")
Back to Journals » International Journal of Nanomedicine » Volume 20
Biodegradable Electrospun PLGA Nanofibers-Encapsulated Trichinella Spiralis Antigens Protect from Relapsing Experimental Autoimmune Encephalomyelitis and Related Gut Microbiota Dysbiosis
Authors Sabljić L , Radulović N, Đokić J, Stojanovic DB , Radojević D, Glamočlija S, Dinić M, Golić N, Vasilev S , Uskoković P, Sofronić-Milosavljević L, Gruden-Movsesijan A, Tomić S
Received 21 October 2024
Accepted for publication 18 January 2025
Published 12 February 2025 Volume 2025:20 Pages 1921—1948
DOI https://doi.org/10.2147/IJN.S499161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. RDK Misra
Ljiljana Sabljić,1 Nataša Radulović,2 Jelena Đokić,3 Dusica B Stojanovic,4 Dušan Radojević,3 Sofija Glamočlija,1 Miroslav Dinić,3 Nataša Golić,3 Saša Vasilev,1 Petar Uskoković,4 Ljiljana Sofronić-Milosavljević,1 Alisa Gruden-Movsesijan,1,* Sergej Tomić1,*
1Institute for the Application of Nuclear Energy, University of Belgrade, Belgrade, Serbia; 2Institute for Biological Research “Siniša Stanković”, University of Belgrade, Belgrade, Serbia; 3Institute for Molecular Genetics and Genetical Engineering, University of Belgrade, Belgrade, Serbia; 4Faculty of Metallurgy and Technology, University of Belgrade, Belgrade, Serbia
*These authors contributed equally to this work
Correspondence: Sergej Tomić, Principal Research Fellow, Institute for the Application of Nuclear Energy, University of Belgrade, Banatska 31b, Zemun, Belgrade, 11080, Serbia, Tel +381 11 2610 126, Fax +381 11 2618 724, Email [email protected]
Purpose: Trichinella spiralis has evolved complex immunomodulatory mechanisms mediated by excretory-secretory products (ESL1) that enable its survival in the host. Consequently, ESL1 antigens display excellent potential for treating autoimmune diseases such as multiple sclerosis (MS). However, whether timely controlled delivery of ESL1 antigens in vivo, as in natural infections, could enhance its therapeutic potential for MS is still unknown.
Methods: To test this, we encapsulated ESL1 antigens into biodegradable poly (lactide-co-glycolic) acid (PLGA) nanofibers by emulsion electrospinning as a delivery system and assessed their release dynamics in vitro, and in an animal MS model, experimental autoimmune encephalomyelitis (EAE), induced 7 days after PLGA/ESL1 subcutaneous implantation. PLGA/ESL1 effects on EAE symptoms were monitored along with multiple immune cell subsets in target organs at the peak and recovery of EAE. Gut barrier function and microbiota composition were analyzed using qPCR, 16S rRNA sequencing, and metabolomic analyses.
Results: ESL1 antigens, released from PLGA and drained via myeloid antigen-presenting cells through lymph nodes, protected the animals from developing EAE symptoms. These effects correlated with reduced activation of myeloid cells, increased IL-10 expression, and reduced accumulation of proinflammatory natural killer (NK) cells, T helper (Th)1 and Th17 cells in the spleen and central nervous system (CNS). Additionally, CD4+CD25hiFoxP3+ regulatory T cells and IL-10-producing B cells were expanded in PLGA/ESL1-treated animals, compared to control animals. The migration of ESL1 to the guts correlated with locally reduced inflammation and gut barrier damage. Additionally, PLGA/ESL1-treated animals displayed an unaltered microbiota characterized only by a more pronounced protective mevalonate pathway and expanded short-chain fatty acid-producing bacteria, which are known to suppress inflammation.
Conclusion: The delivery of T. spiralis ESL1 antigens via biodegradable electrospun PLGA nanofiber implants efficiently protected the animals from developing EAE by inducing a beneficial immune response in the spleen, gut, and CNS. This platform provides excellent grounds for further development of novel MS therapies.
Keywords: electrospinning, PLGA nanofibers, drug delivery, tolerogenic cells, immune modulation, gut microbiota
Graphical Abstract:

Introduction
Multiple sclerosis (MS) is a chronic inflammatory life-threatening autoimmune disease of the central nervous system (CNS), causing progressive neurological disability and considerably undermining the quality and longevity.1 Molecular mimicry has been implicated in the initiation of MS,2 but the underlying cause and the complex immune regulation of MS pathology remain elusive.3 Although available disease-modifying therapies can alter the course of the disease, their efficacy over a long-term period is still insufficient and followed by adverse effects,4 emphasising the need for developing more efficient immunotherapies for MS. The driving mechanisms of MS pathogenesis, and its animal model experimental autoimmune encephalomyelitis (EAE), were shown to include a disbalance in the effector and regulatory arms of multiple immune cell subsets, including T, B, natural killer (NK) cells, and particularly myeloid cells.3,5,6 Moreover, a dysregulated interplay between gut microbiota and the immune system was described as critical for the regulation of MS and EAE pathogenesis.7,8 The gut microbiome is a community of about 1014 different populations of microorganisms inhabiting the intestinal tract (bacteria, viruses, fungi, etc). with all its interactions and activities,9 thus representing the largest environmental factor for autoimmune diseases in genetically susceptible individuals.10 Accordingly, the hygiene hypothesis may well frame an increase in autoimmune disease incidence in environments with less infectious agents and poorer microbial diversity, causing dysregulated immune responses.10
Lack of exposure to infectious agents, like parasite worms - helminths, has been proposed as one of the important causes for dysregulation of the immune system in MS.11,12 Trichinella spiralis is a unique parasitic worm that completes its whole life cycle in one host, residing in the Nurse cell (transformed muscle cell) and communicating with the host organism through excretory-secretory products of the muscle larvae (ESL1).11 Through ESL1 antigens, this parasite creates an environment dominated by anti-inflammatory and regulatory immune responses, unfavourable for developing autoimmune diseases.13–15 We have found previously that the infection with T. spiralis, by applying its ESL1 products, alone or via dendritic cells (DCs), can effectively ameliorate EAE.14,16,17 By triggering the regulatory cytokines (IL-10 and TGF-β) in tolerogenic DCs via different innate receptors and lowering the pro-pathogenic T helper (Th)1 and Th17 cells responses while increasing the number and functions of regulatory T cells (Tregs),18,19 ESL1 antigens can restore dysregulated immune response.14,16,17,20 ESL1 products represent a complex mixture of at least 43 major glycoproteins, including their post-translationally modified isoforms.21 The three glycoproteins (53, 49 and 43 kDa) bearing immunodominant epitopes were demonstrated as potent anti-inflammatory proteins able to induce tolerogenic DCs,22 via toll-like receptor (TLR)2, TLR418 and DC-SIGN.19 ESL1 also contains galectin-1-like proteins which, similar to Galectin-1, increase the capacity of DCs to induce Tregs.23 Furthermore, we were the first to demonstrate that ESL1 also contains 30–80 nm extracellular vesicles (EVs) with a potent immunoregulatory capacity in human cell cultures in vitro.24 T. spiralis-derived EVs alone contain complex structure,25 and they demonstrated the capacity to suppress inflammation in a mouse model of colitis.26 These results illustrate that ESL1 antigens contain different complex components that could be harnessed to effectively ameliorate inflammation in autoimmune diseases, including MS.20 However, this complexity of ESL1 antigens represents a great challenge for their massive production, which is required for their clinical use. Therefore, new innovative approaches are needed to enhance the therapeutic potency of ESL1 products.
Advances in nanotechnologies enabled the development of highly biocompatible drug delivery systems, which can enhance the efficacy of drugs while minimising their toxicity, overall improving drug formulation, delivery efficiency, and manufacturing processes.27 Thereby, biodegradable polymer-based nanomaterials, such as poly (lactic-co-glycolic) acid (PLGA), were shown to improve biodistribution and absorption of therapeutics to the diseased site.28 In contrast to widely studied PLGA micro/nanoparticles (NPs), displaying either anti-inflammatory or proinflammatory effects,29 the immunomodulatory properties of electrospun PLGA nanofibers are less well known. Biodegradable polymers exhibit excellent properties for drug delivery systems, including a high surface-to-volume ratio, high potential for drug encapsulation, easy and low-cost production, and simple surface functionalization chemistry. Nanofibers offer several advantages over nanoparticles, as they have a significantly larger surface area-to-volume ratio, lightweight with small diameters, and controllable pore structures, and generally have better mechanical properties, including modulus elasticity, strength, flexibility, and ease of production in large quantities making nanofibers more accessible for industrial applications.30 Nanofibers with core-shell structure fabricated by emulsion or co-axial electrospinning process31,32 can provide a better controlled and sustained release behaviour than those by emulsification process of the nanoparticles that have a burst drug release profiles.30 Moreover, we showed previously that nanofibers made from poly (ε-caprolactone) and PLGA-based nanofibers prepared by sequential electrospinning are excellent drug delivery systems for timely-controlled drug release, enabling 9% of the drug release within 60 days,33 suggesting the possibility of a continuous drug release for up to 2 years. The approval of PLGA-based biodegradable polymers for human application34 opened the opportunity to prepare an ESL1-based implant that could continuously modulate the autoimmune process in patients. The emulsion electrospinning creates nanofibers with a core-shell structure through a single nozzle. This method uses a stable mixture of two or more immiscible liquids, stabilized by surfactants. In the process, one liquid forms the outer shell of the fibers, while the other forms the core. The processing conditions for emulsion electrospinning differ from blend and co-axial methods, leading to unique nanofiber properties.35,36 Therefore, this study aimed to prepare PLGA/ESL1-based nanofiber implants by emulsion electrospinning and investigate whether they can regulate inflammatory processes in an animal model of relapsing EAE. Additionally, we evaluated the effects of PLGA/ESL1 implants on pathogenic and regulatory immune cell subsets, both at the systemic and CNS levels, as well as their effects on gut microbiota composition, all of which represent promising therapeutic targets of imbalanced immune responses in autoimmune diseases such as MS.
Materials and Methods
Preparation of ESL1 Antigens
Adult male Wistar rats and Dark Agouti (DA) rats were obtained from the Military Medical Academy (MMA, Belgrade, Serbia) and housed under standard conditions of a 12/12 light/dark cycle in an animal facility with ad libitum access to water and food. All procedures and experiments involving animals were conducted approved and performed following the guidelines and regulations of the local ethics committee of the Institute for the Application of Nuclear Energy—INEP, University of Belgrade, Serbia, Belgrade (No. 323–07-11,160/2019-05/1 and No. 323–07-00758/2021-05/1). Wistar rats, 2–4 months old were used to propagate T. spiralis strain ISS 7564. Rats were infected by gastric inoculation with 8500 T. spiralis muscle larvae (L1 larvae). L1 larvae were recovered after 2 months by digesting the carcasses in pre-warmed gastric juice (1% pepsin in 1% HCl, pH 1.6–1.8).37 Muscle larvae were kept under controlled conditions (37 °C, 5% CO2) in complete Dulbecco’s modified Eagle medium (Sigma-Aldrich), supplemented with 10 mm HEPES, 2 mm L-glutamine, 1 mm sodium pyruvate, and 50 U/ mL of penicillin–streptomycin (all from Sigma-Aldrich) for 18 h. ESL1 products were purified from culture supernatants according to a previous procedure,18,19,22–24,38 and kept at −20°C for further use.
For tracking experiments, ESL1 in a quantity of 2 mg was labelled with Fluorescein 5-isothiocyanate (FITC) (Sigma) using 15 µg of FITC per 1 mg of protein. FITC was dissolved in 0.15 M Na2HPO4 x H2O buffer (pH 9.0) and slowly added to the ESL1 solution with continual mixing on the magnetic stirrer device, providing labelling of the protein for 90 min at room temperature. Purification of the labelled ESL1 was performed by gel filtration using a Sephadex G-25 column. Upon separation, the absorbance of FITC-labelled ESL1 (ESL1-FITC) was measured at 280 nm and 495 nm using a spectrophotometer (Ultrospec, Pharmacia Biotech), thus providing information on the protein concentration (Cp=1.4 mg/ mL) and fluorescein/protein ratio (F/P=3.0).
Preparation and Characterization of PLGA/ESL1 Implants
PLGA (average molecular weight MW 30,000–60,000 g/mol), with a mass ratio of lactide: glycolic acid units of 50:50, span-80, N, N-dimethylformamide (DMF), and chloroform (CHCl3) were purchased from Sigma-Aldrich (St. Louis, MO, USA). PLGA 50:50 (PLA/PGA) was chosen because of its predicted degradation rate of 1–2 months,33 which is suitable for the EAE model (lasting up to 1–1.5 months) and can be controlled by the amount of glycolic acid and hydrophilicity of the matrix.33,39 First, PLGA (2.0 g) was dissolved in a chloroform/DMF mixture (8.25/2.75 g) and magnetically stirred at 200 rpm at room temperature for 24 h. After adding biodegradable surfactant Span-80 (33.0 mg) to this solution, ESL1 (or ESL1-FITC) products in 2 mL PBS (2.5 mg/mL) were added, and the mixture was stirred for 2 h. Water-in-oil (W/O) emulsion was prepared as an electrospinning solution, and the oil and water phases were prepared separately. Thus, the emulsion containing 20 wt. % PLGA and 0.25 wt.% ESL1 was prepared (relative to the weight of the polymer). Control PLGA implants were prepared similarly, but without the ESL1 products, by dissolving the polymer in a suitable solvent (solution electrospinning). A vertical electrospinning experimental setup (CH-01, Linari Engineering, Pisa, Italy) was used to prepare nanofibers. To obtain fine nanofibers, the electrospinning conditions were optimised as follows: polymer solutions were ejected from a 20 mL plastic syringe into a metallic needle (1 mm inner diameter) at a distance of 10 cm from the collector, with a flow rate of 3 mL/h and an applied voltage of 20 kV. Electrospun nanofibers were collected on a piece of grounded aluminium foil on a surface of ~100 cm2 containing a total of 5 mg ESL1 (50 µg/cm2).
The morphology of the nanofibers was examined using a field-emission scanning electron microscope (FE-SEM) at 20 kV in conjunction with a Tescan Mira3 XMU microanalysis system (Tescan Orsay Holding AS, Brno, Czech Republic). In each system, 50 nanofibers from five FE-SEM images were selected, and the micrographs were analysed using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA) to obtain their diameter distribution.
The surface chemistry of the prepared nanofibrous material and ESL1 was analysed using Attenuated Total Reflection Fourier-Transform Infrared (ATR–FTIR) spectrometry (Nicolet iS10 (Thermo Fisher Scientific Inc., Waltham, MA, USA). The transmittance spectra were measured in the range of 4000–500 cm−1 with a spectral resolution of 4 cm−1 following 32 scans. The results obtained from ESL1 were analysed using the spectral analysis software OMNICTM (Thermo Fisher Scientific Inc). in the range 1800 cm−1–1400 cm−1.
The glass transition temperatures (Tg) of the PLGA and PLGA/ESL1 nanofibers were determined using differential scanning calorimetry (DSC) (Q10, TA Instruments, USA). The measurements were conducted in the temperature range of 30 °C to 70 °C at a heating rate of 10 °C/min. The sample weight used was 4 ± 0.5 mg, and the heat flow was monitored under a nitrogen purge gas flow of 50 mL min−1. The Tg values were obtained by determining the midpoint between the intersection of the two parallel baselines before and after Tg. Universal Analysis 2000 software was used to calculate the Tg, and a minimum of three measurements were performed for each sample to obtain the average value. The presence of ESL1-FITC in the PLGA nanofibers was also analysed by fluorescence microscopy (Carl Zeiss AxioImager A1, Jena, Germany), in which the ESL1-FITC/PLGA nanofibers were collected on microscopic slides during electrospinning.
ESL1-FITC Release From PLGA in vitro
To measure in vitro release of ESL1-FITC from PLGA nanofibers, nanofiber sheets were cut into squares (1 cm × 1 cm) containing approximately 50 µg of FITC-ESL1 and placed in 24-well sterile microplates containing 1 mL of phosphate buffer (PBS, pH 7.4): wound exudate mixed in a 1:1 ratio (1 mL/well) (WE) to model the inflammatory microenvironment of the implant. The wound exudate was isolated from a sterile inflammation site induced by the subcutaneous implantation of polyvinyl sponges into DA rats (1 cm x 1.5 cm x 0.25 cm), as described previously.40 Briefly, two sponges per animal were implanted at the dorsal site of the skin under general ketamine/xylazine anaesthesia. After 3 days, the animals were euthanised, the sponges were harvested and squeezed with a syringe, and cell-free exudates were collected by pelleting the cells by centrifugation at 2000 rpm for 10 min.
The PLGA/ESL1 nanofibers were incubated at 37 °C and every 3 days for a total of 42 days, 0.5 mL of 50% WE from each well was removed for the analysis and an equal volume of fresh WE mixture was replenished. For blank controls, an equivalent amount of control PBS/PLGA nanofibers was incubated and the supernatants were collected. The fluorescence emission was assessed at 519 nm using a spectrofluorometer (Shimadzu). The concentration of ESL1 was calculated based on the measured fluorescence, according to the standard curve of ESL1-FITC. The percentage of ESL1 released from PLGA nanofibers per 3 days and the cumulative release were calculated accordingly.
The amount of ESL1 encapsulated in the PLGA nanofibers was measured to assess the encapsulation efficiency, defined as the ratio of ESL1 encapsulated in the PLGA nanofibers to the initial quantity of ESL1 added to the spinning solution. Furthermore, the drug loading was calculated by determining the amount of ESL1 encapsulated in the PLGA nanofibers relative to the total amount of PLGA ESL1 nanofibers.
ESL1-FITC Uptake in vitro
Splenocytes (1x107) isolated from DA rats were placed in 2 mL of 6-well plates in complete RPMI 1640 medium (supplemented with 10% FCS, l-glutamine, 2-mercaptoethanol (all from Sigma), and antibiotics (penicillin, streptomycin, Pan Biotech)). The cells were incubated at 37 °C in a humidified incubator with a 5% CO2 atmosphere for 3 h and then treated with ESL1-FITC or non-labelled ESL1 (50 µg/ mL) for the next 24 h. After each time point, the were washed in PBS/0.01% NaN3 and stained with anti-CD11b-APC and anti-MHC class II (I-A) (OX-6)-phycoerythrin (PE)(both from Bio-Rad, Hercules, California, USA), as described by flow cytometry. The percentage of ESL1-FITC positive cells within the CD11b+ MHC class II+ population (myeloid antigen-presenting cells (APC)) was evaluated in splenocytes treated with unlabelled ESL1 at each time point.
Experimental Treatments and EAE Induction
PLGA/ESL1 nanofiber sheets were cut into squares of 1×1 cm (thickness ~ 0.1 cm) with approximately 50 µg of ESL1 per membrane piece. Seven pieces were slightly compressed and implanted subcutaneously in the suprascapular region under sterile conditions; therefore, each treated animal received 350 µg ESL1 (PLGA/ESL1 group). Namely, DA rats were completely anaesthetised with ketamine (200 mg/kg) and their backs were shaved and disinfected with 70% alcohol. A small incision was made in the lower back and the implants were placed deep in the suprascapular region with tweezers. The skin was sutured and disinfected and the animals were monitored daily by a veterinarian. Control groups included animals treated with control PBS/PLGA nanofibers (PLGA group), sham-operated animals treated subcutaneously with ESL1 (350 μg/animal in 200 µL PBS) at the same time (ESL1 group), and sham-operated animals (control group).
EAE was induced as described previously,16,41 7 days after the experimental treatments. Briefly, DA rats were treated with a single intradermal injection (0.1 mL) into the right hind footpad with an encephalitogenic emulsion prepared by mixing rat spinal cord (SC) tissue homogenate (50% w/v in saline) emulsified in an equal volume of complete Freund’s adjuvant (CFA; Difco, Detroit, MI) supplemented with 4 mg/ mL Mycobacterium tuberculosis (Difco). Eight days post-immunization (pi), rats were weighed and scored daily for signs of the disease according to a scheme, 0 - no clinical signs, 1 - flaccid tail, 2 - hind limb paresis, 3 - complete bilateral hind limb paralysis and incontinence, and 4 - moribund state or death. Intermediate scores (± 0.5) were assigned if neurological signs were between typically observed symptoms. Several disease parameters were examined to evaluate the severity of EAE: the cumulative index (the sum of the daily mean clinical scores for each animal over a given number of days throughout the monitoring period), maximal clinical score (the maximal clinical score that each animal developed throughout the experiment), day of onset, and duration of the disease (total number of days with EAE symptoms). Additionally, the total incidence of EAE, as well as the incidence of a particular score (ie 2-paresis or 3-paralysis), was calculated using log-rank analysis. Day 14th upon EAE induction showed a peak of the disease, whereas on day 20th the animals entered the remission phase. An additional peak, relapse, was observed between days 24–28. For each experiment, 5 animals per group were euthanized for cell isolation from spleens and CNS, either at the peak of the disease (day 14th) or during the recovery phase (day 20th) for further analysis, and at least two independent experiments were carried out, as indicated in figure legends.
Cell Isolation and Cultivation
Single-cell suspensions from the spleen, lymph nodes, and CNS isolated at the peak and recovery of EAE were prepared. The cells infiltrating CNS were obtained from the spinal cords (SC) of rats perfused with sterile PBS, as described.41 The spinal cords were homogenized, centrifuged at 100 g, 10 min, +4°C, and the pellet was resuspended in 3 mL of 40% Percoll (Sigma-Aldrich) and overlaid on 3 mL of 70% Percoll gradient. Following centrifugation at 850 × g for 50 min, mononuclear cells were recovered from the 40% / 70% Percoll interface and washed with RPMI medium (Sigma-Aldrich). Spleen and lymph node cells were isolated aseptically, and the spleen cells were lysed with a lysis buffer (155 mm NH4Cl, 12 mm NaHCO3, 0.1 mm EDTA in ddH20) for 5 minutes, followed by washing in PBS. The total number and viability of the cells were determined using a Cell Count Viability kit on a Cell Muse analyser (Cytek Biosciences), and the cells were prepared for flow cytometry analysis. Additionally, splenocytes isolated from PLGA/ESL1 or sham-operated control animals at the peak or recovery of EAE were seeded in 24-well plates (2 x 106/ mL) and cultivated in complete RPMI-1640 medium for 48h to assess the spontaneous production of cytokines in culture. After culturing, cell-free supernatants were collected and stored at −20 °C until analysis by ELISA.
ESL1-FITC/PLGA Tracking in vivo
To assess the release of ESL1-FITC from PLGA nanofibers in vivo, DA rats were implanted with either ESL1-FITC/PLGA or PLGA/ESL1 as a control (21 animals/group). After 3, 4, 7 or 14 days, the animals were euthanized for collection of the peri-implant region (on day 4, 2 animals per group), draining lymph nodes from the axillary (AxLN), cervical (CLN), and inguinal (IngLN) regions, spleen, Payer’s patches, and spinal cords (on days 3, 7 and 14 after implantation, 4 animals per group in each term). Additionally, four animals per group were immunised 7 days after implantation with SC homogenate in complete Freund’s adjuvant (CFA) to induce EAE, and the organs were collected 7 days after immunisation (ie 14 days after implantation). Single-cell suspensions were prepared as described previously, and the cells were stained with anti-CD11b-APC and anti-MHC class II (I-A)-PE (Bio-Rad) to monitor ESL1-FITC presence within myeloid antigen-presenting cells by flow cytometry. To compare the trafficking of released ESL1-FITC with the trafficking of free ESL1-FITC, 3 animals/group were injected with either 350 µg/mL of ESL1-FITC or non-labelled ESL1 subcutaneously in the same suprascapular region, and the myeloid APCs were analysed 3, 7 or 14 days after the injection. The percentage of FITC-positive cells within the CD11b+ MHC II+ population was evaluated according to the corresponding cell isolates from PLGA/ESL1- or free ESL1-treated animals.
Flow Cytometry
Single-cell suspensions prepared from the spleen and SCs were analysed by flow cytometry for surface and intracellular antigens. The cells were washed once in PBS containing 0.01% Na-azide (Sigma-Aldrich), followed by blocking Fc receptors with 2% normal rat serum (eBioscience, Frankfurt am Main, Germany) for 15 minutes, followed by labelling with fluorochrome-conjugated monoclonal antibodies (mAbs) at dilutions recommended by the manufacturer. The incubation for surface staining lasted 30 minutes at +4°C. After the surface staining, the cells were fixed and permeabilised using the fixation/permeabilization kit (R&D Systems) according to the manufacturer’s recommendation, followed by intracellular staining. Intracellular staining of IFN-γ, IL-10 and IL-17 was performed after 6-hour activation of the cell with phorbol-12-myristate-13-acetate (PMA, 20 ng/mL) and ionomycin (500 ng/mL) in the presence of monensin (2 µM) (all from Sigma-Aldrich). The following mAbs were used: mouse IgG1 negative control-Alexa Fluor 488 (F8-11-13), mouse IgG1 negative control-phycoerythrin (PE) (F8-11-13), anti CD68-Alexa Fluor 647 (ED1), anti-CD4-biotin (clone W3/25), anti-CD3-Alexa Fluor 647 (1F4), anti-CD11b-FITC (OX-42), anti-CD11b-APC (OX42), anti-MHC class II-PE (OX-6), (all from Bio-Rad, Hercules, California, USA); mouse IgG1 negative control-biotin (AB_2550616), anti-IL-17A-FITC (eBio17B7), anti-CD45R antibody-FITC (HIS24), anti-CD161-Biotin (10/78), CD86-Pe/Cyanin 5 (GL1), mouse IgG1 negative control (MOPC-21)-Alexa Fluor 647 (all form Thermo Fisher Scientific, Waltham, Massachusetts, USA); anti-CD25-PE (OX-39), anti-IL-10-PE (A5-4), anti-IL-4-PE (OX-81), anti-CD103-biotin (OX-62) (all form BD Pharmingen, San Diego, California, USA); anti-FoxP3-APC (FJK-16s), mouse IgG1 kappa negative control-Allophycocyanin (APC) (P3.6.2.8.1) (eBioscience); anti-IFN-γ-FITC (DB-1), anti-CD8-PECy7 (OX-8), anti-CD45-PE (OX1) PE/Cyanine7 Streptavidin, PerCP-Cyanine 5.5 streptavidin, APC/Cyanine7 Streptavidin, 7AAD viability dye (BioLegend, San Diego, California, USA); rabbit polyclonal anti-mouse IgG (whole molecule)-FITC (Sigma-Aldrich). For each analysis, at least 10,000 cells in the leukocytes gate were acquired on an 8-colours BD LSR II, gated according to their specific side-scatter (SSC)/forward-scatter (FSC) properties, 7-AAD- (viable cells), and avoiding cells with low SSC/FSC properties (predominantly dead cell debris). Non-specific fluorescence was determined using fluorescently-labelled isotype antibodies and fluorescent minus one-negative controls and the signal overlap was compensated for before each experiment using single-labelled cells.
Microscopy Analyses
The spinal cords (SCs) were isolated from 4 animals per group at the peak and recovery phases of EAE by opening the spine on the front and caudal sides of euthanised animals and then expulsing the SCs by strong aspiration with PBS. The lumbar region of the spinal cord was collected using a scalpel, submerged in buffered formalin (Sigma) for 48–72h, and then embedded in paraffin using the Modular Tissue Embedding Centre EC 350 (Especialidades Médicas Myr, Spain) after dehydration in ethanol and xylol. In addition, peri-implant regions and draining lymph nodes were isolated during ESL1-FTIC/PLGA tracking and formalin fixation experiments. Thin cross-sections (~ 5μm) were prepared using a Manual Rotary Microtome (Biobase, China) and the tissues collected on the slides were rehydrated and stained with Mayer’s Haematoxylin and Eosin Y (both from Fisher Scientific). Histological quantification of cellular infiltrates in SCs was performed using H&E-stained sections. The number of inflammatory lesions (infiltrates) and cells per infiltrate were counted per cross section. Inflammatory infiltrates were considered positive only if they were present in three or more consecutive cross sections. At least five slides, each containing 3–4 cross-sections, separated by ~50μm, were analysed for each animal.
Additionally, the slides were prepared for epi-fluorescent microscopy by antigen retrieval in citrate buffer (10mM Citric Acid, 0.05% Tween 20, pH 6.0) at 95°C for 20 min, followed by spontaneous cooling to room temperature. Rabbit anti-myelin base protein (MBP, 1:100; Abcam) and anti-rat CD45-biotin (clone 30-F11, BioLegend, 1:50) were incubated on slides overnight at 4°C, the slides were washed in PBS, and then incubated with secondary anti-rabbit IgG (H+L) -Alexa 488 (1:500) for 2 h and streptavidin – Alexa 546 (1:500) for 30 minutes (both from Thermo Fisher). Alternatively, peri-implant tissues from PLGA/ESL1- and PLGA/ESL1-FITC-treated animals were either H&E stained or incubated with anti-FITC antibody-biotin (BioLegend) (1:200) overnight and after washing in PBS, with streptavidin-Alexa 546. The slides for epifluorescent microscopy were then incubated with DAPI nuclear stain (Thermo Fisher 1:10000) for 15 minutes, washed in PBS, and mounted in a fluorescence mounting medium (Sigma) before analysis. The slides were analysed using a Zeiss AxioImager A1 under light microscopy or under a UV filter set for DAPI (UV-2B, ex: 330–380 nm, DM 400, BA 435), a green filter set for detection of MBP (B-2A, ex: 450–490 nm, DM 505, BA 520), and a red filter set for detection of CD45 or ESL1 antigens (G-2A, ex: 510–560 nm, DM 575, BA 590) in epifluorescent microscopy. The images for epifluorescent microscopy were acquired as monochromatic and later merged and analyzed offline using the ImageJ software (National Institutes of Health, Bethesda, Maryland, USA).
Cytokines Measurements
Cytokines were measured in the supernatants of 48h spleen cell cultures isolated from animals at the peak and recovery phases of EAE. The levels of IL-17, IL-6, IFN-γ, TNF-α, IL-4, and IL-10 were determined using sandwich ELISA kits (R&D Systems) according to the manufacturer’s protocol. The unknown concentrations of cytokines were determined from standard curves using 5-parameter non-linear fit curves (GraphPad Prism).
RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
Total RNA extraction from intestinal samples collected at the peak of EAE was performed using liquid nitrogen homogenization of tissue, followed by resuspension in 500 μL denaturing solution as described previously.42 The next steps included phenol/chloroform extraction, repeated twice, and RNA precipitation with isopropanol. The isolated RNA was treated with the Rapid Out DNA Removal Kit according to the manufacturer’s protocol (Thermo Fisher Scientific). The Revert Aid Reverse Transcriptase (Thermo Fisher Scientific) with random hexamers (Thermo Fisher Scientific) and Ribo Lock RNase inhibitor (Thermo Fisher Scientific) were used to transcribe 0.5 µg of isolated RNA. The target gene expression analysis was performed using 7500 real-time PCR system (Applied Biosystems) with IC Green qPCR Universal Kit (NIPPON Genetics) under the following conditions: 2 min at 95 °C activation, 40 cycles of 5s at 95 °C and 30s at 60 °C. All results were normalized against the β-actin (Actb) gene and expressed as relative target abundance by using the 2−ΔΔCt method.43 All primers for gene expression analysis, presented in Supplementary Table S1, were purchased from Thermo Fisher Scientific.
DNA Extraction, Determination of Segmented Filamentous Bacteria Abundance and Sequencing
The stool samples were collected from all rats before the EAE induction (ie 7 days after the implantation), and at the peak of the disease, and they were stored at −80 °C before extraction. Genomic DNA was extracted using ZR Fecal DNA MiniPrep Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s instructions. The integrity and concentration of extracted DNA were measured using a BioSpec-nano spectrophotometer (Shimadzu, Kyoto, Japan) and Qubit fluorometer (Thermo Fisher Scientific, USA). DNA concentrations were uniformed to evaluate the relative content of segmented filamentous bacteria (SFB) across animals. To assess the relative quantification of amplicons specific for SFB from isolated DNA, we performed qPCR analysis using primers listed in Supplementary Table S1. The relative SFB abundance was normalized to the total bacteria and expressed as a fold change of SFB in each animal before the induction of EAE and at the peak of the disease. Hypervariable V3-V4 regions of the bacterial 16S rRNA gene were amplified using the 341 F/806 R primer set as previously described,41 followed by library preparation, quality checking, and sequencing on the Illumina NovaSeq 6000 platform at Novogene (Beijing, China) in PE250 mode with 30000 tags per sample.
Microbiota Data Processing and Analyses
Based on their barcode sequences, paired-end raw reads were demultiplexed and truncated by removing barcodes and primers. High-quality demultiplexed raw reads were processed using QIIME2 v2020-844 to obtain clean reads for taxonomy analysis. We performed taxonomy assignment using the classify-sklearn plugin with SILVA 132 database as reference (99% sequence identity). The resulting files, together with the metadata, were imported into R Studio (v2022.07.2) (RStudio team) using the functions from package qiime2R (github.com/jbisanz/qiime2R). The function set seed was set at 999 to ensure reproducibility of the results. Phyloseq45 functions were used to calculate the alpha (Shannon index) diversity metric on a rarified sample size of 9400. Significant differences between groups were examined using one-way ANOVA followed by Tukey’s post-hoc test in GraphPad version 9.0.0 software for Windows (San Diego, California, USA).
Analysis of the composition of microbiomes (ANCOM) protocol46 was used to identify differentially abundant taxa at the different taxonomy levels. Species and genera with W scores above 1 were chosen for additional testing with a paired T-test or Pairwise Wilcoxon test in R Studio, and only statistically significant differences are shown.
Predictive functional profiles of the gut microbiota were generated using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)247 pipeline, based on the feature table and representative sequences with default options. To unveil discriminative predicted functional profiles before immunisation and at the peak of disease, PICRUSt2 results were examined using the linear discriminant analysis Effect Size (LEfSe) pipeline on the Galaxy platform,48 with parameters set to α<0.05, for the factorial Kruskal–Wallis test and all-against-all strategies for analysis between classes and only predicted pathways above the LDA threshold 2.5 were plotted. The normality of the distribution, for continuous variables, was checked using the Shapiro–Wilk test. All p values less than 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). The Benjamini-Hochberg method was used to correct the p-values for multiple comparisons. All plots were generated using the ggplot2 (v3.3.5) package and GraphPad software unless otherwise stated.
Raw sequencing data were deposited in the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena) under the accession number PRJEB79235 and secondary accession number ERP163422.
Statistical Analysis
The distribution of data points was analysed using the Shapiro–Wilk test, and then either one-way ANOVA with Tukey’s multiple comparison test or Kruskal–Wallis test was used to compare the relative mRNA expression and the percentage of immune cells within the tested groups at the peak or recovery of EAE. The relative distribution of immune cells was shown as obtained from flow cytometry analysis or normalized (in stack bars) to total gated cells for each analysis panel. The statistical significance in clinical scores between the time points across the tested groups was determined using two-way ANOVA with Dunnett’s post-hoc test. Disease severity parameters between the groups were examined using one-way ANOVA with Tukey’s multiple comparison test. The Log rank test was used to compare the individual EAE scores between the experimental groups. All statistical analyses were performed using GraphPad Prism (v9.1.0) (GraphPad Software).
Results
Preparation, Characterisation and Release of ESL1 from PLGA Nanofibers
Due to easily controllable release properties33 and good biocompatibility,49 PLGA nanofibers have been used as platforms to deliver immunomodulatory ESL1 antigens. Therefore, according to our previous findings on the release dynamics of a model drug,33,50 the PLA: PGA 50:50 mixture was chosen for emulsion electrospinning with the aqueous phase of ESL1 antigens (2.5 mg/mL in PBS) for a targeted dose of 50 µg ESL1 per cm2 of PLGA.
To analyse the morphology of PLGA/ESL1 and control PLGA nanofibers, the diameters were measured from FE-SEM images (Figure 1A), as indicated previously.50 The data suggested that the average diameter of PLGA/ESL1 nanofibers was 0.431 ± 0.02 µm, with a size range between 0.1 µm and 1.2 µm. The microstructure of the control PLGA nanofibers consisted of a relatively uniform cylindrical shape with an average diameter of 0.387 ± 0.03 µm, with the same size range. A comparison of the ATR-FTIR spectra between PLGA/ESL1 and PLGA confirmed the presence of ESL1 within the nanofibers. The PLGA nanofibers spectrum showed low-intensity signals at 2998 and 2926 cm−1, which were attributed to asymmetric stretching of the C−H bond of the −CH2− and −CH−groups of the aliphatic chain of PLGA, respectively. The intense signal at 1747 cm−1 was attributed to the C=O stretching of the ester, and the signals at 1166 and 1087 cm−1 corresponded to the C−O single bond stretching of the ester group.51 The spectra of the ESL1 antigens showed a characteristic FT-IR spectrum with a broad signal at 3366 cm−1 attributed to the OH stretching of water. The low-intensity signals at 2955 cm−1 and 2929 cm−1 were attributed to the asymmetric and symmetric stretching of methylene C-H bonds, respectively. The absorption signal at 1637 cm−1 is representative of amide I owing to the stretching vibrations of the protein carbonyl group C=O groups. The signal at 1545 cm−1 is attributed to amide II due to N−H mixed with C−N bending. The weaker signal centred at 1263 cm−1 represents the COO stretching vibration of proteins. The symmetric stretching vibration of the O–C group at 1186 cm−1 was attributed to carbohydrates such as glycogen.52 The FT-IR of PLGA/ESL1 spectra did not reveal any significant difference between the functional groups of PLGA nanofibers. However, low-intensity signals at 1637 and 1545 cm−1 due to amides I and II confirmed the successful incorporation of ESL1 antigens in the nanofibers without losing their structural nature during emulsion electrospinning (Figure 1B). The DSC results were used to examine changes in the thermal behaviour of the PLGA nanofibers (Figure 1C). The results indicated that the glass transition temperature of PLGA nanofibers decreased from 47.5 ± 0.21 °C to 43.7 ± 0.42 °C compared to PLGA/ESL1 nanofibers. Thermal analysis of the emulsion nanofibers showed a small decrease in Tg values which demonstrated the plasticising properties of a non-ionic surfactant and the possibility of interactions between Span-80 and PLGA.53 This phenomenon also confirms the filling of PLGA nanofibers during coaxial electrospinning, according to previous findings.54
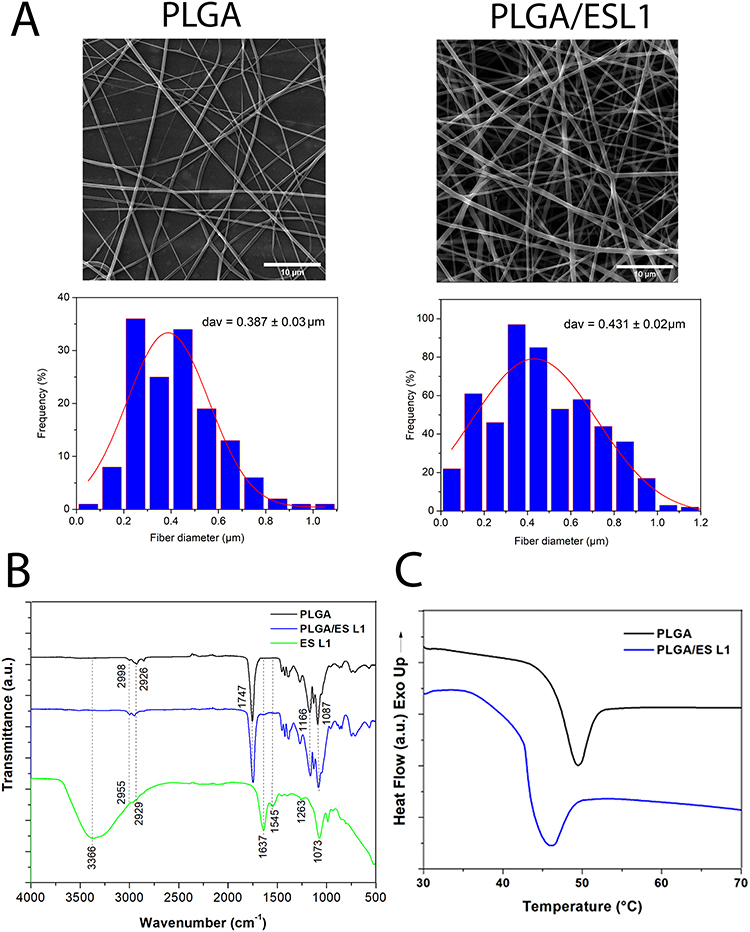 |
Figure 1 Characteristics of PLGA/ESL1 nanofibers and release dynamics of ESL1. (A) Representative FEI-SEM images of PLGA/ESL1 and PBS/PLGA nanofibers are shown, which were used for the measurements of fibre diameter (upper row). The frequency distribution of nanofiber diameters is shown with the fitting curve (lower row). (B) FTIR spectra of PLGA/ESL1 and PLGA are shown with peak transmittance indicated for relevant bonds. (C) DSC analysis of PLGA/ESL1 and PLGA are shown. |
To evaluate the release dynamics of ESL1 from PLGA nanofibers, ESL1-FITC was encapsulated into PLGA fibres as described to obtain 50 µg of ESL1-FITC /cm2 of PLGA. The presence of ESL1-FITC in nanofibers was confirmed using epifluorescence microscopy (Figure 2A). The fluorometry measurements of WE collected every 3 days indicated that the released ESL1 was detectable after the first measurement, and its release increased during the first 27 days, up to a level of 1μg/ mL/day (Figure 2B). Subsequently, ESL1 was still released from PLGA but at a slower rate. Cumulatively, ~90% of the encapsulated ESL1 was released from PLGA in WE during the 40 days of monitoring, with a greater slope during the first period of monitoring (Figure 2C). Based on the obtained results, it was determined that the encapsulation efficiency and drug loading was 99 ± 0.5% and 0.2 ± 0.03%, respectively.
 |
Figure 2 Release dynamics of ESL1 from PLGA in vitro. (A) Representative phase-contrast (upper) and fluorescence (lower) images of PLGA loaded with ESL1-FITC (40x magnification) are shown. (B) Release dynamics of ESL1-FITC (50 µg) from PLGA (1 cm2) incubated in 50% wound exudate in PBS over 44 days. Each dot represents the mean amount ± SD (from triplicates of one out of four independent experiments) of ESL1 collected every 3 days during the incubation. The amount of ESL1 was calculated from the standard curve (inset in the upper right corner). (C) A cumulative release of ESL1-FITC over 44 days is shown as mean ± SD (n=3). |
ESL1 Released from PLGA Effectively Ameliorate EAE
We previously demonstrated that ESL1 antigens were most effective in alleviating relapse-remitting EAE symptoms induced in DA rats, when administered prophylactically in multiple doses before disease induction, compared to single-dose treatment.14 Therefore, to test whether the delivery of ESL1 antigens via degradable PLGA fibres could reproduce this phenomenon, a group of DA rats was implanted subcutaneously with PLGA/ESL1 fibres, containing a total of 350 μg ESL1 /implant, 7 days before disease induction (EAE_PLGA/ESL1). Control groups of animals were at the same time sham-operated and then treated with the equivalent amount of ESL1 subcutaneously (350 μg/rat) (EAE_ESL1), or implanted with control PLGA nanofibers (EAE_PLGA), or just sham-operated (EAE_ctrl), 7 days before disease induction.
After immunisation, the symptoms and weights of the animals were monitored daily for 38 days (Figure 3A and B). The control group (EAE_ctrl) developed a relapsing course of EAE, with initial symptoms appearing between 7 and 10 days after immunisation. The peak of the disease was evident on day 14, followed by partial remission by day 20th, and an additional relapse with a peak on days 24–27. In line with this, the greatest weight loss was observed in the EAE_ctrl group during the first 8 days following immunisation. A similar course of disease and weight changes were observed in animals from the EAE_ESL1 and EAE_PLGA groups. In contrast, the group of animals implanted with PLGA/ESL1 displayed the lowest reduction in weight %, and the lowest severity of EAE scores (Figure 3A and B). According to the area under the curve, only the EAE_ESL1/PGLA group displayed a significant difference compared with the other tested groups. A cumulative index score, as a measure of the average EAE score over the number of days of monitoring, also suggested that the EAE_ESL1/PGLA group displayed significantly lower EAE symptoms than the EAE_ctrl, EAE_ESL1, and EAE_PLGA groups (Figure 3C). The animals from the EAE_PLGA/ESL1 group displayed the lowest maximal clinical score, initial EAE symptoms appearance (day of EAE onset), and total duration of the disease (Figure 3C), all of which suggested a protective effect of PLGA/ESL1 implants in this EAE model.
 |
Figure 3 Effects of PLGA/ESL1 implants on EAE symptoms. The soluble ESL1 (350 µg/dose), or implants of PLGA/ESL1 (containing 350 µg ESL1 / 7 cm2 PLGA) or control PBS/PLGA (7 cm2), were applied subcutaneously in the suprascapular region of DA rats. Seven days after the implantation, the rats were immunised to develop EAE, as described. (A) The weight of each animal was monitored for 18 days, and the data is shown as mean weight % ± SE (N (EAE_PLGA, EAE_ESL1) = 10; N (EAE_PLGA/ESL1, EAE_ctrl) = 20), from two or four independent experiments, respectively. The area under the curve (AUC) for weight %, calculated for each animal, is shown as mean ± SEM. (B) EAE scores over 38 days of monitoring are shown as mean ± SEM (N= as in (A)), and AUC of EAE scores for each animal is presented as mean ± SEM. (C) Cumulative index, maximal clinical score, day of disease onset and duration of the disease are shown as mean ± SEM (N= as in (A), or 10 for duration of disease) and each animal is presented by a dot. (A-C) *p<0.05, **p<0.01, ***p<0.005, ****p<0.001, as indicated (Kruskal–Wallis test with Dunn’s multiple comparison test). (D) Incidence of EAE symptoms (limp tail ≥1, paresis ≥2, paralysis ≥3) in each group during the first 22 days is presented as the log-rank curve. Likewise, the incidence of relapse (score ≥1 after day 22) is shown. *p<0.05 Long-Rank (Mantel-Cox) test vs EAE_ctrl group. |
Log-rank analyses of incidence were performed to assess differences in EAE severity between the tested groups. The results showed that the incidence of EAE (any score) in the EAE_ctrl group, EAE_PLGA, and EAE-ESL1 groups was 100%, whereas only ~75% of animals developed the symptom in the EAE_PLGA/ESL1 group. The incidences of paresis (score ≥2) and paralysis (score ≥3) were also significantly lower in the EAE_PLGA/ESL1 group than in the EAE_ctrl group, whereas the other groups did not differ significantly from EAE_ctrl (Figure 3D). Moreover, the incidence of relapse (ie second EAE peak ≥1) was below 40% in the EAE_PLGA/ESL1 group, as compared to over 80% in animals from other tested groups. These results suggest that the PLGA/ESL1 implant, but not PLGA implantation alone or ESL1 antigens in a single dose, could protect the animals from developing relapsing EAE symptoms.
ESL1 Released from PLGA Migrates Through Draining Lymph Nodes into the CNS and Guts
Implanted materials are sampled by local phagocytic APCs and drained to regional lymph nodes.55 Moreover, we showed previously that ESL1 antigens critically modulate myeloid APC in vitro (in particular DCs), thereby inducing immune tolerance in EAE.14,16 Indeed, when the total splenocytes were cultivated in the presence of FITC-labelled ESL1 antigens for 24h in vitro, the highest uptake of ESL1-FITC (~75%) and the highest fluorescence of FITC-positive cells were found in myeloid APCs (CD11b+ I-A+ cells), followed by CD11b+I-A− and non-myeloid cells (Figure 4A). Therefore, to investigate the migration of ESL1 released from the PLGA fibres in vivo, animals were implanted with ESL1-FITC/PLGA implants or control non-labelled PLGA/ESL1, 7 days before EAE induction to collect the target tissues 4, 7, or 14 days after the implantation for histological and epifluorescent microscopy (Figure 4B, Supplementary Figure S1) and flow cytometry analyses (Figure 4C, Supplementary Figure S2).
 |
Figure 4 Tracking of ESL1-FITC released from PLGA in vivo. (A) ESL1 was labelled with FITC and used in 24h- cultures with rat splenocytes at 50 µg/mL. The live (7AAD-) cells were analysed for CD11b-APC and I-A-PE expression and the Q2 region (CD11b+I-A+) showed the highest fluorescence of ESL1-FITC. Ctrl represents the signal from CD11b/I-A-stained cells not treated with ESL1-FITC. (B) Epi-fluorescent microscopy analyses are shown of the peri-implant region collected 4 days after the implantation of PLGA/ESL1-FITC (upper row); axillary lymph nodes collected 7 days (middle row) after the implantation; the spinal cords collected on 14 days after the implantation and 7 days after EAE induction (lower row). To avoid bleaching and increase ESL1-FITC detection, ESL1-FITC was detected with anti-FITC/Alexa Fluor (AF) 546 mAb, and the nuclei were stained with DAPI (see Supplementary Figure S1). White arrows point to nucleated cells positive for ESL1. (C) The percentage of ESL1-FTIC+ myeloid (CD11b+I-A+) cells from cervical, axillary, inguinal lymph nodes, spleen, Payer’s patches and spinal cord (CNS) were analysed 7 days after the implantation of ESL1-FITC/PLGA, or 14 days after the implantation in immunised (EAE) or non-immunized (healthy) animals, as described. Non-specific fluorescence was determined by using FMO-controls and PLGA/ESL1-treated animals, and the data is shown as mean ± SEM (n=4 animals per each group) (see Supplementary Figure S2 for a representative analysis) from one out of three independent experiments. n.d.-not determined/detected *p<0.05, as indicated (Kruskal–Wallis test with Dunn’s multiple comparison test). |
H&E staining of peri-implant regions of skin and subcutaneous tissues suggested the presence of diffuse immune infiltrates around the implanted material, composed of inflammatory cells, including polymorphonuclear cells, and giant cells, in contrast to the equivalent subcutaneous regions of control skin samples (Supplementary Figure S1A). Epi-fluorescent microscopic analyses of the peri-implant regions 4 days after implantation suggested that ESL1-FITC was released into the surrounding area and could be found within the local infiltrating cells (Figure 4B). The cells in the axillary lymph nodes (AxLN), which drain the suprascapular region of the implant, stained positive in the medullary sinuses and cortex 7 days after the implantation of PLGA/ESL1-FITC, unlike control samples from animals treated with non-fluorescent PLGA/ESL1. Thereby, 0.7% - 5.5% of CD11b+ I-A+ cells were stained over the background in CLN, AxLN, IngLN, spleen, and the Payer’s patches when analysing these lymphoid organs 7 days after the implantation (Figure 4C, Supplementary Figure S2), but not when analysing them 3 days after the implantation. In contrast, when soluble ESL1-FITC was injected directly in the suprascapular region, 0.5%-6.3% of CD11b+I-A+ cells stained positive to ESL1-FITC 3 days after the injection, particularly within CLN and AxLN. In contrast, insignificant level of these cells could be detected 7 or 14 days after the injection (Supplementary Figure S3). The number of mononuclear cells from the spinal cords of non-immunised animals was too low for ESL1 tracking analysis.
Target organ analyses were performed 14 days after implantation in immunised and non-immunized (healthy) animals. In the latter, ESL1 antigen further accumulated in the CLN, AxLN, and spleen (Figure 4C). In contrast, immunised animals displayed significantly higher accumulation of ESL1 within the CLN and AxLN compared to healthy controls and displayed an additional accumulation of ESL1 in the Payer’s patches. The accumulation of ESL1 in the spleens of PLGA/ESL1-FITC-treated animals was not significantly higher compared to healthy animals. Additionally, up to 9.5% of myeloid APCs within the spinal cords of immunised animals were found positive for ESL1 antigen. These results suggested that ESL1 antigens released from PLGA migrate through implant-draining lymph nodes and the spleen in healthy animals in a delayed and continuous pattern as compared to soluble ESL1, and that EAE-induced inflammation potentiated additional migration of the released ESL1 towards the gut and CNS.
PLGA/ESL1 Reduces Splenic Inflammation During EAE
As neither PLGA nor the ESL1 antigens alone displayed any protective effects against EAE in this model (Figure 3), only animals from the EAE_PLGA/ESL1 and EAE_ctrl groups were used in further experiments. Considering the effects of PLGA/ESL1 implants on EAE and the critical role of the spleen in the systemic immune response during EAE,56,57 we next investigated how immune cell subsets are affected in the spleen during EAE. Key cell populations in the spleen were analysed by flow cytometry (Supplementary Figure S4) at the peak of EAE (day 14th) and during the recovery phase (day 20th). It was found that the treated EAE_PLGA/ESL1 animals contained significantly lower proportions of OX62+ I-Ahi DCs at the peak and recovery phases, as well as macrophages (CD68+I-A+) in the recovery phase, compared to EAE_ctrl animals (Figure 5A). Moreover, myeloid cell populations demonstrated lower expression of CD86 and increased expression of IL-10 upon isolation from the EAE_PLGA/ESL1 group compared to the control EAE_ctrl group (Figure 5B). The proportions of NK and T cells (both CD4 and CD8), and B cells did not differ significantly between the groups. A reduced proportion of NK cells and an increased proportion of B cells were observed during the recovery phase, relative to the peak of EAE (Figure 5A and C, Supplementary Figure S5).
 |
Figure 5 Effects of PLGA/ESL1 on immune response in spleen during EAE. Seven days before immunisation with spinal cord homogenate in CFA, DA rats were implanted with PLGA/ESL1 (350µg ESL1 / 7 cm2 PLGA) subcutaneously in the suprascapular region (EAE_PLGA/ESL1) or just sham-operated (EAE_ctrl). Splenocytes collected from animals at the peak of EAE (day 14), or in the recovery phase (day 20) were collected and analysed by flow cytometry, as described in Supplementary Figure S3. (A) The proportion of DCs (OX62+I-Ahi), macrophages (CD68+I-A+), T (CD3+CD161−) and NK cells (CD3−CD161+) cells is shown. (B) The expression of CD86 within DCs and macrophages, and IL-10 expression within total myeloid (CD11b+) cells are shown as mean fluorescence intensity (MFI). (C) The relative proportion of cell types is shown after the normalisation of the gated cells to 100%. See Supplementary Figure S4A for data on the proportion of B cells, CD4+ and CD8+ T cells. (D) The proportion of Th1 (IFN-γ+), Th17 (IL-17+), Th2 (IL-4+) cells within CD4+ T cells subsets, and IFN-γ producing NK cells are shown, as well as CD25hiFoxP3+ Tregs and IL-10-producing T cells. All T cell subsets in the spleen were gated as demonstrated in Figure 7A and 7E) The level of cytokines spontaneously produced by the splenocytes (2 x 106/ mL) in 48h-cultures upon the isolation from EAE_PLGA/ESL1 or EAE_ctrl animals at the EAE peak or recovery from one, out of two experiments is shown. (A, B, D, E) *p<0.05, **p<0.001, ***p<0.005, as indicated (Kruskal–Wallis test with Dunn’s multiple comparison test). |
The functional properties of NK and T cells were significantly modulated by PLGA/ESL1 treatment irrespective of their unaltered proportions in the spleen. Namely, PLGA/ESL1-treated animals displayed a reduced proportion of IFN-γ-producing CD4+ (Th1) and NK cells, and IL-17-producing CD4+ T cells at the peak of EAE, as well as lower expression of CD86 on B cells, compared to EAE_ctrl animals (Figure 5D). These results correlated with the reduced levels of IFN-γ and IL-17 in 48 h cultures of ex vivo splenocytes isolated from PLGA/ESL1 animals at the peak of the disease (Figure 5E). In addition to the reduced proportion of pro-inflammatory lymphocytes in the spleen of these animals, an increased proportion of IL-10-producing CD8+ T cells was observed at the peak of EAE (Figure 5D). The recovery phase of EAE was generally characterised by a reduced proportion of Th1 and Th17 cells and an increased proportion of CD4+CD25hiFoxP3+ Tregs in the spleens of both groups. However, PLGA/ESL1-treated animals displayed a significantly increased proportion of Tregs during the recovery phase compared to the control EAE_ctrl group (Figure 5D). The recovery phase also displayed an increased proportion of IL-4-producing CD8 cells (ie Tc2), as well as IL-10-producing NK and B cells, although there were no differences between the groups (Supplementary Figure S5B). These results, along with the reduced levels of IL-6 in ex vivo splenocyte cultures (Figure 5D), indicated that PLGA/ESL1 implants reduced splenic inflammation in EAE and potentiated regulatory lymphocytes.
PLGA/ESL1 Implants Reduce Immune Cell Infiltration into the CNS
EAE symptoms in DA rats are critically linked to immune cell infiltration into the CNS, particularly in the spinal cords (SCs), which are the most commonly used pathological markers in this experimental model.58 Therefore, the SCs of animals from the EAE_ctrl and EAE_PLGA/ESL1 groups were collected at the peak and recovery phase of EAE and further analysed by microscopy and flow cytometry (Figures 6 and 7). Histological analysis of the H&E-stained lumbar SC cross-sections indicated a significantly higher number of infiltrates in the EAE_ctrl group than in the EAE_ PLGA/ESL1 group at the peak of EAE, although the average number of cells per infiltrate was similar between the groups (Figure 6A and B). Infiltrates were predominantly found in the white matter of SCs on the ventral, dorsal, and lateral horns, and their number was reduced during the recovery phase (Figure 6B). Epifluorescence microscopy analysis of CD45 / myelin-based protein (MBP) staining confirmed that the infiltrates in SCs were of leukocyte origin (Figure 6C). Moreover, the infiltrating areas showed reduced MBP expression at the infiltration sites, indicating tissue damage caused by the infiltrates (Figure 6C).
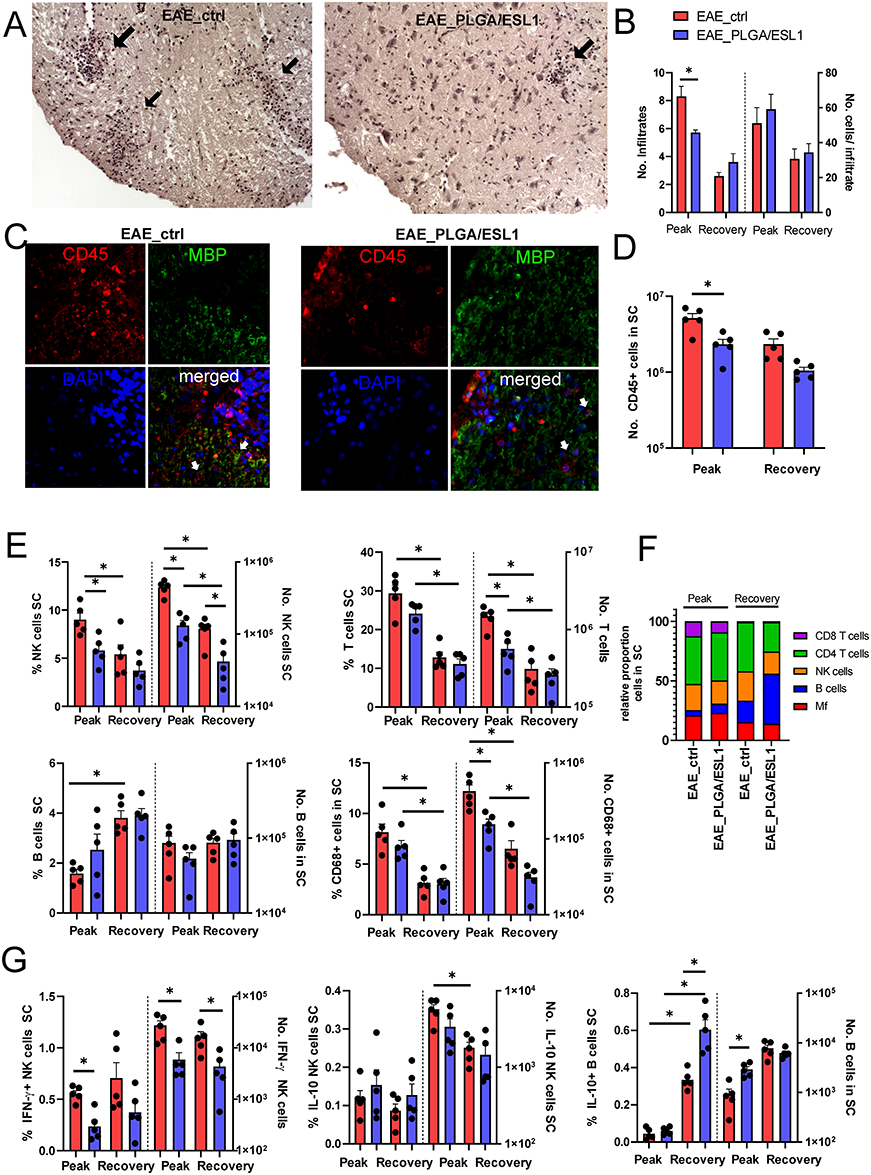 |
Figure 6 Effects of PLGA/ESL1 on the infiltration of leukocytes to the spinal cord. Seven days before the immunisation, animals were implanted with either PLGA/ESL1 or sham-operated, as described. (A) Representative images at 10x obj. mag. are shown of HE-stained spinal cord (lumbar part) cross-sections from EAE_ctrl and EAE_PLGA/ESL1 animals at the peak of EAE, with indicated immune infiltrates (black arrows). (B) The average number of infiltrates and the number of cells/infiltrates were counted from HE-stained slides as described in Materials and Methods. (C) Representative images at 20x obj. mag. are shown after staining the cross-sections (as in (A) with anti-CD45-Alexa Fluor 546 (red) and anti-MBP-Alexa Fluor 488 (green), while nuclei were stained with DAPI. (D) The total number of CD45+ cells in spinal cord infiltrates is shown, as determined according to the Muse Cell Count viability kit, and CD45 staining by flow cytometry. (E) The proportion and the absolute number of NK, T, B cells and macrophages in the spinal cord infiltrates from the EAE peak or recovery are shown. (F) The summarised data indicating the relative proportions of these cells normalised to all gated leukocytes (100%) is shown. (G) The proportions and the number of IFN-γ+ or IL-10+ NK (CD161+CD3−) cells are shown, as well as IL-10+ B cells in the total cells isolated from SC at the EAE peak and recovery from a representative experiment out of two. (B, D, E, G) *p<0.05 as indicated (Kruskal–Wallis test with Dunn’s multiple comparison test). |
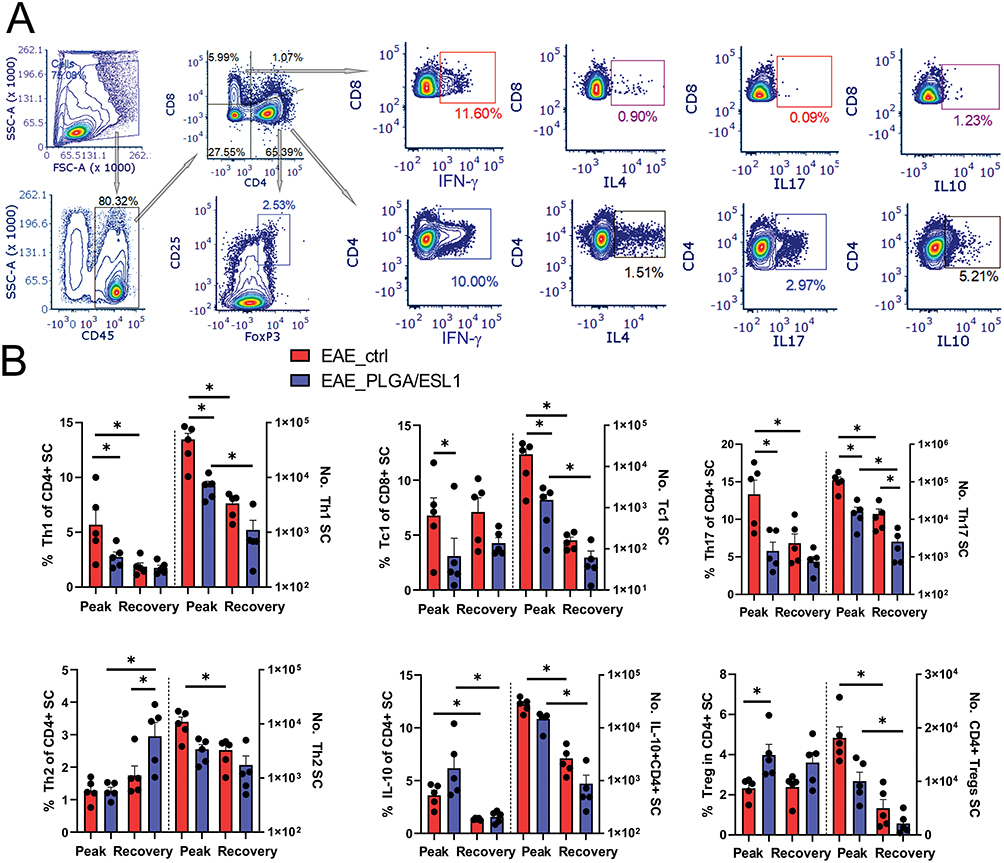 |
Figure 7 Effects of PLGA/ESL1 on the T cell profile in the spinal cord infiltrates. Seven days before the immunisation, animals were implanted with either PLGA/ESL1 or sham-operated, as described, and then isolated from CNS and activated with PMA (20ng/mL) /Ca Ionophore (500ng/mL) and Monensin 3 µM for 4h before staining for flow cytometry. (A) The gating strategy for identification of CD8+T cell subsets producing IFN-γ (Tc1), IL-4 (Tc4), IL-17 (Tc17) and IL-10 (IL-10+ CD8+ T cells) and equivalent subsets of CD4+ Th cells are shown. CD4+ T cells were gated to CD25hi FoxP3+ to identify Tregs. (B) The summarised data from a representative experiment out of two, on the relative proportion and the total number of T cell subsets are shown. See Supplementary Figure S5, for data on total CD4+, CD8+ T cells and IL-10-producing CD8+T cells. *p<0.05 as indicated (Kruskal–Wallis test with Dunn’s multiple comparison test). |
To quantify immune cell infiltrates in SCs, total mononuclear cells were isolated and CD45+ cells were enumerated by flow cytometry. The data showed that EAE_ctrl animals contained significantly more CD45+ cells in SCs than EAE_PLGA/ESL1 animals (Figure 6D). Accordingly, a significantly lower number of NK cells, T cells, and CD68+ macrophages, but not B cells, were found in the SCs of animals from the EAE_PLGA/ESL1 group compared to the EAE_ctrl group at the peak of the disease (Figure 6E and F). The reduced number of NK cells in the EAE_PLGA/ESL1 group was partially due to the reduced proportion of IFN-γ-producing NK cells but not IL-10-producing NK cells, whose relative abundance in the EAE_PLGA/ESL1 group was even higher than that in the EAE_ctrl group (Figure 6G). Additionally, a higher number of IL-10-producing B cells was found in the EAE_PLGA/ESL1 group at the peak of EAE, as compared to the EAE_ctrl group (Figure 6G). In the recovery phase, a lower number of NK cells, T cells, and macrophages were observed in the SC infiltrates in both groups, but the total number of NK cells was still lower in the EAE_PLGA/ESL1 group than in the EAE_ctrl group.
Since T cells are the major regulators of neuroinflammation in MS and EAE,59 we analysed T cell subsets in more detail by measuring the intracellular expression of IFN-γ, IL-4, IL-17, IL-10, and FoxP3 using flow cytometry (Figure 7). Both the relative proportion and the total number of encephalitogenic Th1, Th17, and Tc1 cells were reduced in the SCs of the EAE_PLGA/ESL1 group compared to the control EAE_ctrl group (Figure 7B). Moreover, a significantly increased proportion of Tregs, although not the total number, was found in this group at the peak of the EAE. The animals from the EAE_PLGA/ESL1 group also displayed a tendency of increased proportion of IL-10-producing CD4+ (Figure 7B) and CD8+ (Supplementary Figure S6) T cells in the SCs, but no significant difference was observed between the groups. Additionally, the total number of CD86+ B cells in SCs were not modified by PLGA/ESL1 treatment during the EAE (Supplementary Figure S6). The recovery phase was generally characterised by reduced encephalitogenic T cells, but the number of Th17 cells was still significantly lower in the EAE_PLGA/ESL1 group than in the EAE_ctrl group (Figure 7B). Additionally, an increased proportion of Th2 cells, although not the total number, was found in the recovery phase in the EAE_PLGA/ESL1 group compared to the EAE_ctrl group. Overall, the results suggested that PLGA/ESL1 implants reduced immune cell infiltration into SCs, especially the infiltration of encephalitogenic T cells during relapsing EAE, thereby increasing the relative proportion of anti-inflammatory and regulatory T, B, and NK cells.
PLGA/ESL1 Implants Prevented Intestinal Inflammation, Gut Microbiota Dysbiosis in EAE
The protective effects of PLGA/ESL1 implants on EAE development were related to the downregulation of systemic and CNS inflammation (Figure 5–7), and increased accumulation of ESL1 antigens in the CNS and Payer’s patches (Figure 4). Moreover, the gut-brain axis is considered critical for the regulation of immune responses in MS and EAE models.8,41,60 Therefore, we analysed the gene expression of the pro-inflammatory cytokine TNF-α and tight-junction proteins in intestinal tissues as measures of gut inflammation and intestinal barrier integrity, respectively. In addition to the EAE_PLGA/ESL1 and EAE_ctrl groups, we included sham-operated (healthy) animals as controls (Figure 8). The mRNA levels of TNF-α in animals from the EAE_PLGA/ESL1 group at the peak of the disease were maintained at the same level as in healthy animals and were significantly lower than in the control EAE_ctrl group (Figure 8A). Furthermore, we found preserved mRNA expression levels of tight junction proteins contributing to gut barrier function, Claudin and Occludin, in the EAE_PLGA/ESL1 group, unlike the reduced levels in the EAE_ctrl group (Figure 8A).
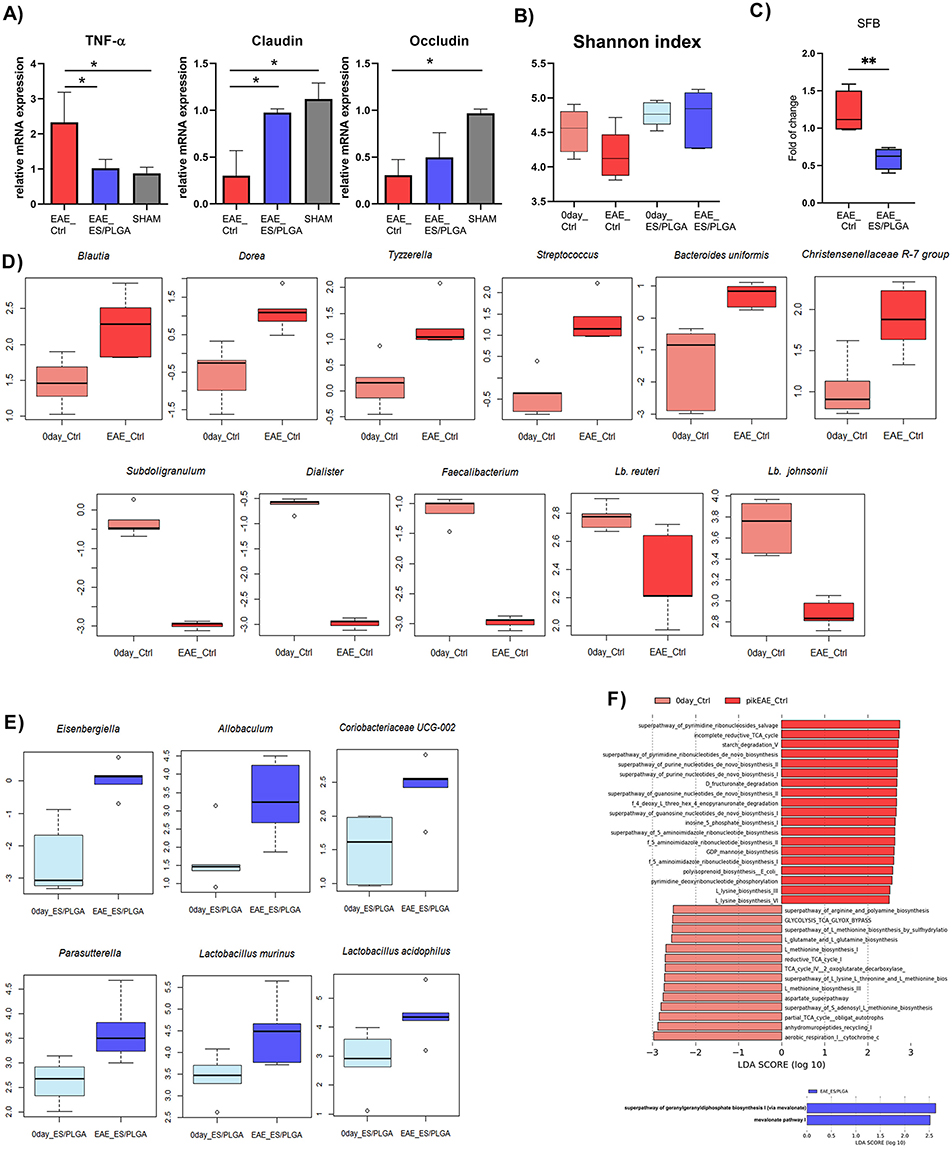 |
Figure 8 Effects of PLGA/ESL1 on gut inflammation and microbiota features. (A) Relative mRNA expression of TNF-α, Claudin and Occludin was determined in gut samples collected from EAE_ctrl, EAE_PLGA/ESL1, and sham-operated animals at the peak of the disease from a representative experiment out of two are shown. *p<0.05 as indicated (one-way ANOVA with Tukey’s multiple comparisons test). The faecal material was collected right before the EAE induction in control (0day_Ctrl) and PLGA/ESL1-treated animals (0day_PLGA/ESL1) and at the peak of disease (EAE_Ctrl, EAE_PLGA/ESL1). DNA was isolated, and 16s rRNA sequencing (B, D, E and F) or qPCR (C) was performed. (B) The changes in the Shannon index between 0day and peak of the disease in the control (red) and PLGA/ESL1-treated (blue) groups are shown by boxplots. (C) The fold changes in SFB-specific sequence expression were calculated by dividing the values measured at the peak of the disease and the values at 0day for each animal. Control (red) and PLGA/ESL1- treated (blue) groups were compared by Student T test (**p<0.01). The changes in the relative abundances of Taxa (ANCOM W > 1, Pairwise Wilcoxon test or paired t-test followed by Benjamini-Hochberg p-value correction) at the EAE peak compared to 0day in (D) control group of animals, (E) PLGA/ESL1- treated group. (F) The most differentially abundant predictive metabolic pathways of gut microbiota at 0day compared to the EAE peak in control (red) and PLGA/ESL1- treated (blue) groups analysed by Linear discriminant analysis (LDA) effect size (LEfSe) (LDA score above 2.5, alpha value of <0.05 for factorial Kruskal–Wallis). Pathways with positive LDA scores (darker bars) are more prominent at the EAE peak of the disease, whereas negative LDA scores (brighter bars) indicate pathways enriched at 0day, in both groups (control (red) and PLGA/ESL1- treated (blue)). |
Microbiota composition is strongly associated with gut barrier functions and immune responses in EAE,8,41,60 so we further investigated how PLGA/ESL1 implants affect changes in the gut microbiota. Therefore, the microbiota composition was analysed before EAE induction in both groups (0day_Ctrl or 0day_PLGA/ESL1), as well as at the peak of EAE (Figure 8B). The gut microbiota diversity, expressed as the Shannon diversity index, was not significantly affected 7 days after PLGA/ESL1 implantation (0 day) as compared to the control group. However, EAE induction somewhat reduced the gut microbiota diversity in the EAE_ctrl group, unlike in the EAE_PLGA/ESL1 group (Figure 8B), suggesting that PLGA/ESL1 implantation provided protection of gut microbiota diversity after EAE induction.
Next, we analysed the changes in bacterial taxa composition in faecal samples before EAE induction in both groups and at the peak of the disease. Analysis of the DNA amplicons specific for segmented filamentous bacteria (SFB) showed a lower level of SFB in the PLGA/ESL1 group compared to the EAE_ctrl group (Figure 8C). Further analyses of taxa composition based on the bacterial 16S rRNA gene indicated the enrichment of species Bacteroides uniformis, Christensenellaceae R-7 group, genus Streptococcus, and several genera from the family Lachnospiraceae (Blautia, Tyzzerella, Dorea) in the EAE_ctrl group of animals (Figure 8D). Also, the decreases in relative abundance of taxa such as Lactobacillus johnsonii, Lactobacillus reuteri, genera Dialister, Faecalibacterium, and Subdoligranulum were observed in the EAE_ctrl group at the peak of the disease. In contrast, only slight changes in the gut microbiota composition of PLGA/ESL1-treated animals were observed at the peak of EAE (Figure 8E). Namely, the relative abundances of four genera, Eisenbergiella, Allobaculum, Coriobacteriaceae UCG-002, and Parasutterella, and two species, Lactobacillus murinus and Lactobacillus acidophilus were significantly increased in this group at the time of the EAE peak.
To further examine the extent of changes in the microbiota of the control EAE animals and EAE_PLGA/ESL1 animals, we performed prediction of metagenome functions based on the 16S rRNA gene at the peak of the disease in comparison to day 0 (ie before EAE induction) (Figure 8F). This analysis pointed to the extensive changes in microbial metabolism at the peak of the disease in control animals, marked by the enrichment of pathways involved in nucleotide, purine, and pyrimidine biosynthesis. In contrast, only two pathways, mevalonate pathway I and the super pathway of geranylgeranyl diphosphate biosynthesis I (via mevalonate), were predicted to be enriched in the PLGA/ESL1 treated group.
Discussion
Here, we tested electrospun PLGA nanofibers loaded with immunomodulatory ESL1 antigens of T. spiralis, as a novel platform for regulating neuroinflammation in a relapsing EAE model in DA rats. PLGA is a widely used biodegradable polymer with excellent biocompatibility, surface chemistry, and controllable release properties.29,33 PLGA micro/nanoparticles (NPs) have been extensively explored as drug delivery systems in autoimmune diseases,29 although their immunological effects are still controversial. The delivery of disease-specific antigens (ie MOG,61,62 PLP63 or insulin64) via PLGA NPs alone was shown to be effective in ameliorating the symptoms in animal models of multiple sclerosis61,63 or type 1 diabetes.64 Accordingly, Li et al62 showed that the subcutaneous delivery of PEGylated MOG-loaded PLGA nanoparticles is sufficient for the effective suppression of EAE, not via direct actions on APCs, but rather by lowering complement activation and neutrophil recruitment to the application site. Additional studies have indicated that immunoregulatory molecules (ie IL-1065) or specific ligands (ie ICAM-1 ligand66) are required, in addition to antigens, to protect mice from EAE development. Thereby, the co-delivery of multiple molecules via PLGA NPs potentiated tolerogenic APCs, particularly DCs, which are predominantly involved in the regulation of Th1 and Th17 responses in EAE.63,66 These promising results encourage the further development of PLGA-based NPs for autoimmunity therapy, especially when rationally designed. However, several studies have suggested that PLGA particles can display intrinsic adjuvant properties, which could induce adverse immunological effects in autoimmunity. Namely, PEGylated or bare PLGA-based particles were increasingly taken up by DCs, followed by activation of NLRP3, production of IL-1β, and T cell activation initiating the inflammatory response.29,67–71 Moreover, a potentially toxic effect of PLGA NPs was described in epithelial cells, wherein these particles displayed a high capacity to induce apoptosis, autophagy, endoplasmic reticulum stress, DNA damage, as well as proinflammatory cytokines (ie CXCL1, IL1A, IL1β, calprotectin, and TNF-α).72 Therefore, although promising, PLGA NPs may display a potential risk for autoimmunity therapy. Besides, many challenges persist in NP-based antigen-specific therapies in humans, including antigen selection, epitope changes during disease progression, and the complexity of autoimmune reactions,73 justifying disease-modifying therapies as better therapeutic alternatives.
In contrast to PLGA NPs, the immunological properties of PLGA nanofibers have been less well studied, and no data available have described their potential application in the treatment of autoimmune diseases. Previous studies have suggested that PLGA nanofibers are highly efficient in supporting cell growth and wound healing due to their excellent biomimetic properties resembling the extracellular matrix.51,74 The immunomodulatory properties of PLGA nanofibers depend greatly on the loaded biomolecules. For example, asiaticoside-loaded PLGA nanofibers reduced host inflammation and promoted M2 macrophage polarisation.75 Likewise, cyclosporin A-loaded PLGA nanofibers attenuated the production of proinflammatory cytokines IL-2, IFN-γ and IL-17,76 whereas vanillin-incorporated PLGA nanofibers displayed anti-oxidative and anti-inflammatory effects in vivo.77 Even the empty PLGA nanofibers displayed acceptable biocompatibility upon implantation. Namely, Chor et al78 described a granulomatous reaction upon implantation of PLGA nanofibers, which resolved over time. It was characterised by the infiltration of epithelioid cells, lymphocytes, Langhans cells, and multinucleated giant cells, but without the formation of a fibrous capsule.78 That immune reaction somewhat resembles the one T. spiralis induces during muscle invasion. Namely, the initial muscle phase of T. spiralis infection shows an increased cellular infiltrate around the nurse cells, following its reduction over the 30-day course due to active modulation of the local immune response by the parasite.79,80 The release rate of ELS1 from PLGA nanofibers in vitro displayed multiple phases over 40 days of monitoring, with an increasing release over the first 27 days, followed by a slower release due to PLGA degradation. By using the emulsion electrospinning method for PLGA/ESL1 preparation, it is possible that the hydrophilic ESL1 avoid contact with organic solvents, which favours its localization within the core of the fibers contributing to the minimization of burst release effects. The degradation rate of PLGA was found to depend on glycolic acid proportion, which is a critical parameter in tuning the hydrophilicity of the matrix.39 The release speed of ESL1 in this study was somewhat higher than what we observed previously for a model drug, Rhodamine B (RhB), in PBS surrounding medium.33 This discrepancy could be due to different diffusion rates of the biomolecules within PLGA, conditions for the in vitro release, and particularly the presence of wound exudate in this study. In line with this, we found previously that the wound exudate significantly enhances the release of Se nanoparticles from poly (ε-caprolactone) polymer microspheres compared to PBS,40 which could also be the case here. However, the release dynamics from PLGA under static conditions can significantly differ from the in vivo release only based on the change in pH during PLGA degradation.81 Therefore, it remains to be investigated which components of the inflammatory exudate and infiltrating cells critically affect the release dynamics of ESL1 from PLGA in vivo.
Here we demonstrated that ESL1 antigens are released from PLGA nanofibers in the local tissue, taken up by infiltrating myeloid cells, which then migrate through the draining lymph nodes and spleen. Although we could not rule out the drainage of free ESL1 through the lymph nodes, the critical effects of ESL1 seem to include modulation of myeloid cells. Namely, we have previously shown that ESL1 products, administered prophylactically in multiple doses (500 μg/dose intraperitoneally), possess the capacity to reduce the severity of EAE, by inducing anti-inflammatory and regulatory mechanisms that prevent the formation of Th1 and Th17 cells in EAE.14,16 These effects could be completely reproduced by transferring ESL1-treated myeloid cells to immunised animals,17 suggesting that the modulation of myeloid cells, such as DCs-CD11b+ I-Ahi and macrophages CD11b+ I-Alow, is a critical mechanism for ESL1 tolerogenic potential in EAE. Thereby, we demonstrated previously that ESL1 were able to induce human tolerogenic DCs, limit their costimulatory potential,18 and lower their capacity to induce Th1 and Th17 cells, while increasing the number and function of Tregs.18–20 These results are in line with the findings obtained in this study. However, this is the first study to link the migration of tolerogenic ESL1 antigens through the lymph nodes and spleen via myeloid cells, and their modulation in vivo. Zhang et al82 showed previously that treatment of mice with all-trans-retinoic acid (ATRA) modulates DC functions in vivo within the draining lymph nodes and spleen, following the amelioration of EAE symptoms. Additionally, the migration of myeloid regulatory cells and EVs through the lymph nodes was also found to be critical for the suppression of EAE.41,83 The increased migration of ESL1+ myeloid cells to draining lymph nodes after EAE induction could be explained by EAE-induced and ESL1-induced expression of CCR7 on myeloid cells, as demonstrated previously,18,82,84,85 although this hypothesis requires a direct proof in further investigations. The accumulation of ESL1+ myeloid cells in the cervical lymph nodes upon EAE induction is of particular significance, considering the role of these lymph nodes in the priming of T cells in EAE and MS.86–88 Therefore, the strategic positioning of the tolerogenic drug-releasing system might be of critical importance for an efficient immunotherapy of MS. The recirculation of inflammatory myeloid cells into CNS was shown to be crucial for restimulation of co-infiltrating encephalitogenic T cells.89 Although less well appreciated,90 many data,91 including our own,41 demonstrated that the migration of regulatory myeloid cells into CNS correlates with the suppression of EAE. Namely, myeloid regulatory cells were shown to induce different subsets of regulatory B92 and T cells in the CNS,41 and suppress the proinflammatory actions of NK and T cells via multiple mechanisms,6,93 as observed in this study. An increased proportion of IL-10-producing B cells in SLAMF5KO mice in the CNS to 10% of total B cells was demonstrated critical for the regulation of EAE.94 Although, similar increase of IL-10-producing B cells was observed in the recovery phase of EAE_PLGA/ESL1-treated animals, further investigations are necessary to delineate the exact role of these cells in the suppression of EAE in DA rats. In addition to myeloid cells, ESL1 released from PLGA may even directly induce IL-10-producing B cells and regulatory T cells. Okada et al95 showed that TLR4 and CD40 co-stimulation synergistically increased the frequency of IL-10-producing B cells in patients with MS. On the other hand, TLR2-ligation on purified Tregs was shown to induce their proliferation and functions.96 In line with these studies, we previously demonstrated, by using TLR4- and TLR2-transfected reporter cell line and human DCs cultures, that ESL1 triggers TLR4 and TLR2 signalling and upregulates CD40 expression.18 Therefore, it is possible that multiple mechanisms of modulation by ESL1 were involved in the amelioration of EAE upon its release from PLGA.
One of the key findings of this study is that released ESL1 prevented EAE-induced gut inflammation (ie TNF-α expression), gut barrier dysfunction (ie Claudin expression), and microbiota dysbiosis. Moreover, our preliminary experiments suggested an increased expression of immunoregulatory genes within the Payer’s patches of ESL1/PLGA-treated animals compared to EAE_ctrl (data not shown), upon the migration of ESL1+ myeloid APCs (Figure 4). An increasing number of studies suggest that changes in the gut microbiota could play a significant role in the progression and regulation of MS.7,97 It was shown that EAE-induced inflammation and the migration of autoreactive T cells to the gut induce gut barrier damage.98 Moreover, encephalitogenic Th17 cells were found to be licenced in the gut before they migrated to the CNS to induce EAE symptoms.99 Accordingly, we showed previously that early migration of regulatory myeloid cells in Payer’s patches corresponded to the reduction of EAE symptoms and preservation of the gut barrier in DA rats.41 The findings that ESL1+ myeloid cells migrate increasingly to Payers’ patches upon EAE induction, suggest that ESL1 may have also modulated EAE symptoms by suppressing inflammation in the gut, preserving the gut barrier and gut microbiota. Accordingly, we found a lower abundance of SFB in PLGA/ESL1-treated animals, which are known for their potential to induce Th17, intestinal inflammation, and EAE.100–102 Moreover, several taxa were found to be enriched in the gut of MS patients,97,103,104 or diseases associated with disturbance of gut barrier integrity,105,106 including Bacteroides uniformis, Christensenellaceae R-7 group, genus Streptococcus, and members of the family Lachnospiraceae (Blautia, Tyzzerella, Dorea), were all found to be enriched in the control EAE group but not in the EAE_PLGA/ESL1 group. The increased abundance of pro-pathogenic taxa in the control EAE group was also followed by a decreased abundance of health-promoting bacteria, such as Lactobacillus johnsonii, Lactobacillus reuteri, and genus Dialister, which were previously described as protective in EAE107 and MS.108 Furthermore, the decrement of the relative abundances of butyric-producing bacteria, Faecalibacterium and Subdoligranum, could be related to the increased TNF-α expression and reduced expression of tight-junctions proteins in the gut of control EAE animals, considering that butyrate is critical for gut homeostasis.109 Based on extensive changes in gut microbiota composition induced by EAE in control animals, the predicted microbial metabolic pathways were marked by enrichment in the nucleotide, purine, and pyrimidine biosynthesis pathways. Considering the pro-inflammatory effects of extracellular nucleotides, primarily ATP and UTP,110 such disturbance in the metabolism of gut microbiota could contribute to intestinal inflammation and EAE development.
In sharp contrast to the control EAE animals, we observed only slight changes in the microbiota of animals treated with PLGA/ESL1, which showed preserved microbiota diversity similar to healthy animals. Interestingly, the abundance of bacteria with potential anti-inflammatory and neuroprotective effects was increased in the PLGA/ESL1 group at the peak of EAE. Namely, Allobaculum, a butyrate-producing commensal, was shown to activate GPR43-expressing Tregs and keep the gut immune homeostasis.111 Coriobacteriaceae UCG-002, which was also found to be enriched in PLGA/ESL1-treated animals, were previously described as an important participants in neuroprotective bile acid metabolism.112 Bile acid metabolism is commonly altered in MS, and bile acid supplementation could prevent the polarization of astrocytes and microglia to neurotoxic phenotypes.113 Ju et al114 showed that the colonization of host microbiota with the genus Parasutterella strongly affects the faecal metabolome, increasing bile acid metabolites, succinate production, and purines concentration (inosine, hypoxanthine, and xanthine). Thereby, succinate and hypoxanthine were described as important regulators of energy homeostasis in the guts.115,116 Furthermore, Lactobacillus murinus was also enriched in PLGA/ESL1-treated animals, and this species was previously described to prevent intestinal injury by stimulating IL-10 production from M2 macrophages, reducing the activation of NLRP3 in microglia, thus preventing neuroinflammation.117 Similarly, Lactobacillus acidophilus was previously demonstrated to exert neuroprotective effects in mice by restraining systemic inflammation and increasing the expression of gut Occludin.118 Therefore, the enrichment of Lactobacillus species, in addition to lower levels of SFB, could be associated with the protective effects of PLGA/ESL1 treatment in EAE. Enrichment of the genus Eisenbergiella, previously associated with MS,119 in the PLGA/ESL1-treated group was the only feature associated with microbiota in the EAE group. The metabolic pathways predicted to be altered in the PLGA/ESL1 group were associated with the production of isopentenyl diphosphate used by some bacteria in the downstream pathways for the production of undecaprenol, menaquinones, ubiquinones, and carotenoids.120 The stimulation of undecaprenol production involved in cell membrane and cell wall biosynthesis, and carotenoids important for the resistance of bacteria to oxidative stress, could serve as protective mechanisms enabling the preservation of microbiota in the inflammatory environment after EAE induction. These mechanisms may have been activated by microbial changes induced in PLGA/ESL1-treated animals, contributing to diminished gut inflammation and EAE symptoms. Menaquinones, belonging to the vitamin K family, are involved in electron transport in bacterial metabolism. Interestingly, this type of microbial vitamin K could be involved in the regulation of the host immune response, as the immunoregulatory role of other molecules of vitamin K has been described, and their deficiency has been associated with intestinal inflammation,121 as well as MS.122 However, these predictions opened further investigations on how PLGA/ESL1 treatment induces the described changes in gut microbiota after EAE induction and what mechanisms critically operate in the regulation of EAE by ESL1-modification of microbiota.
Conclusion
Here we demonstrated for the first time the potential of electrospun PLGA nanofibers to deliver ESL1 antigens originating from T. spiralis into the host upon timely-controlled release in vivo, subsequently protecting the animals from developing an induced autoimmune reaction. This delivery system was able to regulate neuroinflammation in a relapsing EAE model by modulating multiple pathogenic and regulatory immune cell subsets in the spleen and CNS. Moreover, the beneficial effects of PLGA/ESL1 implants in EAE correlated with the preservation of the gut barrier and gut microbiota composition, by enriching the beneficial taxa in the host microbiome while preventing the expansion of EAE-induced pro-pathogenic species. Further investigations are necessary to delineate the key mechanisms of ESL1 actions in vivo among the different promising targets identified, as well as to untangle the complex interactions between microbiota composition, their metabolism, and the host immunity in EAE. The described approach for the controlled delivery of complex disease-modifying agents, such as ESL1, via biodegradable polymers shows great promise for the future development of immunotherapy for autoimmune diseases such as MS.
Abbreviations
7-AAD, 7-Aminoactinomycin D; Actb, β-actin; ANCOM, Analysis of composition of microbiomes; ANOVA, Analysis of variance; APC, Allophycocyanin; APC, antigen-presenting cell; ATRA, all-trans retinoic acid; ATR–FTIR, Attenuated Total Reflection Fourier-Transform Infrared; AUC, area under the curve; Ax, axillary; C, cervical; CD, cluster of differentiation; CFA, complete Freund’s adjuvant; CNS, central nervous system; Cy, Cyanine; DA, Dark Agouti; DAPI, 4’,6-diamidino-2-phenylindole; DC, dendritic cells; DMF, N, N-dimethylformamide; DNA, deoxyribonucleic acid; EAE, experimental autoimmune encephalomyelitis; EDTA, Ethylenediaminetetraacetic Acid; ELISA, Enzyme linked immunosorbent assay; ENA, European Nucleotide Archive; ESL1, excretory-secretory products of Trichinella spiralis L1 larvae; EV, extracellular vesicle; FE-SEM, Field Emission Scanning Electron Microscope; FITC, Fluorescein 5-isothiocyanate; FoxP3, forehead box P3; FSC, forward scatter; GPR, G-protein receptor; HEPES, 4- (2-hydroxyethyl)-1-piperazineethanesulfonic acid; IFN, interferon; IgG, immunoglobulin G; IL, interleukin; INEP, Institute for the Application of Nuclear Energy; Ing, inguinal; LDA, Linear discriminant analysis; LEfSe, Linear discriminant analysis Effect Size; LN, lymph node; mAb, monoclonal antibody; MBP, myelin base protein; MHC, major histocompatibility complex; MMA, Military Medical Academy; MOG, myelin oligodendrocyte protein; MS, multiple sclerosis; MW, molecular weight; NK, natural killer; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; NP, nanoparticle; PBS, phosphate buffered saline; PE, phycoerythrin; PEG, polyethylene glycol; PerCP, Peridinin-Chlorophyll-Protein; PICRUSTt, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States; PLGA, poly (lactic-co-glycolic) acid; PMA, phorbol-12-myristate-13-acetate; qPCR, quantitative polymerase chain reaction; RhB, Rhodamine B; RNA, ribonucleic acid; SC, spinal cord; SFB, segmented filamentous bacteria; SSC, side scatter; Tg, glass transition temperature; TGF, transforming growth factor; Th, T helper; TLR, Toll-like receptor; TNF, tumor necrosis factor; Treg, regulatory T cell; UCG, unclassified group; UV, ultraviolet; WE, wound exudate; wt. %, weight percentage.
Data Sharing Statement
All data generated or analysed during this study are included in this article or in the supplementary data. The raw data that support the findings of this study are available from the corresponding author, upon reasonable request. The datasets generated and analysed for 16S rRNA sequencing are available in the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena) repository under accession number PRJEB79235 and secondary accession number ERP163422.
Ethics Approval
The experimental procedures within this work involving animals were approved and carried out following the guidelines and regulations by the local ethics committee of the Institute for the Application of Nuclear Energy—INEP, University of Belgrade, Serbia, Belgrade (No. 323–07-11,160/2019-05/1 and No. 323-07-00758/2021-05/1).
Acknowledgments
The authors are also grateful to Milan Marković, Milica Lazarević, Bojan Jevtić and Đorđe Miljković for their kind help in immunohistochemical analyses, Marija Gnjatović for her kind help in ESL1 tracking experiments, and Marina Bekić and Aleksandra Todorović for their kind help during the EAE experiments.
Author Contributions
All the authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Ministry of Science, Technological Development, and Innovation of the Republic of Serbia under Contract No. 451-03-68/2024-14/200042, No. 451-03-68/2024-14/200019, No. 451-03-65/2024-03/200135), and by the Science Fund of the Republic of Serbia, PROMIS, #6062673, Nano-MDSC-Thera.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi:10.1016/S0140-6736(08)61620-7
2. Lanz TV, Brewer RC, Ho PP, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. 2022;603:7900):321–327. doi:10.1038/s41586-022-04432-7
3. Bar-Or A, Li R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 2021;20(6):470–483. doi:10.1016/S1474-4422(21)00063-6
4. Vargas DL, Tyor WR. Update on disease-modifying therapies for multiple sclerosis. J Investig Med. 2017;65(5):883–891. doi:10.1136/jim-2016-000339
5. Amorim A, De Feo D, Friebel E, et al. IFNγ and GM-CSF control complementary differentiation programs in the monocyte-to-phagocyte transition during neuroinflammation. Nat Immunol. 2022;23(2):217–228. doi:10.1038/s41590-021-01117-7
6. Bekić M, Tomić S. Myeloid‐derive suppressor cells in the therapy of autoimmune diseases. Eur J Immunol. 2023;53(12):2250345. doi:10.1002/eji.202250345
7. Correale J, Hohlfeld R, Baranzini SE. The role of the gut microbiota in multiple sclerosis. Nat Rev Neurol. 2022;18(9):544–558. doi:10.1038/s41582-022-00697-8
8. Parodi B, Kerlero de Rosbo N. The gut-brain axis in multiple sclerosis. is its dysfunction a pathological trigger or a consequence of the disease? Front Immunol. 2021;12. doi:10.3389/fimmu.2021.718220
9. Berg G, Rybakova D, Fischer D, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8(1):103. doi:10.1186/s40168-020-00875-0
10. Murdaca G, Greco M, Borro M, Gangemi S. Hygiene hypothesis and autoimmune diseases: a narrative review of clinical evidences and mechanisms. Autoimmunity Rev. 2021;20(7):102845. doi:10.1016/j.autrev.2021.102845
11. Maizels RM, McSorley HJ, Smyth DJ. Helminths in the hygiene hypothesis: sooner or later? Clin Exp Immunol. 2014;177(1):38–46. doi:10.1111/cei.12353
12. Bach JF. The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol. 2018;18(2):105–120. doi:10.1038/nri.2017.111
13. Yang X, Yang Y, Wang Y, et al. Excretory/secretory products from trichinella spiralis adult worms ameliorate DSS-induced colitis in mice. PLoS One. 2014;9(5):e96454. doi:10.1371/journal.pone.0096454
14. Radovic I, Gruden-Movsesijan A, Ilic N, et al. Immunomodulatory effects of Trichinella spiralis-derived excretory–secretory antigens. Immunol Res. 2015;61(3):312–325. doi:10.1007/s12026-015-8626-4
15. Eissa MM, Mostafa DK, Ghazy AA, El Azzouni MZ, Boulos LM, Younis LK. Anti-arthritic activity of schistosoma mansoni and trichinella spiralis derived-antigens in adjuvant arthritis in rats: role of FOXP3+ treg cells. PLoS One. 2016;11(11):e0165916. doi:10.1371/journal.pone.0165916
16. Sofronic-Milosavljevic LJ, Radovic I, Ilic N, Majstorovic I, Cvetkovic J, Gruden-Movsesijan A. Application of dendritic cells stimulated with Trichinella spiralis excretory-secretory antigens alleviates experimental autoimmune encephalomyelitis. Med Microbiol Immunol. 2013;202(3):239–249. doi:10.1007/s00430-012-0286-6
17. Gruden-Movsesijan A, Ilic N, Mostarica-Stojkovic M, Stosic-Grujicic S, Milic M, Sofronic-Milosavljevic L. Mechanisms of modulation of experimental autoimmune encephalomyelitis by chronic Trichinella spiralis infection in Dark Agouti rats. Parasite Immunol. 2010;32(6):450–459. doi:10.1111/j.1365-3024.2010.01207.x
18. Ilic N, Gruden-Movsesijan A, Cvetkovic J, et al. Trichinella spiralis excretory–secretory products induce tolerogenic properties in human dendritic cells via Toll-like receptors 2 and 4. Front Immunol. 2018;9:11. doi:10.3389/fimmu.2018.00011
19. Cvetkovic J, Ilic N, Gruden-Movsesijan A, et al. DC-SIGN signalling induced by Trichinella spiralis products contributes to the tolerogenic signatures of human dendritic cells. Sci Rep. 2020;10(1):1–14. doi:10.1038/s41598-020-77497-x
20. Ilić N, Kosanović M, Gruden-Movsesijan A, et al. Harnessing immunomodulatory mechanisms of Trichinella spiralis to design novel nanomedical approaches for restoring self-tolerance in autoimmunity. Immunol Lett. 2021;238:57–67. doi:10.1016/j.imlet.2021.04.012
21. Robinson MW, Connolly B. Proteomic analysis of the excretory-secretory proteins of the L1 larva, a nematode parasite of skeletal muscle. PROTEOMICS. 2005;5(17):4525–4532. doi:10.1002/pmic.200402057
22. Cvetkovic J, Sofronic-Milosavljevic L, Ilic N, Gnjatovic M, Nagano I, Gruden-Movsesijan A. Immunomodulatory potential of particular Trichinella spiralis muscle larvae excretory–secretory components. International Journal for Parasitology. 2016;46(13):833–842. doi:10.1016/j.ijpara.2016.07.008
23. Ilic N, Bojic-Trbojevic Z, Lundström-Stadelmann B, Cujic D, Mitic I, Gruden-Movsesijan A. IMMUNOMODULATORY COMPONENTS OF TRICHINELLA SPIRALIS EXCRETORY-SECRETORY PRODUCTS WITH LACTOSE-BINDING SPECIFICITY. EXCLI J. 2022;21:793–813. doi:10.17179/excli2022-4954
24. Kosanović M, Cvetković J, Gruden-Movsesijan A, et al. Trichinella spiralis muscle larvae release extracellular vesicles with immunomodulatory properties. Parasite Immunol. 2019;41(10). doi:10.1111/pim.12665
25. Gao X, Yang Y, Liu X, et al. Extracellular vesicles from Trichinella spiralis: proteomic analysis and protective immunity. PLoS Negl Trop Dis. 2022;16(6):e0010528. doi:10.1371/journal.pntd.0010528
26. Gao X, Yang Y, Liu X, et al. Extracellular vesicles derived from Trichinella spiralis prevent colitis by inhibiting M1 macrophage polarization. Acta Trop. 2021;213:105761. doi:10.1016/j.actatropica.2020.105761
27. Sahu T, Ratre YK, Chauhan S, Lvks B, Nair MP, Verma HK. Nanotechnology based drug delivery system: current strategies and emerging therapeutic potential for medical science. J Drug Delivery Sci Technol. 2021;63:102487. doi:10.1016/j.jddst.2021.102487
28. Pardeshi SR, Nikam A, Chandak P, Mandale V, Naik JB, Giram PS. Recent advances in PLGA based nanocarriers for drug delivery system: a state of the art review. Int J Polym Mater Polym Biomater. 2023;72(1):49–78. doi:10.1080/00914037.2021.1985495
29. Cappellano G, Comi C, Chiocchetti A, Dianzani U. Exploiting PLGA-based biocompatible nanoparticles for next-generation tolerogenic vaccines against autoimmune disease. Int J mol Sci. 2019;20(1):204. doi:10.3390/ijms20010204
30. Shan X, Liu C, Li F, Ouyang C, Gao Q, Zheng K. Nanoparticles vs. nanofibers: a comparison of two drug delivery systems on assessing drug release performance in vitro. Des. Monomers Polym. 2015;18(7):678–689. doi:10.1080/15685551.2015.1070500
31. Nikmaram N, Roohinejad S, Hashemi S, et al. Emulsion-based systems for fabrication of electrospun nanofibers: food, pharmaceutical and biomedical applications. RSC Adv. 2017;7(46):28951–28964. doi:10.1039/C7RA00179G
32. Khan J, Khan A, Khan MQ, Khan H. Applications of co-axial electrospinning in the biomedical field. Next Materials. 2024;3:100138. doi:10.1016/j.nxmate.2024.100138
33. Milosevic M, Stojanovic D, Simic V, et al. A computational model for drug release from PLGA implant. Materials. 2018;11(12):2416. doi:10.3390/ma11122416
34. Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. doi:10.1016/s0142-9612(00)00115-0
35. Radisavljevic A, Stojanovic DB, Perisic S, et al. Cefazolin-loaded polycaprolactone fibers produced via different electrospinning methods: characterization, drug release and antibacterial effect. Eur. J. Pharm. Sci. 2018;124:26–36. doi:10.1016/j.ejps.2018.08.023
36. Buzgo M, Mickova A, Rampichova M, Doupnik M. 11 - Blend electrospinning, coaxial electrospinning, and emulsion electrospinning techniques. In: Focarete ML, Tampieri A editors. Core-Shell Nanostructures for Drug Delivery and Theranostics. Woodhead Publishing Series in Biomaterials. Woodhead Publishing; 2018:325–347. doi:10.1016/B978-0-08-102198-9.00011-9.
37. Gamble HR, Bessonov AS, Cuperlovic K, et al. International commission on Trichinellosis: recommendations on methods for the control of Trichinella in domestic and wild animals intended for human consumption. Vet Parasitol. 2000;93(3–4):393–408. doi:10.1016/s0304-4017(00)00354-x
38. Ilic N, Worthington JJ, Gruden-Movsesijan A, Travis MA, Sofronic-Milosavljevic L, Grencis RK. Trichinella spiralis antigens prime mixed Th1/Th2 response but do not induce de novo generation of Foxp3+ T cells in vitro. Parasite Immunol. 2011;33(10):572–582. doi:10.1111/j.1365-3024.2011.01322.x
39. Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel). 2011;3(3):1377–1397. doi:10.3390/polym3031377
40. Filipović N, Veselinović L, Ražić S, et al. Poly (ε-caprolactone) microspheres for prolonged release of selenium nanoparticles. Mater Sci Eng C. 2019;96:776–789. doi:10.1016/j.msec.2018.11.073
41. Radojević D, Bekić M, Gruden-Movsesijan A, et al. Myeloid-derived suppressor cells prevent disruption of the gut barrier, preserve microbiota composition, and potentiate immunoregulatory pathways in a rat model of experimental autoimmune encephalomyelitis. Gut Microbes. 2022;14(1):2127455. doi:10.1080/19490976.2022.2127455
42. Xia WW, Zhao J, Zheng Y, et al. Active soil nitrifying communities revealed by in situ transcriptomics and microcosm-based stable-isotope probing. Appl Environ Microbiol. 2020;86(23):e01807–20. doi:10.1128/AEM.01807-20
43. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
44. Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi:10.1038/s41587-019-0209-9
45. McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi:10.1371/journal.pone.0061217
46. Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. 2015;26:27663. doi:10.3402/mehd.v26.27663
47. Douglas GM, Maffei VJ, Zaneveld JR, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38(6):685–688. doi:10.1038/s41587-020-0548-6
48. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
49. Ji W, Yang F, Seyednejad H, et al. Biocompatibility and degradation characteristics of PLGA-based electrospun nanofibrous scaffolds with nanoapatite incorporation. Biomaterials. 2012;33(28):6604–6614. doi:10.1016/j.biomaterials.2012.06.018
50. Milosevic M, Stojanovic DB, Simic V, et al. Preparation and modeling of three‐layered PCL/PLGA/PCL fibrous scaffolds for prolonged drug release. Sci Rep. 2020;10(1):11126. doi:10.1038/s41598-020-68117-9
51. Guzmán-Soria A, Moreno-Serna V, Canales DA, García-Herrera C, Zapata PA, Orihuela PA. Effect of Electrospun PLGA/Collagen Scaffolds on Cell Adhesion, Viability, and Collagen Release: potential Applications in Tissue Engineering. Polymers. 2023;15(5):1079. doi:10.3390/polym15051079
52. de AFG, Gallardo-Velazquez T, Osorio-Revilla G, et al. Feasibility study for the detection of Trichinella spiralis in a murine model using mid-Fourier transform infrared spectroscopy (MID-FTIR) with attenuated total reflectance (ATR) and soft independent modelling of class analogies (SIMCA). Vet Parasitol. 2012;190(3):496–503. doi:10.1016/j.vetpar.2012.07.004
53. Vasita R, Mani G, Agrawal CM, Katti DS. Surface hydrophilization of electrospun PLGA micro-/nano-fibers by blending with Pluronic® F-108. Polymer. 2010;51(16):3706–3714. doi:10.1016/j.polymer.2010.05.048
54. Nagiah N, Murdock CJ, Bhattacharjee M, Nair L, Laurencin CT. Development of tripolymeric triaxial electrospun fibrous matrices for dual drug delivery applications. Sci Rep. 2020;10(1):609. doi:10.1038/s41598-020-57412-0
55. Schudel A, Francis DM, Thomas SN. Material design for lymph node drug delivery. Nat Rev Mater. 2019;4(6):415–428. doi:10.1038/s41578-019-0110-7
56. Alexandre YO, Mueller SN. Splenic stromal niches in homeostasis and immunity. Nat Rev Immunol. 2023;23(11):705–719. doi:10.1038/s41577-023-00857-x
57. Melero-Jerez C, Alonso-Gómez A, Moñivas E, et al. The proportion of myeloid-derived suppressor cells in the spleen is related to the severity of the clinical course and tissue damage extent in a murine model of multiple sclerosis. Neurobiol Dis. 2020;140:104869. doi:10.1016/j.nbd.2020.104869
58. Burrows DJ, McGown A, Jain SA, et al. Animal models of multiple sclerosis: from rodents to zebrafish. Mult Scler. 2019;25(3):306–324. doi:10.1177/1352458518805246
59. Kurschus FC. T cell mediated pathogenesis in EAE: molecular mechanisms. Biomed J. 2015;38(3):183–193. doi:10.4103/2319-4170.155590
60. Ghezzi L, Cantoni C, Pinget GV, Zhou Y, Piccio L. Targeting the gut to treat multiple sclerosis. J Clin Invest. 2021;131(13):e143774. doi:10.1172/JCI143774
61. Gholamzad M, Ebtekar M, Shafiee Ardestani M. Intravenous injection of myelin oligodendrocyte glycoprotein-coated PLGA microparticles have tolerogenic effects in experimental autoimmune encephalomyelitis. Iran J Allergy Asthma Immunol. 2017;16(3):271–281.
62. Li PY, Bearoff F, Zhu P, et al. PEGylation enables subcutaneously administered nanoparticles to induce antigen-specific immune tolerance. J Controlled Rel. 2021;331:164–175. doi:10.1016/j.jconrel.2021.01.013
63. Saito E, Kuo R, Kramer KR, et al. Design of biodegradable nanoparticles to modulate phenotypes of antigen-presenting cells for antigen-specific treatment of autoimmune disease. Biomaterials. 2019;222:119432. doi:10.1016/j.biomaterials.2019.119432
64. Liu M, Feng D, Liang X, et al. Old dog new tricks: PLGA microparticles as an adjuvant for insulin peptide fragment-induced immune tolerance against type 1 diabetes. Mol Pharm. 2020;17(9):3513–3525. doi:10.1021/acs.molpharmaceut.0c00525
65. Cappellano G, Woldetsadik AD, Orilieri E, et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. 2014;32(43):5681–5689. doi:10.1016/j.vaccine.2014.08.016
66. Wang H, Shang J, He Z, et al. Dual peptide nanoparticle platform for enhanced antigen-specific immune tolerance for the treatment of experimental autoimmune encephalomyelitis. Biomater Sci. 2022;10(14):3878–3891. doi:10.1039/D2BM00444E
67. Yoshida M, Babensee JE. Poly(lactic-co-glycolic acid) enhances maturation of human monocyte-derived dendritic cells. J Biomed Mater Res A. 2004;71(1):45–54. doi:10.1002/jbm.a.30131
68. Sharp FA, Ruane D, Claass B, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106(3):870–875. doi:10.1073/pnas.0804897106
69. Ding T, Sun J, Zhang P. Immune evaluation of biomaterials in TNF-alpha and IL-1beta at mRNA level. J Mater Sci Mater Med. 2007;18(11):2233–2236. doi:10.1007/s10856-007-3014-9
70. Nicolete R, dos SDF, Faccioli L. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int Immunopharmacol. 2011;11(10):1557–1563. doi:10.1016/j.intimp.2011.05.014
71. Ma S, Feng X, Liu F, Wang B, Zhang H, Niu X. The pro-inflammatory response of macrophages regulated by acid degradation products of poly(lactide-co-glycolide) nanoparticles. Eng. Life Sci. 2021;21(10):709–720. doi:10.1002/elsc.202100040
72. Wagner RD, Johnson SJ, Danielsen ZY, et al. Polyethylene glycol-functionalized poly (lactic acid-co-glycolic acid) and graphene oxide nanoparticles induce pro-inflammatory and apoptotic responses in Candida albicans-infected vaginal epithelial cells. PLoS One. 2017;12. doi:10.1371/journal.pone.0175250.
73. Derdelinckx J, Cras P, Berneman ZN, Cools N. Antigen-specific treatment modalities in MS: the past, the present, and the future. Front Immunol. 2021;12:624685. doi:10.3389/fimmu.2021.624685
74. Liu SJ, Kau Y, Chou C, Chen JK, Wu R, Yeh W. Electrospun PLGA/collagen nanofibrous membrane as early-stage wound dressing. J Membr Sci. 2010;355(1–2):53–59. doi:10.1016/J.MEMSCI.2010.03.012
75. Huang J, Zhou X, Shen Y, et al. Asiaticoside loading into PLGA electrospun nanofibers attenuates host inflammatory response and promotes M2 macrophage polarization. J Biomed Mater Res Part A. 2019. doi:10.1002/jbm.a.36793
76. Holan V, Chudickova M, Trošan P, et al. Cyclosporine A-loaded and stem cell-seeded electrospun nanofibers for cell-based therapy and local immunosuppression. J Controlled Rel. 2011;156(3):406–412. doi:10.1016/j.jconrel.2011.07.022
77. Lee Y, Kwon J, Khang G, Lee D. Reduction of inflammatory responses and enhancement of extracellular matrix formation by vanillin-incorporated poly(lactic-co-glycolic acid) scaffolds. Tissue Eng Part A. 2012;18(19–20):1967–1978. doi:10.1089/ten.tea.2012.0001
78. Chor A, Takiya CM, Dias ML, et al. In Vitro and in vivo cell-interactions with electrospun poly (lactic-co-glycolic acid) (PLGA): morphological and Immune Response Analysis. Polymers (Basel). 2022;14(20):4460. doi:10.3390/polym14204460
79. Fabre MV, Beiting DP, Bliss SK, Appleton JA. Immunity to Trichinella spiralis muscle infection. Vet Parasitol. 2009;159(3–4):245–248. doi:10.1016/j.vetpar.2008.10.051
80. Beiting DP, Gagliardo LF, Hesse M, Bliss SK, Meskill D, Appleton JA. Coordinated control of immunity to muscle stage Trichinella spiralis by IL-10, regulatory T cells, and TGF-beta. J Immunol. 2007;178(2):1039–1047. doi:10.4049/jimmunol.178.2.1039
81. ying HY, Qi M, Zhang M, ze LH, Zhi YD. Degradation mechanisms of poly (lactic-co-glycolic acid) films in vitro under static and dynamic environment. Trans Nonferrous met Soc China. 2006;16:s293–s297. doi:10.1016/S1003-6326(06)60194-5
82. Zhan XX, Liu Y, Yang JF, et al. All-trans-retinoic acid ameliorates experimental allergic encephalomyelitis by affecting dendritic cell and monocyte development. Immunology. 2013;138(4):333–345. doi:10.1111/imm.12040
83. Turano E, Scambi I, Bonafede R, et al. Extracellular vesicles from adipose mesenchymal stem cells target inflamed lymph nodes in experimental autoimmune encephalomyelitis. Cytotherapy. 2024;26(3):276–285. doi:10.1016/j.jcyt.2023.12.007
84. Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17(1):30–48. doi:10.1038/nri.2016.116
85. Brandum EP, Jørgensen AS, Rosenkilde MM, Hjortø GM. Dendritic cells and CCR7 expression: an important factor for autoimmune diseases, chronic inflammation, and cancer. Int J mol Sci. 2021;22(15):8340. doi:10.3390/ijms22158340
86. Podkowa A, Miller RJ, Motl R, Fish R, Oelze M. Focused ultrasound treatment of cervical lymph nodes in rats with EAE: a pilot study. Ultrasound Med Biol. 2016;42(12):2957–2964. doi:10.1016/j.ultrasmedbio.2016.08.007
87. Zwam M, Huizinga R, Melief M, et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med. 2009;87(3):273–286. doi:10.1007/s00109-008-0421-4
88. van ZM, Huizinga R, Heijmans N, et al. Surgical excision of CNS‐draining lymph nodes reduces relapse severity in chronic‐relapsing experimental autoimmune encephalomyelitis. J Pathol. 2009;217. doi:10.1002/path.2476.
89. Sagar D, Lamontagne A, Foss CA, Khan ZK, Pomper MG, Jain P. Dendritic cell CNS recruitment correlates with disease severity in EAE via CCL2 chemotaxis at the blood–brain barrier through paracellular transmigration and ERK activation. J Neuroinflammation. 2012;9(1):245. doi:10.1186/1742-2094-9-245
90. Mansilla MJ, Hilkens CMU, Martínez-Cáceres EM. Challenges in tolerogenic dendritic cell therapy for autoimmune diseases: the route of administration. Immunother Adv. 2023;3(1). doi:10.1093/immadv/ltad012
91. Owens T, Benmamar-Badel A, Wlodarczyk A, et al. Protective roles for myeloid cells in neuroinflammation. Scandinavian J Immunol. 2020;92(5):e12963. doi:10.1111/sji.12963
92. Knier B, Hiltensperger M, Sie C, et al. Myeloid-derived suppressor cells control B cell accumulation in the central nervous system during autoimmunity. Nat Immunol. 2018;19(12):1341–1351. doi:10.1038/s41590-018-0237-5
93. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485–498. doi:10.1038/s41577-020-00490-y
94. Radomir L, Kramer MP, Perpinial M, et al. The survival and function of IL-10-producing regulatory B cells are negatively controlled by SLAMF5. Nat Commun. 2021;12(1):1893. doi:10.1038/s41467-021-22230-z
95. Okada Y, Ochi H, Fujii C, et al. Signaling via toll-like receptor 4 and CD40 in B cells plays a regulatory role in the pathogenesis of multiple sclerosis through interleukin-10 production. J Autoimmun. 2018;88:103–113. doi:10.1016/j.jaut.2017.10.011
96. Sutmuller R, den BMD, Kramer M, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116(2):485–494. doi:10.1172/JCI25439
97. Chen J, Chia N, Kalari KR, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. 2016;6(1):28484. doi:10.1038/srep28484
98. Nouri M, Bredberg A, Weström B, Lavasani S. Intestinal barrier dysfunction develops at the onset of experimental autoimmune encephalomyelitis, and can be induced by adoptive transfer of auto-reactive T cells. PLoS One. 2014;9(9):e106335. doi:10.1371/journal.pone.0106335
99. Duc D, Vigne S, Bernier-Latmani J, et al. Disrupting myelin-specific Th17 cell gut homing confers protection in an adoptive transfer experimental autoimmune encephalomyelitis. Cell Rep. 2019;29(2):378–390.e4. doi:10.1016/j.celrep.2019.09.002
100. Ivanov II, Atarashi K, Manel N, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell. 2009;139(3):485–498. doi:10.1016/j.cell.2009.09.033
101. Metwaly A, Jovic J, Waldschmitt N, et al. Diet prevents the expansion of segmented filamentous bacteria and ileo-colonic inflammation in a model of Crohn’s disease. Microbiome. 2023;11(1):66. doi:10.1186/s40168-023-01508-y
102. Berer K, Krishnamoorthy G. Microbial view of central nervous system autoimmunity. FEBS Lett. 2014;588(22):4207–4213. doi:10.1016/j.febslet.2014.04.007
103. Tremlett H, Fadrosh DW, Faruqi AA, et al. Gut microbiota in early pediatric multiple sclerosis: a case-control study. Eur J Neurol. 2016;23(8):1308–1321. doi:10.1111/ene.13026
104. Miller PG, Bonn MB, Franklin CL, Ericsson AC, McKarns SC. TNFR2 deficiency acts in concert with gut microbiota to precipitate spontaneous sex-biased central nervous system demyelinating autoimmune disease. J Immunol. 2015;195(10):4668–4684. doi:10.4049/jimmunol.1501664
105. Olaisen M, Flatberg A, van B GA, et al. Bacterial mucosa-associated microbiome in inflamed and proximal noninflamed ileum of patients with Crohn’s disease. Inflamm Bowel Dis. 2021;27(1):12–24. doi:10.1093/ibd/izaa107
106. Schirmer M, Smeekens SP, Vlamakis H, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016;167(4):1125–1136.e8. doi:10.1016/j.cell.2016.10.020
107. Dm JI, Goertz JE, Marin IA, Costello J, Overall CC, Gaultier A. Experimental autoimmune encephalomyelitis is associated with changes of the microbiota composition in the gastrointestinal tract. Sci Rep. 2020;10(1). doi:10.1038/s41598-020-72197-y
108. Ventura RE, Iizumi T, Battaglia T, et al. Gut microbiome of treatment-naïve MS patients of different ethnicities early in disease course. Sci Rep. 2019;9(1):16396. doi:10.1038/s41598-019-52894-z
109. Van Hul M, Le Roy T, Prifti E, et al. From correlation to causality: the case of Subdoligranulum. Gut Microbes. 2020;12(1):1849998. doi:10.1080/19490976.2020.1849998
110. Inami A, Kiyono H, Kurashima Y. ATP as a pathophysiologic mediator of bacteria-host crosstalk in the gastrointestinal tract. Int J mol Sci. 2018;19(8):2371. doi:10.3390/ijms19082371
111. Balakrishnan B, Luckey D, Bodhke R, et al. Prevotella histicola protects from arthritis by expansion of allobaculum and augmenting butyrate production in humanized mice. Front Immunol. 2021;12:609644. doi:10.3389/fimmu.2021.609644
112. Zhao X, Zhang Z, Hu B, Huang W, Yuan C, Zou L. Response of gut microbiota to metabolite changes induced by endurance exercise. Front Microbiol. 2018;9:765. doi:10.3389/fmicb.2018.00765
113. Bhargava P, Smith MD, Mische L, et al. Bile acid metabolism is altered in multiple sclerosis and supplementation ameliorates neuroinflammation. J Clin Invest. 2020;130(7):3467–3482. doi:10.1172/JCI129401
114. Ju T, Kong JY, Stothard P, Willing BP. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019;13(6):1520–1534. doi:10.1038/s41396-019-0364-5
115. Fischbach MA, Sonnenburg JL. Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe. 2011;10(4):336–347. doi:10.1016/j.chom.2011.10.002
116. Lee JS, Wang RX, Alexeev EE, et al. Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J Biol Chem. 2018;293(16):6039–6051. doi:10.1074/jbc.RA117.000269
117. Hu J, Deng F, Zhao B, et al. Lactobacillus murinus alleviate intestinal ischemia/reperfusion injury through promoting the release of interleukin-10 from M2 macrophages via Toll-like receptor 2 signaling. Microbiome. 2022;10(1):38. doi:10.1186/s40168-022-01227-w
118. Ma Y, Liu T, Fu J, et al. Lactobacillus acidophilus exerts neuroprotective effects in mice with traumatic brain injury. J Nutr. 2019;149(9):1543–1552. doi:10.1093/jn/nxz105
119. Cox LM, Maghzi AH, Liu S, et al. Gut microbiome in progressive multiple sclerosis. Ann. Neurol. 2021;89(6):1195–1211. doi:10.1002/ana.26084
120. Wilding EI, Brown JR, Bryant AP, et al. Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J Bacteriol. 2000;182(15):4319–4327. doi:10.1128/jb.182.15.4319-4327.2000
121. Lai Y, Masatoshi H, Ma Y, Guo Y, Zhang B. Role of Vitamin K in intestinal health. Front Immunol. 2022;12. doi:10.3389/fimmu.2021.791565
122. Lasemi R, Kundi M, Moghadam NB, Moshammer H, Hainfellner JA. Vitamin K2 in multiple sclerosis patients. Wien Klin Wochenschr. 2018;130(9–10):307–313. doi:10.1007/s00508-018-1328-x
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Role of Gut Microbiota in the Development of Some Autoimmune Diseases
Cui X, Cong Y
Journal of Inflammation Research 2025, 18:4409-4419
Published Date: 25 March 2025