathe S, Ji N, et al. Formulation development of itraconazole PEGylated nano-lipid carriers for pulmonary aspergillosis using hot-melt extrusion technology. Int J Pharm X. 2021:3. doi:10.1016/j.ijpx.2021.100074.
135. Mozaffar S, Radi M, Amiri S, McClements DJ. A new approach for drying of nanostructured lipid carriers (NLC) by spray-drying and using sodium chloride as the excipient. J Drug Deliv Sci Technol. 2021;61. doi:10.1016/j.jddst.2020.102212
136. Ganesan P, Narayanasamy D. Lipid nanoparticles: different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain Chem Pharm. 2017;6:37–56. doi:10.1016/j.scp.2017.07.002
137. Viegas C, Patrício AB, Prata JM, Nadhman A, Chintamaneni PK, Fonte P. Solid lipid nanoparticles vs. nanostructured lipid carriers: a comparative review. Pharmaceutics. 2023;15(6). doi:10.3390/pharmaceutics15061593
138. Dumont C, Jannin V, Miolane C, et al. A proof-of-concept for developing oral lipidized peptide Nanostructured Lipid Carrier formulations. J Drug Deliv Sci Technol. 2019:54. doi:10.1016/j.jddst.2019.101394.
139. Ajiboye AL, Nandi U, Galli M, Trivedi V. Olanzapine loaded nanostructured lipid carriers via high shear homogenization and ultrasonication. Sci Pharm. 2021;89(2). doi:10.3390/scipharm89020025
140. Khan S, Sharma A, Jain V. An overview of nanostructured lipid carriers and its application in drug delivery through different routes. Adv Pharm Bull. 2023;13(3):446–460. doi:10.34172/apb.2023.056
141. Subramaniam B, Siddik ZH, Nagoor NH. Optimization of nanostructured lipid carriers: understanding the types, designs, and parameters in the process of formulations. J Nanopart Res. 2020;22(6). doi:10.1007/s11051-020-04848-0
142. Xia D, Shrestha N, van de Streek J, Mu H, Yang M. Spray drying of fenofibrate loaded nanostructured lipid carriers. Asian J Pharm Sci. 2016;11(4):507–515. doi:10.1016/j.ajps.2016.01.001
143. Chauhan G, Shaik AA, Kulkarni NS, Gupta V. The preparation of lipid-based drug delivery system using melt extrusion. Drug Discov Today. 2020;25(11):1930–1943. doi:10.1016/j.drudis.2020.07.025
144. Muhindo D, Ashour EA, Almutairi M, Joshi PH, Repka MA. Continuous production of raloxifene hydrochloride loaded nanostructured lipid carriers using hot-melt extrusion technology. J Drug Deliv Sci Technol. 2021;65. doi:10.1016/j.jddst.2021.102673
145. Gomes GVL, Sola MR, Rochetti AL, Fukumasu H, Vicente AA, Pinho SC. β-carotene and α-tocopherol coencapsulated in nanostructured lipid carriers of murumuru (Astrocaryum murumuru) butter produced by phase inversion temperature method: characterisation, dynamic in vitro digestion and cell viability study. J Microencapsul. 2019;36(1):43–52. doi:10.1080/02652048.2019.1585982
146. Waghule T, Dabholkar N, Gorantla S, Rapalli VK, Saha RN, Singhvi G. Quality by design (QbD) in the formulation and optimization of liquid crystalline nanoparticles (LCNPs): a risk based industrial approach. Biomed Pharmacother. 2021;141. doi:10.1016/j.biopha.2021.111940
147. Politis SN, Colombo P, Colombo G, Rekkas DM. Design of experiments (DoE) in pharmaceutical development. Drug Dev Ind Pharm. 2017;43(6):889–901. doi:10.1080/03639045.2017.1291672
148. Tavares Luiz M, Santos Rosa Viegas J, Palma Abriata J, et al. Design of experiments (DoE) to develop and to optimize nanoparticles as drug delivery systems. Eur J Pharm Biopharm. 2021;165:127–148. doi:10.1016/j.ejpb.2021.05.011
149. Jankovic A, Chaudhary G, Goia F. Designing the design of experiments (DOE) – an investigation on the influence of different factorial designs on the characterization of complex systems. Energy Build. 2021;250. doi:10.1016/j.enbuild.2021.111298
150. Benedetti B, Caponigro V, Ardini F. Experimental design step by step: a practical guide for beginners. Crit Rev Anal Chem. 2022;52(5):1015–1028. doi:10.1080/10408347.2020.1848517
151. Rampado R, Peer D. Design of experiments in the optimization of nanoparticle-based drug delivery systems. J Control Release. 2023;358:398–419. doi:10.1016/j.jconrel.2023.05.001
152. Beg S, Rahman M, Kohli K. Quality-by-design approach as a systematic tool for the development of nanopharmaceutical products. Drug Discov Today. 2019;24(3):717–725. doi:10.1016/j.drudis.2018.12.002
153. Fukuda IM, Pinto CFF, Moreira CDS, Saviano AM, Lourenço FR. Design of experiments (DoE) applied to pharmaceutical and analytical quality by design (QbD). Braz J Pharm Sci. 2018;54(Special Issue). doi:10.1590/s2175-97902018000001006
154. Patel P, Trivedi A, Prajapati BG, Kapoor DU. QbD- driven development and characterization of Lactoferrin-loaded nanoparticles in in-situ gel for treating postmenopausal osteoporosis. Next Res. 2025;2(1):100142. doi:10.1016/j.nexres.2025.100142
155. Patel P, Thanki A, Kapoor DU, Prajapati BG. QbD decorated ellagic acid loaded polymeric nanoparticles: factors influencing desolvation method and preliminary evaluations. Nano-Struct Nano-Objects. 2024;40. doi:10.1016/j.nanoso.2024.101378
156. Cunha S, Costa CP, Moreira JN, Sousa Lobo JM, Silva AC. Using the quality by design (QbD) approach to optimize formulations of lipid nanoparticles and nanoemulsions: a review. Nanomedicine. 2020;28. doi:10.1016/j.nano.2020.102206
157. Kurata K, Shimada K, Takamatsu H. Application of the Taguchi method to explore a robust condition of tumor-treating field treatment. PLoS One. 2022;17(1). doi:10.1371/journal.pone.0262133
158. Vasantharaj K, Jerold M, Deepanraj B, Velan M, Sivasubramanian V. Assessment of a sulfidogenic system utilizing microalgal biomass of Chlorella pyrenoidosa as an electron donor: Taguchi based grey relational analysis. Int J Hydrogen Energy. 2017;42(42):26545–26554. doi:10.1016/j.ijhydene.2017.08.001
159. Mendes M, Basso J, Silva J, et al. Biomimeting ultra-small lipid nanoconstructs for glioblastoma treatment: a computationally guided experimental approach. Int J Pharm. 2020:587. doi:10.1016/j.ijpharm.2020.119661.
160. Maretti E, Rustichelli C, Romagnoli M, et al. Solid lipid nanoparticle assemblies (SLNas) for an anti-TB inhalation treatment—A design of experiments approach to investigate the influence of pre-freezing conditions on the powder respirability. Int J Pharm. 2016;511(1):669–679. doi:10.1016/j.ijpharm.2016.07.062
161. Gujral G, Kapoor D, Jaimini M. An updated review on design of experiment (DOE) in pharmaceuticals. J Drug Delivery Ther. 2018;8(3). doi:10.22270/jddt.v8i3.1713
162. Singh B, Kapil R, Nandi M, Ahuja N. Developing oral drug delivery systems using formulation by design: vital precepts, retrospect and prospects. Expert Opin Drug Deliv. 2011;8(10):1341–1360. doi:10.1517/17425247.2011.605120
163. Vitorino C, Silva S, Gouveia F, Bicker J, Falcão A, Fortuna A. QbD-driven development of intranasal lipid nanoparticles for depression treatment. Eur J Pharm Biopharm. 2020;153:106–120. doi:10.1016/j.ejpb.2020.04.011
164. Correia AC, Moreira JN, Sousa Lobo JM, Silva AC. Design of experiment (DoE) as a quality by design (QbD) tool to optimise formulations of lipid nanoparticles for nose-to-brain drug delivery. Expert Opin Drug Deliv. 2023;20(12):1731–1748. doi:10.1080/17425247.2023.2274902
165. Tran T, Gonzalez Perdomo ME, Haghighi M, Amrouch K. Effects of cationic and anionic surfactants on the stability, rheology and proppant suspension of nanoparticle-stabilized fracturing foams at elevated temperature. Geoenergy Sci Eng. 2023;228. doi:10.1016/j.geoen.2023.212041
166. Bandi SP, Kumbhar YS, Venuganti VVK. Effect of particle size and surface charge of nanoparticles in penetration through intestinal mucus barrier. J Nanopart Res. 2020;22(3). doi:10.1007/s11051-020-04785-y
167. Sarwar A, Katas H, Zin NM. Antibacterial effects of chitosan-tripolyphosphate nanoparticles: impact of particle size molecular weight. J Nanopart Res. 2014;16(7). doi:10.1007/s11051-014-2517-9
168. Liu W, Pan H, Zhang C, et al. Developments in methods for measuring the intestinal absorption of nanoparticle-bound drugs. Int J Mol Sci. 2016;17(7). doi:10.3390/ijms17071171
169. Shah MK, Madan P, Lin S. Preparation, in vitro evaluation and statistical optimization of carvedilol-loaded solid lipid nanoparticles for lymphatic absorption via oral administration. Pharm Dev Technol. 2014;19(4):475–485. doi:10.3109/10837450.2013.795169
170. Patel M, Desai A, Kansara V, Vyas B. Core shell lipid-polymer hybrid nanoparticles for oral bioavailability enhancement of ibrutinib via lymphatic uptake. AAPS Pharm Sci Tech. 2023;24(6). doi:10.1208/s12249-023-02586-9
171. Niu Q, Gao J, Wang L, Liu J, Zhang L. Regulation of differentiation and generation of osteoclasts in rheumatoid arthritis. Front Immunol. 2022;13. doi:10.3389/fimmu.2022.1034050
172. Nabi B, Rehman S, Baboota S, Ali J. Insights on oral drug delivery of lipid nanocarriers: a win-win solution for augmenting bioavailability of antiretroviral drugs. AAPS Pharm Sci Tech. 2019;20(2). doi:10.1208/s12249-018-1284-9
173. Dening TJ, Rao S, Thomas N, Prestidge CA. Oral nanomedicine approaches for the treatment of psychiatric illnesses. J Control Release. 2016;223:137–156. doi:10.1016/j.jconrel.2015.12.047
174. Jawahar N, Hingarh PK, Radhakrishnan A, et al. Enhanced oral bioavailability of an antipsychotic drug through nanostructured lipid carriers. Int J Biol Macromol. 2018;110:269–275. doi:10.1016/j.ijbiomac.2018.01.121
175. Vora LK, Gholap AD, Jetha K, Thakur RRS, Solanki HK, Chavda VP. Artificial intelligence in pharmaceutical technology and drug delivery design. Pharmaceutics. 2023;15(7). doi:10.3390/pharmaceutics15071916
176. Simões MF, Silva G, Pinto AC, et al. Artificial neural networks applied to quality-by-design: from formulation development to clinical outcome. Eur J Pharm Biopharm. 2020;152:282–295. doi:10.1016/j.ejpb.2020.05.012
177. Nagy B, Galata DL, Farkas A, Nagy ZK. Application of artificial neural networks in the process analytical technology of pharmaceutical manufacturing—a review. AAPS J. 2022;24(4). doi:10.1208/s12248-022-00706-0
178. Grumbach C, Czermak P. Process analytical technology for the production of parenteral lipid emulsions according to good manufacturing practices. Processes. 2022;10(6). doi:10.3390/pr10061174
179. Thapa RK, Kim JO. Nanomedicine-based commercial formulations: current developments and future prospects. J Pharm Investig. 2023;53(1):19–33. doi:10.1007/s40005-022-00607-6
180. Caputo F, Favre G, Borchard G, et al. Toward an international standardisation roadmap for nanomedicine. Drug Deliv Transl Res. 2024;14(9):2578–2588. doi:10.1007/s13346-024-01646-2
181. Alghamdi MA, Fallica AN, Virzì N, Kesharwani P, Pittalà V, Greish K. The promise of nanotechnology in personalized medicine. J Pers Med. 2022;12(5). doi:10.3390/jpm12050673
182. Maojo V, Fritts M, de la Iglesia D, et al. Nanoinformatics: a new area of research in nanomedicine. Int J Nanomed. 2012;7:3867–3890. doi:10.2147/IJN.S24582
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.