")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Exploring the Potential of Pyridine Carboxylic Acid Isomers to Discover New Enzyme Inhibitors
Authors Yaqoob S, Khan FA , Tanveer N, Ali S, Hameed A, El-Seedi H, Jiang ZH , Wang Y
Received 25 January 2025
Accepted for publication 1 May 2025
Published 20 May 2025 Volume 2025:19 Pages 4039—4091
DOI https://doi.org/10.2147/DDDT.S513461
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Sana Yaqoob,1– 3,* Farooq-Ahmad Khan,1– 3,* Nimra Tanveer,2,3 Shujaat Ali,2,3 Abdul Hameed,4 Hesham El-Seedi,5 Zi-Hua Jiang,6 Yan Wang1,3
1Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (Ministry of Education of China), Guangxi Key Laboratory of Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, Guilin, Guangxi, People’s Republic of China; 2Third World Center for Science and Technology, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Sindh, Pakistan; 3H. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Sindh, Pakistan; 4Department of Chemistry, University of Sahiwal, Sahiwal, Punjab, Pakistan; 5Department of Chemistry, Faculty of Science, Islamic University of Madinah, Madinah 42351, Saudi Arabia; 6Department of Chemistry, Lakehead University, Thunder Bay, ON, Canada
*These authors contributed equally to this work
Correspondence: Farooq-Ahmad Khan, Email [email protected] Yan Wang, Email [email protected]
Abstract: Pyridine carboxylic acid isomers — picolinic acid, nicotinic acid, and isonicotinic acid — have historically resulted in a plethora of drugs against tuberculosis, cancer, diabetes, Alzheimer’s, angina, dementia, depression, allergy, respiratory acidosis, psoriasis, acne, hypertension, hyperlipidemia, HIV/AIDS (specifically HIV-1), among others. Despite the large number of therapeutic agents derived from these isomers, the research involving these scaffolds is still exceptionally active. The current surge in enzyme inhibitory activities by the compounds derived from them has further created space for the discovery of new drug candidates. This review focuses on the medicinal relevance of these isomers by analyzing structure-activity relationships (SARs) and highlighting emerging trends from patents filed over the last decade. Notably, pharmaceutical giants like Bayer, Bristol-Myers Squibb, Novartis, Curis, and Aurigene have developed enzyme inhibitors based on these scaffolds with nanomolar potency. The role of these isomers in the development of antiviral agents, including protease inhibitors, is also discussed. Overall, this review brings to the readers, a pragmatic opportunity to comprehend the recent literature, highlighting the scaffolds’ importance in the design of new enzyme inhibitors. Furthermore, it discusses the structure-activity relationship of pyridine carboxylic acid-derived compounds and highlights the current patenting trends in medicinal chemistry.
Keywords: pyridine, enzyme inhibitors, nitrogen heterocycles, patents, pharmaceuticals, current trend, substituent effect
Graphical Abstract:

Introduction
Heterocyclic chemistry is a rich source of unique and innovative drug formulations.1–3 Among various nitrogen-containing heterocycles, pyridine has found extensive use in contemporary drug design due to its ability to accommodate diverse substitution patterns on the ring, thereby allowing its incorporation into countless molecules with a wide range of biological activities.4–9 Pyridine stands out as the second most widely utilized nitrogen heterocycle in FDA-approved pharmaceuticals, securing the leading position among aromatic compounds.10–12 Pyridine continues to fascinate researchers, as shown by our database analysis.9,13–17 Throughout the last decade from 2013 to 2023, there was a consistent stream of research papers and patents featuring the word “pyridine” in their titles (Figure 1). These numbers not only highlight the enduring fascination of the scientific community but are also a testament to pyridine’s relevance in technological patents.
 |
Figure 1 Number of research papers and patents featuring the word “pyridine” in their titles over the past one decade (2014–2024); Source: SciFindern. |
Analysis of the United States FDA database reveals a notable presence of pyridine, as 14% of the total drugs contain this moiety (Figure 2). Pyridine’s substantial representation in the database not only highlights its key role in the pharmaceutical landscape but also the potential of this heterocyclic moiety in shaping the future of therapeutics development.
 |
Figure 2 N-Heterocyclic drugs distribution in FDA database. |
Center for Drug Evaluation and Research (CDER) is a vital part of the FDA and plays a crucial role in ensuring the safety and efficacy of drugs in the United States. Over the past decade (2013–2023), CDER has approved a myriad of New Molecular Entities (NMEs) with a marked presence of pyridine-containing NMEs in the annual approvals (Figure 3). Year after year, this continued inclusion of pyridine-containing NMEs emphasizes the integral role of this heterocyclic moiety in the pharmaceutical landscape, thereby emphasizing its potential and value in the realms of therapeutic advancements.18
 |
Figure 3 New molecular entities (NMEs) containing pyridine moiety, which were approved by FDA over the past one decade (2013–2023). |
Figure 4 offers an in-depth analysis of the FDA database, which gives a remarkable insight into the substitution patterns of pyridine-containing drugs approved during the last decade. In terms of substitution types, a substantial proportion of these drugs contain di-substituted pyridine, closely followed by tri-substituted and mono-substituted pyridines. While some drugs contained tetra-substituted and hexa-substituted pyridine, penta-substituted variants were notably absent from the database (Figure 4a).
 |
Figure 4 (a) Analysis of substitution type; and (b) examination of substitution pattern in pyridine containing drugs approved by FDA over the past one decade (2013–2023). |
Upon careful examination of substitution patterns within pyridine-containing pharmaceuticals approved between 2013 and 2023, a distinct trend emerges. There is a notable inclination towards substitution at position 2 of the pyridine ring, followed by significant instances of substitution at positions 5 and 3, especially when multiple substituents are present. Interestingly, some drugs feature direct substitution on the nitrogen atom within the ring structure (Figure 4b). These observations highlight the dynamic nature of the drug design and the adaptability of this heterocyclic moiety in pharmaceutical developments over the years.
In this comprehensive review, we delve into the intricate realm of pharmaceuticals derived from pyridine carboxylic acid isomers—an exploration critical to the contemporary understanding of medicinal chemistry. Through meticulous analysis, this study unveils the therapeutic significance and diverse applications of these compounds within the realm of approved drugs as well as patent communications. Pyridine carboxylic acid derivatives display a broad spectrum of biological activities and many of them have been approved for use in the clinic to treat various conditions including infections, inflammation, and cancers. This broad therapeutic potential is closely linked with their structural features. For example, the pyridine ring, which is aromatic in nature and electron deficient, facilitates π-π stacking and hydrogen bond interactions with biological targets, thereby enhancing the binding affinity. The carboxylic group contributes additional polarity and can co-ordinate with metal ions, a property particularly useful in enzyme inhibition. Moreover, the ease of substitution at various positions of pyridine ring allows for structural flexibility and fine-tuning of activity and selectivity. Together, these features make pyridine carboxylic acid derivatives highly versatile scaffolds in medicinal chemistry. In recent years, there have been a good number of patents communicating the role of various pyridine carboxylic acid derivatives in this area of enzyme inhibition. Despite the significant impact of pyridine carboxylic acid-containing compounds on the pharmaceutical industry, there have been no review articles published specifically based on pyridine carboxylic acid derivatives. In the following sections, we provide a comprehensive review of approved drugs derived from pyridine carboxylic acid isomers and highlights of patents published in the last decade (2013–2023) which have disclosed a great number of pyridine carboxylic acid derivatives as potent enzyme inhibitors and potential therapeutic agents to treat various diseases.
Approved Drugs Derived from Pyridine Carboxylic Acid Isomers
The intricate interplay between molecular structures and pharmacological effects underscores the pivotal role played by pyridine carboxylic acid isomers in shaping the landscape of modern pharmaceuticals. The compounds stemming from pyridine carboxylic acids hold potential; likely due to the presence of nitrogen in the aromatic ring. The core is famous for its three isomers: picolinic acid (1), nicotinic acid (2), and isonicotinic acid (3) with carboxylic acid substitution at 2nd, 3rd, and 4th positions on the pyridine, respectively (Figure 5a). A detailed representation of natural products, which are categorized based on specific pyridine carboxylic acid isomers is also shown in Figure 5b. Many of them have garnered recognition as approved therapeutics by the European Medicines Agency (EMA) or the Food and Drug Administration (FDA) of the United States. This includes natural drugs containing the picolinic acid moiety, like streptonigrin (4) with notable antitumor and antibacterial properties, along with antibiotics like fusaric acid (5) and its derivative, (+)-S-fusarnolic acid, both obtained from Fusarium heterosporium.19,20 In this category, another group of antibiotics, promothiocin (6) isolated from Streptomyces sp. SF2741, and pyridomycin (7) obtained from Dactylosporangium fulvum were found promising against tuberculosis.21,22 Nicotinic acid-derived natural products include several noteworthy examples, like Ilicifoliunine A (8), sourced from Maytenus ilicifolia, plagiochianin B (9) from Plagiochila duthiana, and clivimine (10) from Clivia miniate, all contributing towards the arsenal of natural pharmacological agents.23–25 It’s worth noting that the figure presented in this context highlights plantagonine (11) from Plantago psyllium, which is a unique alkaloid containing the isonicotinic acid structure.26 Notably, the ubiquitous niacin (vitamin B) is part of nicotinuric acid (12), trigonelline (13) sourced from Trigonella foenumgraecum, and elexacaftor (14) having anti-cystic fibrosis properties.27–30 All these examples highlight the importance of isomeric variations of pyridine carboxylic acids, their derivatives, and related natural products in the pursuit of pharmacologically active agents.
 |
Figure 5 (a) Isomeric variants of pyridine carboxylic acid; (b) Representative examples of natural products and EMA/FDA approved natural product drugs. |
Representative FDA-approved drugs stemming from different isomers of pyridine carboxylic acids are shown in Figure 6. Regorafenib (15) can tackle certain cancers as well.31 Sorafenib (16) was approved in 2005 to help with liver, kidney, and thyroid cancers.32 Lasmiditan (17) marked a milestone in migraine management, securing approval in 2019.33 Picolinic acid-derived Quinupristin (18) emerged in 1999 as a new antibiotic for tough infections.34 Nevirapine (19) combated the challenges of HIV with a significant impact in 2016.35 Ubrogepant (20) was sanctioned in 2019 as a CGRP and 5HT1F antagonist to tackle migraines.36 Flotufolastat F-18 (21) with cutting-edge imaging capabilities aligned with the dynamic needs of prostate cancer diagnostics.37 Tazarotene (22) got approval for treating skin issues in 2017.38 Serdexmethylphenidate (23) signified a breakthrough in addressing attention deficit hyperactivity disorder (ADHD).39 By 2019, elexacaftor (24) was approved to target cystic fibrosis.29 In 1983, Nicotinamide (25) gained recognition as a nutraceutical, sanctioned for the treatment of pellagra, highlighting its vital role in nutritional therapy.40 Avatrombopag (26) received approval for treating blood issues in 2018.41 Asciminib (27) as a tyrosine kinase inhibitor (TKI) was approved in 2021 to handle leukemia.42 The nicotinic acid-derived FDA-approved drugs include Metyrapone (28), used to treat a hormone-related condition. Isoniazid (29) initially emerged as an anti-tuberculosis agent, along with protionamide (30) and ethionamide (31), to fight hard-to-treat tuberculosis.43–46
 |
Figure 6 FDA approved drugs derived from the isomers of pyridine carboxylic acid, their indication and year of approval. |
Ethionamide (31), a second-line tuberculosis treatment, is only used in combination therapy or the treatment of drug-resistant tuberculosis.47 Nialamide (32) is a drug with anti-thrombotic properties. Its effects on the rabbit ear were observed when the internal surface of the arteries and veins were damaged, and a powerful thrombotic effect was observed by preventing intravascular thrombosis without inhibition of extravascular clotting.48 Nialamide also had antidepressant properties, but it was withdrawn by the US, Canada, and UK in the 1960s due to serious side effects caused by tyrosine-containing foods.49 Before 1950, Iproniazid (33), a medication renowned for its anti-tuberculosis properties, demonstrated significant improvements over earlier treatments.50 Notably, it proved more effective than isoniazid in the treatment of bone and joint lesions, leading to its widespread recognition and safe usage during that era.51,52 An experiment at Sea View Hospital Staten, Island N.Y., in 1951 revealed that iproniazid caused certain toxic side effects in patients when compared to the other two tuberculosis medications, including isoniazid. Despite this, the use of iproniazid continued by controlling and encountering its toxicity, and benefits exhibited in the case of osseous-tuberculous, were transferred to use in other lesions.53 Iproniazid was originally developed to treat tuberculosis, but its mood-relaxing properties were discovered in 1952, making it the first commercialized antidepressant drug. This drug was used for decades for antidepressant treatment but was withdrawn in several countries, including the USA, due to hepatotoxic side effects. The drug has been replaced by other medication but it has an assigned place in therapy.54,55 The discontinuation of nialamide (32), iproniazid (33), and methaniazide (34) reflects the dynamic nature of pyridine-4-carboxylic acid in pharmaceutical evolution.
The compounds 35–48 presented in Figure 7 comprehend a spectrum of analgesic effects, accentuating their significance across diverse inflammatory conditions. Nicomorphine (35), a potent opioid analgesic, and nicocodeine (36), an opioid receptor agonist, offer robust pain relief.56 Dexamethasone isonicotinate (37) stands out as a notable anti-inflammatory agent, while the 2021-introduced CGRP antagonist,57 (38), demonstrates promise in inhibiting neurogenic inflammation. Clonixin (39) and morniflumate (40), both COX-1 and COX-2 inhibitors, exemplify the NSAID class, displaying their prowess in alleviating inflammation.58,59 Menthyl nicotinate (41) serves to foster circulation and relief in musculoskeletal conditions.60 Noteworthy is niflumic acid (42) selectivity in addressing rheumatoid arthritis-associated inflammation through COX-2 inhibition.61 Other compounds, including nifenazone (43), a broad-spectrum analgesic, and derivatives like nicoboxil (44), propyl nicotinate (45), and methyl nicotinate (46), contribute distinct pain management perspectives. Particularly, 2-(p-tolyl)ethyl nicotinate (47) — an FDA orphan drug — offers promise against primary biliary cholangitis.62–64 Finally, amlexanox (48), an inhibitor of TBK1 and IKK-e, exhibits potential in modulating inflammatory responses for aphthous ulcer treatment.65
 |
Figure 7 Pyridine carboxylic acid derived anti-inflammatory drugs. |
Figure 8 encapsulates a compilation of antimicrobial agents, originating from different pyridine carboxylic acid isomers. Among antibiotics derived from picolinic acid, streptonigrin (49) is a noteworthy representative. Recent studies suggest that this isomer has the potential to block the release of free fatty acids from fat cells and increase the activity of lipoprotein lipase, offering promise as both a combination therapy and an economical standalone treatment for lowering lipid levels.66–68 Notably, saquinavir (50), approved by the FDA in 1997, exhibited protease inhibition activity against HIV.69 Further, pristinamycin (51) is another significant antibiotic in this context.70 In the domain of antibiotics featuring a nicotinic scaffold, the array includes cefpiramide (52), a third-generation antibiotic, and the well-established nalidixic acid (53), introduced in 1986 by FDA.71,72 Concurrently, enoxacin (54), approved by FDA in 2017, demonstrates broad-spectrum antibacterial potential.73 Additionally, Gemifloxacin (55), established in 2003, further enriches this spectrum.74 Finally, the category of antibiotics incorporating an isonicotinoyl moiety includes cefalonium (56), as a first-generation antibiotic.75 Equally, cefsulodin (57) exhibits specificity against Pseudomonas aeruginosa. In addressing tuberculosis, aconiazide (58) assumes a pivotal role.76–79 Notably, enisamium (59), a Russian introduction, offers antiviral efficacy against influenza.80 Thus, the figure illuminates the extensive repertoire of antibiotic drugs emerging from pyridine carboxylic acid derivatives, delineating their expansive therapeutic potential across diverse infectious conditions.
 |
Figure 8 Antimicrobial drugs originating from pyridine carboxylic acid isomers. |
Figure 9 provides a comprehensive assemblage of antidyslipidemic agents derived from pyridine carboxylic acid isomers, thereby revealing innovative therapeutic interventions to treat dyslipidemia. Notably, niceritrol (60) and nicofuranose (61) function as cholesterol reducers, contributing to lipid management.81,82 The introduction of inositol nicotinate (62) in 2005 adds a vasodilatory agent to the spectrum, while etofibrate (63) serves as a dyslipidemic agent.83–85 Furthermore, the recognition of nicorandil (64) by the EMA in 2015 underscores its vasodilatory properties.86,87 In contrast, within the domain of picolinic acid derivatives, bupicomide (65) is a singular example of an antidyslipidemic agent enriching the repertoire of antidyslipidemic options.88,89
 |
Figure 9 Antidyslipidemic drugs derived from pyridine carboxylic acids. |
The vibrant potential of pyridine carboxylic acid-derived drug candidates is highlighted in Figure 10, which shows their clinical trial stages. For instance, picolinic acid-derived drug candidates include verubecestat (66) — a BACE2 inhibitor with a Ki of 0.38 nM — which is in Phase 2 of clinical trials.90 Avoralstat (67) — a PKK inhibitor, and tetomilast (68), a PDE4 inhibitor are in Phase 3 trials.91,92 GSK-269984A (69), a PGE2 EP1 inhibitor with an IC50 of 7.9 nM, and SKLB610 (70), a VEGFR2 inhibitor, have demonstrated their potential in pre-clinical stages.93,94 Similarly, the spectrum of nicotinic acid-derived drugs under development presents a cohort of candidates with distinct attributes. Farudodstat (71), a DHODH inhibitor with an IC50 of 35 nM, and Motesanib (72), a VEGFR1 inhibitor with an IC50 of 2 nM, are undergoing phase 2 trials.95,96 Basmisanil (73), a GABAA-a5 inhibitor (IC50 = 8 nM) is also in phase 2, while apatinib (74), a VEGFR2 inhibitor with an IC50 of 1 nM is undergoing phase 3 trials.97,98 Ninerafaxstat (75), a pFOX inhibitor, is in Phase 2, while flunixin meglumine (76), a COX-1 inhibitor with an IC50 of 0.55 mm, is in the pre-clinical stage.99,100 Moreover, GSK189254A (77), an H3R antagonist with a pKi range of 9.59–9.90, is in Phase 1, and losmapimod (78), a p38 MAPK inhibitor with a pKi of 7.6 for p38β, is progressing through phase 2 trials.101,102 BMS-688521 (79), an LFA/ICAM inhibitor with an IC50 of 2.5 nM, has shown promise in the pre-clinical stage.103 Notably, the isonicotinic acid-derived drug candidates include KDOAM-20 (80), which is a KDM5 inhibitor (IC50 = 40 nM) and is in pre-clinical stage.104 Furthermore, KDM5-C70 (81) — a JARID1B inhibitor with an IC50 of 49 nM, and OPRT inhibitor DFP11207 (82) are in phase 1 of clinical trials.105,106
 |
Figure 10 Pyridine carboxylic acid derivatives in drug development pipeline. |
The Effect of Pyridine Substitution on Key Pharmacological Parameters
The physicochemical properties of molecules are significantly impacted by replacing the phenyl group with a ring-containing nitrogen atom, which can translate into improved pharmacological parameters.107 Herein, we describe some key parameters, such as biochemical potency, target selectivity, cellular potency, and binding affinity by comparing the results of pyridine and non-pyridine ring-containing analogues. Figure 11 summarizes exciting examples of the pyridine effect on improving some key pharmacological parameters. For example, Dandapani et al evaluated nearly 100,000 different chemical structures and found that compound 83 could effectively stop the growth of Trypanosoma cruzi — a parasite responsible for Chagas disease. However, its isonicotinoyl analogue was 500-fold more potent than the parent molecule (T. cruzi IC50 = 1.0 nM for 84). Likewise, ponatinib (85) — a multikinase inhibitor — is used to treat a type of highly resistant blood cancer by blocking some proteins that help in the growth of cancer cells. Ponatinib is also being evaluated for other types of cancers, such as lung and bile duct cancers. However, this drug has many side effects because it blocks many other proteins. By adding nitrogen to the structure of ponatinib, the new version (86) of this drug was 28-fold more potent by effectively blocking MNK1 and MNK2 proteins that make some cancers resistant to treatments. Naltrexone (87) is a drug with an antagonistic effect on the μ-opioid receptor (μ-OR) in our body. In 2013, Zhang et al made changes to naltrexone to help it fit into the receptor cavity and discovered that the addition of nitrogen to the drug improved its receptor binding. The docking study for compound 88 showed 880-fold improved binding affinity in comparison to 87 (μ-OR Ki = 120 nM). This new version of naltrexone did not activate the receptor, which is desirable for its intended purpose. When 6-substituted pyrrolo[2,3-d]pyrimidine containing compound 89 was modified by the addition of a nitrogen atom in the phenyl side-chain structure, compound 90 was obtained (Figure 12).
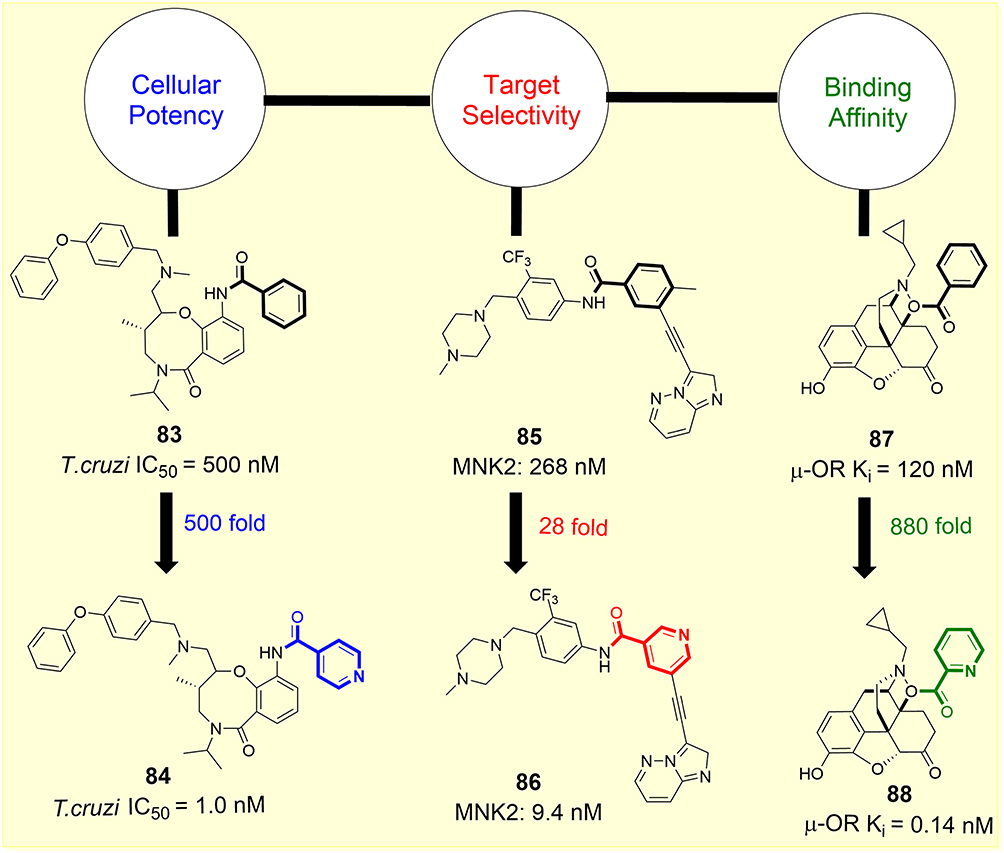 |
Figure 11 Effect of pyridine ring on pharmacology of bioactive molecules. |
 |
Figure 12 Enhancement of antitumor effect by the addition of nitrogen atom. |
Upon testing this compound on isogenic Chinese hamster ovary (CHO) cells expressing folate receptors (FRs) α and β, it had superior anti-proliferative activity due to its competitively binding with [3H] folic acid. In further assays involving KB tumor cells, compound 90 exhibited an IC50 value of 0.37 nM, making it 3 times more potent than the original compound 89. The findings suggest that compound 90 is potentially a powerful antitumor agent having reduced toxicity to normal cells.108
Le et al reported inhibitors of matrix metalloproteinase-13 (MMP-13) — an enzyme involved in osteoarthritis. To block MMP-13, the active compound 91 and its pyridine carboxylic acid derivative 92 were discovered by high-throughput screening (HTS) followed by structure-activity relationship (SAR) studies (Figure 13). It was found that compound 92 could be much more effectively absorbed and remained in the body for a longer period compared to 91. Bioavailability (%F) of 92 in the animal model was 73%, which was significantly higher than that of 91 having 9% only. Similarly, 91 had a clearance rate (CL) of 20 mL/min/kg, while 92 had a slower rate of 7.6 mL/min/kg, meaning 92 stayed in the body longer. Furthermore, compound 91 had an AUC (the area under the plasma drug concentration-time curve) of 380 ng/h/mL, while 92 had a much higher AUC of 13,000 ng/h/mL, indicating a greater exposure of the body to pyridine carboxylic acid derived 92 over time.107,109
 |
Figure 13 Effect of nitrogen atom substitution on in vivo pharmacological profiles. |
Overall, these examples identify the pyridine carboxylic acid isomers as important constituents of drug design for multiparameter optimization. Beyond all of the above-mentioned properties, pyridine derivatives are receiving interest for their ability to act as enzyme inhibitors.110 Pyridine carboxylic acid-derived compounds function as enzyme inhibitors against a wide range of enzymes such as urease,111 synthase,112 tyrosinase,113 myeloperoxidase (MPO), and acetylcholinesterase,114 cyclooxygenase-2 (COX-2),115 histone demethylase,116,117 calpain (calcium-activated protease),118 Bcr-Abl tyrosine kinase,119 and c-Met kinase.120–122 Hence in this perspective, there have been a large number of patents published in recent years communicating the role of various pyridine carboxylic acid derivatives in this area of enzyme inhibition.
Patent Literature Review (2013–2023)
Many enzyme inhibitors are well known for their function as drugs in clinical settings.123 Interestingly, most of the patents unveiled herein depicted the enzyme-inhibitory activity of pyridine carboxylic acid derivatives. Within the diverse range of inhibitors emanating from pyridine carboxylic acid, the focus of this section will be on the patents and papers from 2013 to 2023, as shown in Table 1. These are categorized, depending on the proposed target when specified. The molecular structures of the notable biologically active inhibitors can be seen in Figure 14.
 |
Table 1 Patents Published on Pyridine Carboxylic Acid Derivatives in Different Therapeutic Areas from 2013 to 2023 |
 |
Figure 14 Biologically active pyridine carboxylic acid derived enzyme inhibitors reported in recently published literature. |
IRAK4 Inhibitors
Interleukin-1 receptor-associated kinase 4 (IRAK4) is the most proximal kinase in the Toll-like receptor (TLR)/interleukin (IL)-1 receptor (TLR/IL-1R) signaling cascade, which is associated with autoimmune diseases, inflammation, and cancers.149–151 TLR assembles the MyDDosome complex during signal transduction, which then activates transcriptional factors and the downstream pro-inflammatory cytokines production. Human and rodent genetics support the role of IRAK4 in immunological response. Moreover, clinical trials are ongoing for the evaluation of IRAK4 inhibition for non-Hodgkin lymphoma.151 Bryan et al reported an IRAK4 inhibitor 93 with a clinical code CA-4948 having IC50 less than 50 nM (Figure 15).152 Similarly, Bayer125 and Bristol-Myers Squibb (BMS) have disclosed some noteworthy IRAK4 inhibitors such as 94153,154 with an IC50 value of 3.4 nM. BMS also discovered some potent IRAK4 inhibitors such as 95 and 96 with IC50 values of 3 and 2 nM, respectively. Gilead reported even more potent IRAK4 inhibitors 97‒100 with IC50 values being less than 1 nM.151,155 Remarkably, the compounds 93–100 are built around a substituted pyridine core, which facilitates hydrogen bonding and π-π stacking interactions within the kinase active site. The presence of electron-deficient nitrogen-containing rings enhance binding affinity, whereas rigid linkers like amides or ureas help maintain optimal geometry for target engagement. Together, these features support a strong and selective binding to IRAK4, with several inhibitors demonstrating IC50 values in nanomolar range.
 |
Figure 15 Structures of IRAK4 inhibitors 93‒100. |
ASK1 Inhibitors
Apoptosis signal-regulating kinase 1 (ASK1) is a serine-threonine kinase that activates other kinases in response to a range of stresses like calcium overload, ROS (reactive oxygen species) stress, lipopolysaccharides and endoplasmic reticulum stress156,157 ASK1 has been found to engage in the development of cancer, diabetes, fibrosis, neurodegenerative, and cardiovascular diseases.158–162
Rowbottom disclosed a group of compounds as ASK1 inhibitors. The ability of ASK1 inhibition of 141 compounds was tested using the luminescent ADP-Glo™ Kinase Assay and Myelin Basic Protein (MBP) as a substrate (Promega Corporation, Madison, WI). It was found that the picolinic acid derivatives having different R groups did not show any activity but when the nitrogen atom in the picolinic acid moiety is replaced with a C-F group together with an R group modifications (Figure 16), all of the compounds showed significant inhibitory effect with an IC50 < 300 nM for most compounds.126
 |
Figure 16 Structures of ASK1 inhibitors 108‒115. |
TRPC6 Inhibitors
The superfamily of transient receptor potential (TRP) channels comprises over 28 members, which are categorized into seven subfamilies.163,164 The transient receptor potential cation channel, subfamily C, member 6 (TRPC6) plays a multifaceted role in neuroprotection, particularly in Alzheimer’s disease (AD) and cerebral ischemia. It has the potential to enhance dendritic spine formation, protect neurons from ischemic damage, and promote synaptic growth and cognitive functions.165 However, excessive TRPC6 activity can be detrimental to various body systems, possibly contributing to conditions like breast cancer and glomerular sclerosis. The precise function of TRPC6 in AD and ischemia is debated, with conflicting reports of reduced and increased activity.166,167 The extensive TRP gene family is responsible for encoding transient receptor potential (TRP) proteins, which assemble into unique ion channels selectively permeable to cations. Striking a balance in TRPC6 activity is crucial for brain function preservation, emphasizing the need to investigate molecular irregularities and develop targeted pharmacological treatments.168,169 Applications of pyridine carboxylic acid derivatives as transient receptor potential C6 (TRPC6) ion channel inhibitors for the treatment of pain, cardiac and respiratory conditions, renal disease, liver disease, ischemia or ischemic reperfusion injury, fibrotic disorders, cancer, and muscular dystrophy, have been patented (WO2019081637). To the best of our knowledge, this patent represents the only available report in the patented literature specifically describing pyridine carboxylic acid-based compounds as TRPC6 inhibitors. Considering the important role of ion channels in modulating the capacity for membrane and ion flux in cells, it is of great interest to identify agents that can promote or inhibit specific ion channels as diagnostic tools as well as potential therapeutic agents.168 TRPC6 is one of these channels, which is part of the TRP ion channel family. It is a calcium-permeable channel, more specifically a calcium-permeable cation channel that is non-selective.170 A range of pyridine carboxylic acid derivatives were synthesized via a multistep synthetic route and tested as TRPC6 inhibitors. The generic structure 116‒129 comprising of pyridine carbonyl core is represented in Figure 17.
 |
Figure 17 Structures of TRPC6 inhibitors 116‒129. |
As described in the referenced patent (WO2019081637A1), a total of 95 pyridine carbonyl derivatives were synthesized and evaluated using Fluorescence Imagining Plate Reader (FLIPR) assay in a cell-based system that measures calcium influx as proxy for TRPC6 activity. Among these compounds 116, 117, 125, 126, and 127 demonstrated high potency with IC50 values below 27 nM, while compounds 118–124, 128, and 129 showed moderate potency (~27 Nm). Notably, these compounds displayed excellent selectivity, showing significantly weaker or no inhibitory activity against related TRP channels, including TRPC3, TRPC5 and TRPC7 as indicated in the patent’s comparative selectivity profiling. These compounds can be used in a lower dosage form either alone or in combination with adjuvants that enhance the inhibitor stability, thus reducing the potential cytotoxicity and adverse side effects when used as a monotherapy. Compared to other TRP channels like TRPC3, TRPC5, and TRPC7; these pyridine carboxylic acid derivatives have very strong potency and selectivity for the TRPC6 channel and can be a good starting point for the pharmacological modulation of TRPC6.127
WDR5-MLL1 Inhibitors and Modulators
The protein-protein interaction between WDR5 (WD40 repeat protein 5) and MLL1 (mixed-lineage leukemia 1) is important for maintaining optimal H3K4 methyltransferase activity of MLL1.171,172 Dysregulation of MLL1 catalytic function is relevant to mixed-lineage leukemia, and targeting WDR5-MLL1 interaction could be a promising therapeutic strategy for leukemia harboring MLL1 fusion proteins.172 In cancer research, the dysregulation of histone methylation pathways is a well-established contributor to tumorigenesis.173–176 WDR5-MLL1 inhibitors and modulators offer a promising avenue for intervention by disrupting this crucial interaction, thereby potentially reestablishing normal epigenetic patterns and suppressing cancer progression.174,177 Gogliotti et al disclosed an invention in a patent US 2020/0055824 A1 that relates generally to compositions comprising benzamides and picolinamides to modulate the interaction of WD repeat-containing protein 5 (WDR5) with chromatin, cognate transcription, and other regulatory factors for the treatment of solid cancers, leukemia, and other WDR5 dependent diseases. Mixed lineage leukemia (MLL) is a gene involved in the translocation of chromosomes in acute leukemia subtypes such as acute myeloid leukemia (2.8%) and acute lymphoblastic leukemia (10%).178 Intrinsic histone methyl transferase (HMT) activity of MLL1 is extremely low and requires a complex assembly of WDR5, RbBP5, ASH2L, and DPY30 protein partners for effective H3K4 trimethylation, the so-called WRAD complex. The invention being discussed describes benzamide and picolinamides having guanidino-, imino-, or heterocycle-containing groups as their meta substituents. These compounds disrupt the WDR5-MLL1 protein-protein interaction and are extensively employed in pharmaceutical compositions, treating proliferative disorders and conditions such as cancer. Different assays such as time-resolved fluorescence resonance energy transfer (TR-FRET) assay and fluorescence polarization assay (FPA) were performed to evaluate the potential of the synthesized picolinamide derivatives 130‒139 as inhibitors of WDR5. The data presented in Figure 18 demonstrates the utility of the representative compounds as selective inhibitors of the WDR5 protein to bind peptides from the relevant MLL domain. As described in the supporting patent (US10807959B2), compound 136 and 138 demonstrated strong inhibitory activity values of 0.1 nM and 0.15 nM, respectively, in the TR-FRET assay. Furthermore, the cellular viability of human tumor cell lines was determined for compounds 140‒142 by examining the anti-proliferative activity using MLL harboring cell lines. Concisely, picolinamide derivatives can be an excellent choice in treating proliferative disorders and cancer, especially leukemia.128
 |
Figure 18 Structures of WDR5-MLL1 inhibitors and modulators 130‒142. |
KDM4 Inhibitors
KDM4 inhibitors, a class of compounds gaining increasing attention in the field of epigenetics, target a group of enzymes known as lysine-specific demethylase 4 (KDM4), which play a crucial role in regulating gene expression by removing specific methyl groups from histone proteins.179,180 The significance of KDM4 inhibitors extends into the field of cancer research, as dysregulated histone methylation is a hallmark of many cancer types. These inhibitors offer a promising avenue for therapeutic intervention, potentially reprogramming the epigenetic landscape to suppress tumor growth and metastasis.181,182 Beyond cancer, KDM4 inhibitors hold promise in addressing other diseases linked to aberrant epigenetic modifications, such as neurological disorders and cardiovascular conditions.183 As our understanding of epigenetics continues to expand, KDM4 inhibitors represent a valuable tool in the quest for targeted therapies and precision medicine, offering new possibilities for treating a wide range of complex diseases.184
Pyridine carboxylic acid-derived compounds are also investigated as histone demethylase inhibitors.116,185,186 Histone lysine demethylase 4 (KDM4) catalyzes the removal of methyl marks from histone lysine residues to control chromatin structure and gene expression epigenetically.187,188 Histone demethylase activity can be inhibited by methyllysine histone substrate mimics (Figure 19).189 Epitherapeutics APS filed a patent disclosing histone demethylase inhibitors.131 In AlphaLISA assays, the pyridine-4-carboxylic acid analogues 143–150 presented in Figure 20 demonstrated inhibitory activities against one or more of KDM4A, KDM4B, and KDM4C. Several of these compounds were tested in immunofluorescence assays using U2OS cells, and the representative compounds 143 and 144 were found to be potent KDM4C inhibitors (IC50 < 1 µM).190 Moreover, the histone lysine-demethylase inhibitory effect of the compounds under discussion was tested within the cell, and global levels of tri-methylation on lysine 4 of histone 3 (H3K4me3) were assessed by Western blot in the breast cancer BT474 cell line. Compounds with different R groups exhibited strong biochemical KDM inhibitory as well as cellular activity against all cell lines tested below 100 nM concentration. The structures of the most potent JmjC-KDM inhibitors are displayed in Figure 20. In brief, it is suggested that the use of compounds 145‒150 in therapeutically effective amounts, may help treat diseases associated with JmjC-KDM inhibitors as anticancer and antiviral agents.130
 |
Figure 19 Mechanistic pathway of histone demethylases. |
 |
Figure 20 Structures of KDM inhibitors and modulators 143‒150. |
The ability of pyridine carboxylic acid-derived compounds enclosed in patent WO 2018/149986 A1 to inhibit the activity of KDM5B was determined by employing AlphaLISA technology in vitro using human recombinant proteins. For the determination of IC50 values, the patented inhibitors were tested at eight logarithmic serial dilutions. The invention also relates to pharmaceutical compositions comprising these compounds and to use in cancer therapy. The tested compounds 151‒159 showed IC50 in the range of 1‒3 nM, see Figure 21. 130
 |
Figure 21 Structures of KDM 5 inhibitors and modulators 151‒159. |
A patent application WO2010043866 disclosed a series of pyridine-2,4-dicarboxylic acids for their inhibitory activities against JMJD2E in the FDH-coupled demethylase assay. Among the tested compounds, pyridine-2,4-dicarboxylic acid (160), 3-(4-methoxybenzylamino)pyridine-2,4-dicarboxylic acid (161), 3-(2-fluorophenyl amino)pyridine-2,4-dicarboxylic acid (162), 3-(o-tolylamino)pyridine-2,4-dicarboxylic acid (163), and 3-(2-aminophenylamino)pyridine-2,4-dicarboxylic acid (164) (Figure 22) showed quite potent inhibition with IC50 values of 1.4, 0.3, 1.1, 7.9, and 0.6 µM, respectively. Another test was conducted on 2,2-bipyridyl derivatives (heteroaryl derivatives, cofactor disruptor) in the FDH coupled inhibition assay and the non-denaturing MS binding assay. The compounds (165‒170) in Figure 22, showed strong binding affinity and IC50 values of 6.6, 1.5, 3.6, 3.8, 4.5, and 8.0 µM, respectively.190
 |
Figure 22 Structures of KDM inhibitors and modulators 160‒170. |
In 2014, Quanticel Pharmaceuticals, Inc. filed a patent application WO 2014/100463A1 that described the substituted aminopyridine derivatives (171‒176) and their pharmaceutical compositions as inhibitors of histone demethylase for treating cancer and neoplastic diseases are shown in Figure 23. Most of these compounds induced complete tumor regression via an in vivo approach. The compounds were tested to inhibit JARID1A, JARIDlB, and JMJD2C demethylase activity in vitro employing time-resolved-fluorescence resonance energy transfer (TR-FRET) detection method. Most of the compounds were found to be active and inhibited the activity of a demethylase comprising of a JmjC domain (eg, a histone demethylase such as a JHDM protein) with low IC50 values (≤ 0.10 µΜ). The decrease in JARID1B is connected to a rise in tri-methylated H3K4 levels within tumor suppressor genes. The quantification of tri-methylated histone H3 and cellular inhibition of KDM5A and 5B, was done on ZR-75-1 breast cancer cell line via immuno-blotting assay. The subsequent IC50 values from these cellular assays revealed a correlation between the degree of inhibition of these enzymes in cancer cell lines and the degree of inhibition of these enzymes in a biochemical assay. To further support this claim, the most active compounds were subjected towards in vivo xenographic evaluation into nu/nu mice model. Most of the compounds had an overall favorable profile. In addition, such compounds have the advantage of being fairly soluble in water as well as stable in vivo; a characteristic not frequently shared by most synthetic derivatives. Since they exhibited specific inhibitory potential toward histone demethylases; it has been suggested that they can serve as an effective tool to regulate the modulation of demethylation in a cell, either generally or with respect to one or more specific target genes.132
 |
Figure 23 Structures of KDM (JARID 1A, JARID 1B, JMJD2C) inhibitors and modulators 171–176. |
Quanticel Pharmaceuticals, Inc. submitted a follow-up patent application WO 2015/200709A1 to their 2014 patent application WO 2014/100463A1. The generic structure for compounds 177–194 is given in Figure 24. Screening of these compounds for histone demethylase inhibitory activity was carried out using enzymatic and cellular inhibition assays. The results disclosed a number of substituted pyridine carboxylic acid derivatives as in vitro selective histone demethylase enzyme inhibitors with IC50 values in the sub-nanomolar range for cancer prevention and treatment of neoplastic disease. The enzymatic assays were performed to inhibit JMJD2C activity based upon time resolved-fluorescence resonance energy transfer (TR-FRET) detection. The human KYSE-150 (SMAD4 mut, TP53 mut) esophageal carcinoma cell line was used to test cell proliferation by in vitro cell-based assay. The IC50 values of cellular proliferation were found typically lower than 0.10 µΜ. The most potent inventive compounds were further evaluated for in vivo xenograft study using MCF-7 cells. The subsequent results provided the compounds with good to excellent oral bioavailability and pharmacokinetic profiles as highly active and selective histone demethylase inhibitors.133
 |
Figure 24 Structures of KDM inhibitors and modulators 177‒194. |
WO2022/047230A1 patented by Fibrogen. Inc included 142 pyridine carboxylic acid derivatives (see representative structures of 195–227, Figure 25). Among them, 139 compounds are pyridine-2-carboxylic acid derivatives with different R groups and the remaining three compounds (199‒201) are pyridine-4-carboxylic acid derivatives. All these derivatives were tested for the inhibition of KDM5B and KDM4A. All pyridine-2-carboxylic acid derivatives showed pronounced inhibition against KDM5B with IC50 < 1.0 µM. Three pyridine-4-carboxylic acid derivatives showed weaker inhibition with IC50 of 1.4 and 2.0 µM. Among all compounds, only some of them displayed moderate inhibition against KDM4A with IC50 < 40 µM.
 |
Figure 25 Structures of KDM inhibitors and modulators from WO2022/047230A1 (195‒227). |
Gilead Sciences, Inc. filed a patent (US 2018/0042905A1) disclosing an invention that relates to novel methods of treating Hepatitis B virus (HBV) by administering KDM5 inhibitors. The most auspicious compounds 228–235 (Figure 26) mentioned in the invention are listed in Table 2 with a summary of their HBV antiviral activity. The data in the patent leads us to the conclusion that pyridine carboxylic acid derivatives can be considered promising lead molecules for the development of new antiviral drug candidates for lifelong therapy of HBV.191
 |
Table 2 HBV Antiviral Activity (EC50, µm) of Compounds 228–235 |
 |
Figure 26 Structures of KDM inhibitors from US 2018/0042905 A1 (228‒235). |
High concentrations of sub-viral particles (SVPs) with HBV surface antigen (HBsAg) in chronic HBV infection are a significant obstacle to effective immune responses. Despite significant advances in HBV treatment, only a small percentage of patients achieve the ideal goal of “functional cure”, which is characterized by hepatitis B surface antigen (HBsAg) loss. More specifically, sustained loss of HBsAg is significant because it is linked to better long-term outcomes. HBV surface antigen (HBsAg) is a major obstacle that provides direct evidence in the timing of the decision to discontinue treatment and start off-therapy retreatment during antiviral therapies.192,193
3HAO Inhibitors
As an enzyme of the kynurenine pathway, which is directly responsible for the conversion of 3-hydroxyanthranilic acid into quinolinic acid (QUIN), through the intermediate formation of a semimuconic aldehyde and its subsequent non-enzymatic cyclization, 3-hydroxyanthranilate-3,4-dioxygenase (3HAO) is potentially involved in a series of neurodegenerative disorders and diseases, such as Huntington’s disease, Alzheimer’s disease, HIV-related dementia, and cerebral ischemia. As reported in the supporting patent (WO2012097869A1), compounds 236–238 (Figure 27) demonstrated 3HAO inhibition in rat brain homogenates, showing inhibition of 49%, 22%, and 78% at 10 µM, and 90%, 76%, and 97% at 100 µM, respectively. In human brain homogenates, the same compounds showed 66%, 42%, and 80% inhibition at 10 µM, and 93%, 100%, and 100% at 100 µM, respectively.
 |
Figure 27 Structures of 3HAO inhibitors 236‒238. |
PDGFR Selective Inhibitors
Pyridine carboxylic acid derivatives have also been reported as selective platelet-derived growth factor receptor (PDGFR) inhibitors to treat pulmonary arterial hypertension (PAH). Studies in animal models have revealed that PDGF appears to play a significant role in vascular remodeling.194 Both PDGFRα and PDGFRβ receptors are associated with specific receptor tyrosine kinases. Inhibiting PDGFR kinase has been proposed as an additional therapeutic modality, and this hypothesis appears to be validated by clinical studies with the non-selective PDGFR inhibitor imatinib.195 However, the adverse effects linked to a lack of selectivity imply that clinical utility requires the identification and development of selective PDGFR inhibitors.196 Recently compound 239197 (PK-10453) has been described as a non-selective PDGFR inhibitor. PK-10453 inhibits PDGFRα and PDGFRβ with respective IC50 values of 10.1 and 35 nM. On the other hand, NOVARTIS AG specifically claims the crystalline form of PDGFR inhibitor 240 which is also a pyridine carboxylic acid-derived compound and is considered even more potent and selective than either imatinib or PK-10453 with IC50 of 3.0 nM.135 It is possible that Novartis intends to develop compound 240196,198 for the treatment of PAH.196 The structures of 239 and 240 are given in Figure 28.
 |
Figure 28 Structures of PDGFR inhibitors 239 and 240. |
Trk Inhibitors
Activation of the Tropomyosin-receptor-kinase (Trk) A/B/C receptor promotes growth, survival, and differentiation of discrete neuronal populations during development, adult life, and aging. It also plays a role in the emergence and progression of human disease. Trk-specific inhibitors have therapeutic applications in cancer and pain, making them a burgeoning field of study in oncology and neurology. Pyridine carboxylic acid core-containing compounds are reported as Trk Inhibitors. The compounds 241‒243199,200 with fluorination at position 3 of the piperidine ring are prominent and showed marked potency with IC50 values of 1.7, 1.3, and 1.5 nM respectively, except compound 244 having 3R,4R stereo configuration with adjacent ether, decreases potency depicted from IC50 value (12.3 nM) (Bailey, Schirrmacher, Farrell, and Bernard-Gauthier, 2017; WO 2015/092610 Al, 2015). In another short publication, pyrrolidine analogs were described, which utilized a 6-aminonicotinamide hinge binder adorned with a variety of amide substitutions. Compounds 245 and 246 in this series are presented as sets of enantiomers and trend for a preference of the (R)-enantiomer 245 with IC50 of 11.6 nM.201 The compounds 241‒246 are illustrated in Figure 29.
 |
Figure 29 Structures of Trk inhibitors 241‒246. |
FABP Inhibitors
Numerous studies show that excessive free fatty acid (FFA) is the pathogenic factor for a wide range of disorders such as atherosclerosis, obesity, hypertension, and metabolic syndrome. These are diseases that synergistically compromise human health. Fatty acid binding proteins (FABPs) are in charge of transporting FFA from cytoplasm to different cellular organelles like mitochondria, nucleus, peroxisomes, lipid droplets, and ER (endoplasmic reticulum), and thus play a pertinent role in cellular functions as depicted in (Figure 30). As a result, developing and employing FABP inhibitors could be a viable strategy for controlling atherosclerosis, obesity, diabetes, and metabolic syndrome in humans.68 William W. Bachovchin et al disclosed pyridine-3-carboxylic acid derivatives (niacin) as FABP inhibitors in their two patents.68,137 The derivatives included the representative compound 247 as shown in Figure 31. Compound 247 displayed a significant effect on lipid modulation by chronic administration of compounds compared to niacin in a hamster model. The concentrations of LDL (low-density lipoprotein) and total cholesterol in compound 272-treated trials are at least 1.67 and 1.54 times greater than those in the niacin trials. Additionally, the HDL (high-density lipoprotein) is noticeably higher in the compound 40 trace, in addition to the VLDL and LDL being significantly reduced. Meanwhile, when treated with compound 247, both ABCA1 and ApoAI mRNA levels were higher in the compound 247-treated animals relative to vehicle control animals. Therefore, Compound 247 could be used to develop new drugs to treat FABP-related disorders and metabolic syndrome. Because of their significant impact on the liver, niacin derivatives are also thought to target FABP4.68 Although niacin has been linked to liver toxicity202 and glucose intolerance in chronic dose settings, certain chemical modifications could avoid these side effects. In brief, pyridine-3-carboxylic acid derived-compounds could be good candidates for the development of new FABP inhibitors to treat various ailments.10,203,204
 |
Figure 30 Intracellular transport of free fatty acid (FFA). |
 |
Figure 31 Structure of FABP inhibitor 247. |
FXIa Inhibitors
The most common medications used to prevent and treat thrombotic disorders are anticoagulants. Each anticoagulant used in clinical practice is linked to serious adverse effects, particularly bleeding. It has been proposed that Factor XIa (FXIa), a crucial factor involved in the amplification of the procoagulation signal, is a primary target for the development of anticoagulant drugs.138 The importance of pyridine carboxylic acid derived compound as FXIa inhibitor can be estimated from the fact that aryl boronic acid 249 (IC50 = 1.4 µM) having pyridine carboxylic acid core reported to show better FXIa active site inhibition as compared to its precursor 248 (IC50 = 7.3 µM) deprived of pyridine carboxylic acid core. The compound 249 has also much better selectivity than 248. It was observed that S enantiomer of compound 249 was bound in active site assuming that only S enantiomer was found active against FXIa.138,205 A review by Al Horani also discusses FXIa inhibitors in detail. Among these compounds was pyridine carboxylic acid derivative 250 with FXIa IC50 of 20 nM.206 Furthermore, Bayer Pharma, Germany, filed several patent applications in 2015, disclosing phenyl alanine derivatives as FXIa inhibitors for the prophylaxis of various ailments related to cardiovascular disorders and thrombotic diseases. One of the interesting phenyl alanine derivative 251 having pyridine carboxyl moiety is reported as FXIa inhibitor with IC50 of 0.9 nM and the concentration required to double the clotting time was reported as 0.08 µM in APTT assay.138 The structures of compounds 248‒251 are shown in Figure 32.
 |
Figure 32 Structures of FXIa inhibitors 248‒251. |
TGFβ Inhibitor
Transforming Growth Factor Beta (TGFβ) is a multi-functional cytokine, which is involved in numerous biological processes including cell growth, differentiation, immune regulation, fibrosis, and cancer progression. TGFβ inhibitors are being explored for cancer, fibrotic diseases, autoimmune and inflammatory conditions. Sistla et al disclose a patent unveiling novel crystalline polymorphic and amorphous form of 4-(2-(5-chloro-2-fluorophenyl)-5-isopropylpyridin-4-ylamino)-N-(l,3-dihydroxypropan-2-yl) nicotinamide 252207,208 (Figure 33) known for treating abnormal cell growth, such as cancer, in mammals by acting as a potent and selective ΤGFβ inhibitor.139
 |
Figure 33 Structures of TGFβ inhibitor 252. |
Nav1.8 Inhibitors
Novel derivatives of 2-Amino-N-heteroaryl-nicotinamides as Nav1.8 inhibitors were disclosed in patent WO 2020/092667 A1. Voltage-gated sodium ion channel (Nav1.8) is thought to be involved in a variety of diseases such as inflammatory pain, neuropathic pain, chronic itch disorders, etc. The importance of Nav1.8 is summarized in Figure 34.
 |
Figure 34 Importance of Nav1.8 inhibitors. |
The potency of the patented compounds was assayed using a patch clamp assay, and human Nav1.8 and Nav1.5 channels were stably expressed in human embryonic kidney (HEK) 293, and their inhibition was measured as a function of drug concentration by an offline analysis. Finally, 218 compounds were evaluated for their Nav1.8 channel activity, the majority of which inhibited the flow of sodium through human Nav1.8 and Nav1.5 channels with IC50 values in the single-digit nanomolar range. The presence of a halogen substituent plays an important role in the Nav1.8 activities of this series, particularly when it is located in an ortho-position. The compound 258 5-chloro-2-(4.4-difluoroazepan-l-yl)-6-methyl-N-(2-sulfamoylpyridin-4-yl) nicotinamide having chloro and methyl substitution at the nicotinamide moiety, exhibited an IC50 of 0.3 nM against Nav1.8 channels employing a 1 hertz pulse train stimulation. Compounds 253‒257 and 259‒264 also showed significant activity (Figure 35). It is speculated that these compounds may be beneficial in the prevention, treatment, or management of cough, pain, acute itch, and chronic itch disorders. It was suggested by the inventor that the inhibitory effect of these compounds is dependent on halogen substituents; particularly, the presence of a fluoro group increases the activity in comparison to other groups. As part of a pharmaceutical preparation, these compounds can be used alone or in conjunction with a well-known analgesic.140
 |
Figure 35 Structures of Nav1.8 inhibitor 253–264. |
DHODH Inhibitors
Dihydroorotate dehydrogenase (DHODH) inhibitors act upon dihydroorotate dehydrogenase, thereby hindering pyrimidine biosynthesis.209 This inhibition disrupts DNA and RNA synthesis in rapidly dividing cells, making them promising for cancer and autoimmune disease therapies by suppressing immune cell proliferation.210,211 Pyridine carboxylic acid derivatives are also known for their dihydroorotate dehydrogenase (DHODH) inhibitory activity. DHODH inhibitors have demonstrated their effectiveness in treating a variety of diseases.209 Despite extensive efforts to develop DHODH inhibitors, no FDA approval has yet been obtained.212 Julio Cesar et al described the use of new amino derivatives of nicotinic and isonicotinic acid as inhibitors of the DHODH. The amino(iso)nicotinic acid derivatives were conveniently synthesized as described in Scheme 1. These compounds were subjected to human DHODH activity using a chromogen reduction assay with DCIP (2,6-dichlorophenol indophenol) to observe the inhibition effect.
 |
Scheme 1 General structures of amino nicotinic and isonicotinic acid derivatives as DHODH inhibitors. |
The most significant feature observed for effective DHODH inhibitory activity is their substitution pattern on phenyl ring with flouro or trifluoromethyl group. These compounds 265‒272213 were proved as potent DHODH inhibitors with IC50 values in a range of 3‒8 nM (Figure 36). In addition to efficient inhibition of DHODH, the amino derivative of nicotinic and isonicotinic acid in the patent being discussed, also inhibits the proliferation of cells with a high turnover rate, particularly in lymphocyte cells. In conclusion, these patented amino (iso)nicotinic acid derivatives and their pharmaceutical compositions have been found useful for the diagnosis, prevention, or suppression of diseases and disorders that are susceptible to improvement by treatment with inhibitors of dihydroorotate dehydrogenase. Such diseases include autoimmune diseases, immune diseases, malignant neoplastic diseases, destructive bone disorders, angiogenic-related disorders, infectious, inflammatory, and viral diseases. This discloser also describes the dosage formation and pharmaceutical preparation methods of these amino(iso)nicotinic acid derivatives as potent DHODH inhibitors. These findings suggest new approaches towards the development of DHODH inhibitors involving pyridine carboxylic acid moieties. It highlighted their importance as a target for drug discovery and chemotherapeutics.141
 |
Figure 36 Structures of DHODH inhibitor 265‒272. |
XO Inhibitors
Xanthine oxidase (XO) is a multipurpose molybdoflavoprotein that can produce uric acid and reactive oxygen species via catalysis and thus can lead to various diseases like hyperuricemia, gout, heart failure, cardiovascular diseases, hypertension, diabetes, inflammation, cancers, kidney diseases, and articular diseases.211,214–218 A number of patent publications have reported xanthine oxidase inhibitors (WO 1998/018765, WO 2007/043457, WO 1992/009279, WO 2008/126770, WO 2007/004688, WO 2008/126899, and WO 2008/126,898). Among these, WO 1998/018765 exhibits the inhibitory effects of pyrazoles and phenyl derivatives against xanthine oxidase, and WO 2008/126898 describes the inhibitory effects of indole compounds against xanthine oxidase. In 2014, Soga et al published a patent for deciphering novel compounds that act as xanthine oxidase inhibitors. A total of thirty compounds were reported in this invention, out of which seven compounds 273‒279 were having a pyridine carboxylic acid core (Figure 37). The best XO inhibitory activity was shown by compound 276 with an IC50 of 2.1 nM. The ability to lower uric acid in plasma and the liver was also determined in vivo in a xanthine oxidase assay using an oxonic acid-induced hyperuricemic model, albeit moderate to poor activity was observed for pyridine-derived compounds.142,219
 |
Figure 37 Structures of XO inhibitors 273–279. |
GSK-3 Inhibitors
Glycogen synthase kinase-3 (GSK-3) inhibitors have gained substantial attention in biomedical research and drug development.220 These compounds, designed to target GSK-3, a key enzyme in cellular processes, hold promise for treating various diseases.221,222 In neurological disorders like Alzheimer’s, they may mitigate neurodegeneration.223,224 GSK-3 inhibitors also show potential in cancer therapy, influencing cell cycle regulation and promoting apoptosis in cancer cells. Moreover, they have applications in regenerative medicine and diabetes research. Their versatility and central role in cellular pathways make them valuable tools for understanding and potentially addressing a wide range of medical conditions, offering prospects for novel therapeutic interventions.225
In 2015, Bristol-Myers Squibb disclosed a series of aromatic and heterocyclic derivatives of isonicotinic acid (WO 2015/069594 Al) as inhibitors of glycogen synthase kinase 3 (GSK-3). In the patent under discussion, nicotinamide derivatives were conveniently synthesized by direct amination. To measure GSK-3 inhibitory activity, a kinase assay was developed using fluoresceinated peptide and a β-glycerol phosphate buffer system. A dose-response curve was generated to evaluate the ability to inhibit > 50% of the kinase activity (IC50) at eleven varying concentrations. Their IC50 values ranged from subnanomolar to nanomolar concentrations, depending on the substitution pattern. Hundreds of compounds were prepared inhibiting GSK-3. The most potent activity (IC50 value < 1.0 nM for GSK-3β and < 5 nM for GSK-3α, respectively) was observed for compounds 280‒284, while other compounds displayed moderate activity. The structures of compounds with significant inhibitory potential are given in Figure 38 along with their IC50 values. These findings suggest that the regulation of GSK-3 modulators can also be useful for the treatment of neuropathological and symptomatic aspects of Alzheimer’s disease and other neurodegenerative disorders.143
 |
Figure 38 Structures of GSK-3 inhibitors 280–284. |
PKal Inhibitors
A number of plasma kallikrein (PKal) inhibitors have been identified so far, but only a few of them have entered clinical trials or have been marketed.226,227 Lifesci Pharmaceuticals, Inc. reported a novel series of heterocyclic derivatives with a pyridine carboxylic acid core and their pharmaceutical compositions, which strongly inhibited PKal. Screening of the compounds was carried out using human plasma kallikrein (hPK) (Abeam) in an enzymatic assay. The resultant data showed that 136 among the tested 146 compounds were able to inhibit hPK with IC50 ≤ 1.0 µΜ. Besides, the series were subjected to in vitro cellular assay. The majority of the compounds had EC50 values less than 1.0 µM, making them suitable candidates for the treatment of plasma kallikrein-related metabolic disorders, such as angioedema. The structures of 285–292 are depicted in Figure 39.144
 |
Figure 39 Structures of PKal inhibitors 285–292. |
CA Inhibitors
Carbonic anhydrase (CA) inhibitors suppress the conversion of CO2 and H2O into bicarbonate and block the reabsorption of bicarbonate from the proximal tubules (Figure 40) in the kidneys.228,229 Studies revealed that CA inhibitors reduce aqueous humor secretion, which is found between the lens and cornea of the eyeball, and decrease intraocular pressure.230 The inhibitors have greater use in edema, altitude sickness, glaucoma, and some adjuvant treatment for epilepsies.231 Pyridine carboxylic acid derivatives inhibit the enzyme carbonic anhydrase more effectively. Some novel pyridine carboxamide derivatives 293–297 showed a CA inhibitory profile against various isoforms such as acetylcholinesterase (AChE) (KIS 3.07–87.26 nM), human carbonic anhydrase I and II isoforms (hCA I and II) (KIS 1.47–10.06 nM and KIS 3.55–7.66 nM), respectively.232 Another heterocyclic sulfonamide derivative 298, exhibited CA inhibition activity against three forms of CA isozymes, CA I, II (cytosolic forms), and IV (membrane-bound) form. CA (II) (Ki = 4 nM) and (IV) (Ki = 10 nM) showed inhibitory effects in the lower nanomolar range.233 Nicotinic acid-containing 6-substituted scaffolds 299–312 were developed and evaluated for CA inhibition activity (Figure 41). The activity was profound against isoenzyme CA (III). The docking studies and chromatographic data of nicotinic acid derivatives demonstrated the binding of the carboxylic part to the Zn2+ ion of the enzyme active site. Moreover, a hydrophobic part at position 6 enhanced activity, such as 6-(hexyloxy) pyridine-3-carboxylic acid (Ki = 41.6 µM).234
 |
Figure 40 Pyridine carboxylic acid derivatives with inhibitory effects against carbonic anhydrase. |
 |
Figure 41 Structures of Carbonic anhydrase inhibitors 293–312. |
IspH Inhibitors
IspH is a crucial enzyme for isoprenoid biosynthesis in pathogenic bacteria. This enzyme is absent in humans, which makes it an attractive and selective target for antibacterial drug development. Additionally, IspH inhibition may interfere with tumor cell metabolism, highlighting its potential in cancer therapy. 4-Hydroxy-3-methylbut-2-enyl diphosphate reductase (IspH) inhibitors can play a role in bacterial infections and cancer. The Wistar Institute patented a series of compounds having inhibition against IspH in 2021. Among them, a pyridine-4-carboxylic acid derivative (313, Figure 42) was able to inhibit IspH with an IC50 of 1.44 µM.145
 |
Figure 42 Structure of IspH inhibitor 313. |
CCR3 Inhibitors
Alkahest, Inc. patented a pyridine-4-carboxylic acid derivative235 (314, Figure 43) as a C-C motif chemokine receptor 3 (CCR3) inhibitor, which can be used for neurodegenerative disease. The compound 314 acted as an antagonist of CCR3, the receptor for Eotaxin-1 (CCL11). CCL11 is a protein increasing in blood by age, and the negative correlation between CCL11 and cognitive function has been proved.236 Chronic inhibition against CCR3 was evaluated in vivo by several neurodegenerative animal models, such as the mouse MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of Parkinson’s Disease, the Alzet mini-pump, model 2002, the oxazolone-induced model of eosinophilia, the synuclein transgenic mouse model of Parkinson’s Disease, etc., and indicated a dose-dependent effect.
 |
Figure 43 Structures of CCR3 inhibitor 314. |
α-Amylase and α-Glucosidase Inhibitors
Diabetes mellitus disrupts carbohydrate homeostasis and lipid metabolism due to defects in pancreatic functions and can lead to organ failure, stroke, heart arrest, loss of vision, limb amputation, and damage to the nervous system. The two major enzymes, α-amylase and α-glucosidase, are involved in antidiabetic treatment. Pancreatic inhibition of carbohydrate digestive enzymes such as α-amylase and α-glucosidase inhibitors in the intestine can help in combating secondary diabetes and postprandial hyperglycemia (PPHG). In a recent study, 5-amino nicotinic acid derivatives 315‒326, summarized in Figure 44, have been synthesized and evaluated for inhibition activity against α-amylase and α-glucosidase with IC50 values in the range of 12.17‒37.33 µg/mL for α-amylase and 12.01–38.01 µg/mL for α-glucosidase; see Table 3 for the inhibitory profile.237
 |
Table 3 Inhibition Study of α-Amylase and α-Glucosidase Compounds 315–326 |
 |
Figure 44 Structures of α-amylase and α-glucosidase inhibitors 315–326. |
Type I MetAPs Inhibitors
Methionine aminopeptidases (MetAPs) cleave N-terminal methionine from newly synthesized proteins and peptides distributed in both prokaryotes and eukaryotes, the inhibition of which is important for biological processes, subcellular location, and eventual degradation of proteins. MetAPs serve important physiological functions and disrupting the MetAPs gene in Escherichia coli (Ec MetAP1) or Salmonella typhimurium is a noxious phenomenon. In Saccharomyces cerevisiae, the inhibitor slows the bacterial growth, either hampering map1 or map2. Therefore, MetAPs are potential targets for antibacterial and antifungal drug development. Inhibitors of the enzyme provide more effective treatment for bacterial and fungal infections. Recently, pyridine-2-carboxylic acid thiazol-2-ylamide (PACT) analogues 327‒347 (Figure 45) were checked against type I MetAPs inhibition activity. It is found that substitutions at position 3 of pyridine were found more effective than position 2 substitutions and inhibition activity was enhanced when N and O atoms were connected to the pyridine ring directly, see Table 4.238
 |
Table 4 Inhibitory Activity of Type I MetAPs Inhibitors 327–347 |
 |
Figure 45 Structures of Type I MetAPs inhibitors 327–347. |
KMO Inhibitors
The Kynurenine pathway (KP) has multiple functions including the generation of cellular energy in the form of nicotinamide adenine dinucleotide (NAD+) and catabolize tryptophan. In case of inflammation, KP enzymes, such as kynurenine 3-monooxygenase (KMO), upregulate and produce toxic substances that may cause disorders, such as neurodegenerative diseases.239,240 Diclofenac (348) is known as a human KMO protein binder and inhibitor in cell lysate with low micromolar KD 64.8 μM and IC50 13.6 μM, respectively, and low millimolar cellular IC50 1.35 mm. (Figure 46).241,242
 |
Figure 46 Structure of KMO inhibitor 348. |
COX-1 and COX-2 Inhibitors
Non-steroidal anti-inflammatory drugs (NSAIDs) are used against inflammation, pyrexia, and pain. Many of them follow their action by inhibiting cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) enzymes, which are involved in the biosynthesis of prostaglandins from arachidonic acid through a catalytic reaction. However, toxicity related to NSAIDs, such as ulceration, bleeding, and perforation, limits their use as anti-inflammatory drugs. Nicotinic acid (pyridine-3-carboxylic acid) derivatives have been demonstrated as good options for COX-1 and COX-2 inhibitors, such as drug Niflumic acid (349), and prodrugs Flunixin (350) and Talniflumate (351). It was reported compounds including nicotinic acid portion 352–358 (Figure 47) showed reduced gastric toxicity with potent activities. Some novel nicotinic acid derivatives 359–373 (Figure 47) exhibited COX-2 inhibitory activity with greater selectivity as compared to COX-1 inhibition. Compounds 361 and 364 have shown highly potent anti-inflammatory activity compared to diclofenac and indomethacin and are as potent as celecoxib. The selectivity was 1.8–1.9-fold greater than the reference drug, celecoxib, see Table 5. Histopathological studies on nicotinic acid derivatives were also carried out. The results showed that there was no ulceration and that the gastric profile was safe.243
 |
Table 5 COX-1 and COX-2 Inhibitory Activity of Compounds 359–373 |
 |
Figure 47 Structures of COX-1 and COX-2 inhibitors 349–373. |
Urease Inhibitors
Urease is a nickel-containing metalloenzyme that is involved in the catalytic reaction of urea into ammonia and carbamate. Carbamate further hydrolyzes into bicarbonate and another molecule of ammonia is produced. This pH increase is associated with alkalinity and results in gastric ulcers, kidney stones, pyelonephritis, and hepatic coma. Urease inhibitors are required to regulate urease activity. Nicotinic and isonicotinic thiosemicarbazide derivatives 374–399 illustrated in Figure 48 possess highly potent urease inhibitory activity, showing IC50 values of 1.13 to 19.74 μM.111
 |
Figure 48 Structures of urease inhibitors 374–399. |
Multi-Kinase Inhibitors
The FDA approved Ponatinib (400), a multi-kinase inhibitor developed by Ariad Pharmaceuticals, in 2012. It currently targets a wide range of cancer-driver kinases. Among these are the kinases FGFR1-4, FLT3, ABL1, and RET. Inspired by this, the discussed patent outlines a variation of Ponatinib, where the original benzimide moiety is swapped with a nicotinamide moiety. This change is considered a potentially more efficient and less harmful alternative to Ponatinib, especially in its application for treating diseases like cancer, notably acute myeloid leukemia (AML). Ponatinib (400)244,245 and HSN748 (401)246 shown in Figure 49, were tested against a panel of disease-associated kinases, that have been demonstrated to be inhibited by Ponatinib. Though there were some noticeable discrepancies with some other kinases, the inhibitory profile of HSN748 against ABLl (T3l5I) and FLT3-ITD was interestingly similar to that of Ponatinib. ABL1 and FLT3 are mutated in chronic and acute myeloid leukemias, respectively. Ponatinib and HSN748 have similar activities against ABL1, ABLl (T3l5I). Interestingly HSN748 has a significantly lower IC50 against FLT3 (D835Y) kinase than Ponatinib (compare IC50 of 13.8 nM for HSN748 versus 176 nM for Ponatinib, as shown in Table 6.
 |
Table 6 Ponatinib and HSN748 IC50 Against Several Kinases |
 |
Figure 49 Structures of multi-kinase inhibitor 400 and 401. |
Most FLT3 inhibitors initially worked well but within a few months, patients relapsed due to kinase mutation, which decreased the treatment’s effectiveness. Therefore, HSN748 may be a more effective treatment option for drug-resistant AML (due to kinase mutation) compared to Ponatinib.147
Both vascular endothelial growth factor receptor 2 (VEGFR2) and platelet-derived growth factor receptor (PDGFRβ) are important receptor-type tyrosine kinases (RTKs) involved in vascular disease.247 It was demonstrated that compounds inhibiting both kinases are more effective.248 Allergan Inc. patented two series of compounds synthesized based on Formula I and Formula II (Figure 50). In the series of Formula 1, the most preferred compounds 402‒406 have the greatest potency against both VEGFR2 and PDGFRβ. In the series of Formula II, compounds 407–410 show activity against both the VEGF and PDGF receptors. Compounds 407 and 408 have the highest activity against the VEGFR2 receptor; while compounds 409 and 410 had the best activity against the PDGFRβ receptor (Figure 50).148
 |
Figure 50 Structures of multi-kinase inhibitors 402–410. |
Conclusion
Pyridine is a therapeutically active moiety, which is found in innumerable compounds, of both synthetic and natural origin. While there is a plethora of pyridine derivatives known to date, pyridine carboxylic acid isomers and its derivatives hold a special place in medicinal and pharmaceutical chemistry. This is mainly due to their renowned biological activities, and ease of syntheses. They can be further derivatized into numerous other interesting compounds using well-known and well-established synthetic strategies. Hence, pyridine carboxylic acid and its derivatives can be easily fused to other aryl or heteroaryl moieties, thereby bringing together active pharmacophores and thus opening up new avenues for drug discovery.
This review has highlighted a range of patented pyridine carboxylic acid derivatives with promising activity profile against key biological targets. These include IRAK4 and ASK1 inhibitors with anti-inflammatory and anti-cancer potential, WDR5 inhibitors targeting oncogenic MYC interaction, and selective PDGFR inhibitors for the treatment of pulmonary arterial hypertension (PAH). Other notable applications include inhibitors of Trk, FABPs, FXIa, TGFβ, DHODH, TRPC6, KDM4, XO, GSK-3, PKaI, carbonic anhydrase, α-amylase, α-glucosidase, MetAPs, COX-1/COX-2, urease, and antiviral agents against hepatitis B. Several of these compounds are currently in various phases of clinical trials, and the hopes remain high regarding their success. A comprehensive summary of the enzyme targets, abbreviations, and their disease relevance is provided in supporting information.
Despite this encouraging progress, several challenges remain. Many pyridine carboxylic acid derivatives suffer from suboptimal pharmacokinetic properties, limited target selectivity, or insufficient in vivo validations. Furthermore, the patented literature often lack detailed mechanistic or structural biology data, making it difficult to fully assess therapeutic viability. Future research should focus on enhancing the metabolic stability, improving selectivity via structure-guided designs, and expanding biological validations in relevant disease model. In addition, more integrated studies combining computational modellings, SAR optimizations, and in vivo pharmacology will be essential to translate these compounds into clinically successful therapies. In conclusion, pyridine carboxylic acid isomers represent a class of privileged scaffolds with broad therapeutic potential. This review offers a unique patent-based perspective, capturing a decade of industry efforts and emerging trends that may inform and inspire future academic and translational research.
Acknowledgment
The authors are grateful for the support from the Higher Education Commission of Pakistan (NRPU Project No. 17588) and the “111 Center” (D25019).
Author Contributions
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data, took part in drafting the article or revising it critically for important intellectual content, agreed to submit to the current journal, gave final approval to the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
Supporting Information
Supporting information contains enzymes abbreviation, full version, and disease relevance.
References
1. Niu Z-X, Wang Y-T, Zhang S-N, et al. Application and Synthesis of Thiazole Ring in Clinically Approved Drugs. Eur J Med Chem. 2023;250:115172. doi:10.1016/j.ejmech.2023.115172
2. Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57(24):10257–10274. doi:10.1021/jm501100b
3. Wen X, Wu X, Jin R, Lu X. Privileged Heterocycles for DNA-Encoded Library Design and Hit-to-Lead Optimization. Eur J Med Chem. 2023;248:115079. doi:10.1016/J.EJMECH.2022.115079
4. El-Bana GG, Zoorob HH, Ibrahim ME, Hamama WS. Advances in 4,6‐dimethyl-3-Amino-3 h -Pyrazolo[3,4-b] Pyridine-Based and Their Annulated Systems. Synth Commun. 2020;50(19):2861–2884. doi:10.1080/00397911.2020.1786126
5. Makhoba XH. The role of pyridine derivatives on the treatment of some complex diseases: a review. In: Recent Developments in the Synthesis and Applications of Pyridines. Elsevier; 2023:143–158. doi:10.1016/B978-0-323-91221-1.00015-4
6. Altuna-Urquijo M, Gehre A, Stanforth SP, Tarbit B. A convenient synthesis of pyridine and 2,2′-bipyridine derivatives. Tetrahedron. 2009;65(5):975–984. doi:10.1016/J.TET.2008.11.090
7. Verma V, Schafer LL. One-pot sequential hydroamination protocol for N-heterocycle synthesis: one method to access five different classes of tri-substituted pyridines. J Org Chem. 2023;88(3):1378–1384. doi:10.1021/acs.joc.2c02155
8. Movassaghi M, Hill MD, Ahmad OK. Direct synthesis of pyridine derivatives. J Am Chem Soc. 2007;129(33):10096–10097. doi:10.1021/ja073912a
9. Failla M, Lombardo GW, Orlando P, et al. Late‐stage functionalisation of pyridine‐containing bioactive molecules: recent strategies and perspectives. European J Org Chem. 2023;26(16):22–39. doi:10.1002/EJOC.202300074
10. Henry GD. De novo synthesis of substituted pyridines. Tetrahedron. 2004;60(29):6043–6061. doi:10.1016/j.tet.2004.04.043
11. Chen MZ, Micalizio GC. Three-component coupling sequence for the regiospecific synthesis of substituted pyridines. J Am Chem Soc. 2012;134(2):1352–1356. doi:10.1021/ja2105703
12. Ling Y, Hao Z-Y, Liang D, Zhang C-L, Liu Y-F, Wang Y. The expanding role of pyridine and dihydropyridine scaffolds in drug design. Drug Des Devel Ther. 2021;15(October):4289–4338. doi:10.2147/DDDT.S329547
13. Bull JA, Mousseau JJ, Pelletier G, Charette AB. Synthesis of pyridine and dihydropyridine derivatives by regio- and stereoselective addition to n -activated pyridines. Chem Rev. 2012;112(5):2642–2713. doi:10.1021/cr200251d
14. Dash J, Lechel T, Reissig H-U. Scope of a novel three-component synthesis of highly functionalized pyridines. Org Lett. 2007;9(26):5541–5544. doi:10.1021/ol702468s
15. Stanforth SP, Tarbit B, Watson MD. Synthesis of pyridine derivatives using aza Diels–alder methodology. Tetrahedron Lett. 2002;43(34):6015–6017. doi:10.1016/S0040-4039(02)01215-7
16. Vessally E, Hosseinian A, Edjlali L, Bekhradnia A, Esrafili MD. New Page to access pyridine derivatives: synthesis from N-propargylamines. RSC Adv. 2016;6(75):71662–71675. doi:10.1039/C6RA08720E
17. Khan FA, Yaqoob S, Ali S, et al. Designing functionally substituted pyridine-carbohydrazides for potent antibacterial and devouring antifungal effect on Multidrug Resistant (MDR) Strains. Molecules. 2023;28(1):1–25. doi:10.3390/molecules28010212
18. Mahmoud NFH, El‐Sewedy A. Facile synthesis of novel heterocyclic compounds based on pyridine moiety with pharmaceutical activities. J Heterocycl Chem. 2020;57(4):1559–1572. doi:10.1002/jhet.3881
19. Bringmann G, Reichert Y, Kane VV. The total synthesis of streptonigrin and related antitumor antibiotic natural products. Tetrahedron. 2004;60(16):3539–3574. doi:10.1016/j.tet.2004.02.060
20. Crutcher FK, Puckhaber LS, Stipanovic RD, et al. Microbial resistance mechanisms to the antibiotic and phytotoxin fusaric acid. J Chem Ecol. 2017;43(10):996–1006. doi:10.1007/s10886-017-0889-x
21. Yun BS, Hidaka T, Furihata K, Seto H. Promothiocins A and B novel thiopeptides with a tipA promoter inducing activity produced by Streptomyces Sp. SF2741. J Antibiot. 1994;47(4):510–514. doi:10.7164/antibiotics.47.510
22. Hartkoorn RC, Sala C, Neres J, et al. Towards a new tuberculosis drug: pyridomycin – nature’s isoniazid. EMBO Mol Med. 2012;4(10):1032–1042. doi:10.1002/emmm.201201689
23. Santos VAFFM, Regasini LO, Nogueira CR, et al. Antiprotozoal sesquiterpene pyridine alkaloids from maytenus ilicifolia. J Nat Prod. 2012;75(5):991–995. doi:10.1021/np300077r
24. Han -J-J, Zhang J-Z, Zhu R-X, et al. Plagiochianins A and B, Two Ent-2,3- seco-aromadendrane derivatives from the liverwort plagiochila duthiana. Org Lett. 2018;20(20):6550–6553. doi:10.1021/acs.orglett.8b02888
25. Ieven M, Vlietinck AJ, Berghe DAV, Tottb J. Plant antiviral agents. III. isolation of alkaloids from clivia miniata regel (Amaryl-lidaceae). Plant Antivir Agents. 1982;45(5):564–573.
26. Lumfullin KL, Yuldashev PK, Yunusov SY. Investigation of the alkaloids of pedicularis olgae. Chem Nat Compd. 1965;1(5):287–288. doi:10.1007/BF00563709
27. Huang C-F, Cheng M-L, Fan C-M, Hong C-Y, Shiao M-S. Nicotinuric acid: a potential marker of metabolic syndrome through a metabolomics-based approach. Diabetes Care. 2013;36(6):1729–1731. doi:10.2337/dc12-1067
28. Zhou J, Chan L, Zhou S-W. Trigonelline: a plant alkaloid with therapeutic potential for diabetes and central nervous system disease. Curr Med Chem. 2012;19(21):3523–3531. doi:10.2174/092986712801323171
29. Hoy SM. Elexacaftor/Ivacaftor/Tezacaftor: First Approval. Drugs. 2019;79(18):2001–2007. doi:10.1007/s40265-019-01233-7
30. Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809–1819. doi:10.1056/NEJMoa1908639
31. Ettrich TJ, Seufferlein T. Regorafenib. In: Recent results in cancer research. Fortschritte der Krebsforschung. Progres dans les recherches sur le cancer. Vol. 211. Springer New York LLC; 2018:45–56. doi:10.1007/978-3-319-91442-8_3
32. Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev. 2006;5(10):835–844. doi:10.1038/nrd2130
33. Curto M, Cipolla F, Cisale GY, et al. Profiling lasmiditan as a treatment option for migraine. Expert Opin Pharmacother. 2020;21(2):147–153. doi:10.1080/14656566.2019.1694004
34. Péchère JC. Current and Future management of infections due to methicillin-resistant staphylococci infections: the role of quinupristin/dalfopristin. J Antimicrob Chemother. 1999;44 Suppl A(suppl_1):11–18. doi:10.1093/jac/44.suppl_1.11
35. Milinkovic A, Martínez E. Nevirapine in the treatment of HIV. Expert Rev Anti Infect Ther. 2004;2(3):367–373. doi:10.1586/14787210.2.3.367
36. Curto M, Capi M, Cipolla F, Cisale GY, Martelletti P, Lionetto L. Ubrogepant for the treatment of migraine. Expert Opin Pharmacother. 2020;21(7):755–759. doi:10.1080/14656566.2020.1721462
37. Heo Y-A. Flotufolastat F 18: diagnostic first approval. Mol Diagn Ther. 2023;27(5):631–636. doi:10.1007/s40291-023-00665-y
38. Sharma S, Kanugo A, Gaikwad J. Design and development of solid lipid nanoparticles of tazarotene for the treatment of psoriasis and acne: a quality by design approach. Mater Technol. 2022;37(8):735–744. doi:10.1080/10667857.2021.1873637
39. Braeckman R, Guenther S, Mickle TC, Barrett AC, Smith A, Oh C. Dose proportionality and steady-state pharmacokinetics of serdexmethylphenidate/dexmethylphenidate, a novel prodrug combination to treat attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2022;32(5):288–295. doi:10.1089/cap.2022.0015
40. Goulart Nacácio E Silva S, Occhiutto ML, Costa VP. The use of nicotinamide and nicotinamide riboside as an adjunct therapy in the treatment of glaucoma. Eur J Ophthalmol. 2023;33(5):1801–1815. doi:10.1177/11206721231161101
41. Wan Z, Chen M, Han B. Avatrombopag, a promising novel thrombopoietin receptor agonist for refractory/relapsed/intolerant non-severe aplastic anemia: a phase 2 single-arm clinical trial. Ann Med. 2023;55(1):2224044. doi:10.1080/07853890.2023.2224044
42. Hochhaus A, Réa D, Boquimpani C, et al. Asciminib vs bosutinib in chronic-phase chronic myeloid leukemia previously treated with at least two tyrosine kinase inhibitors: longer-term follow-up of ASCEMBL. Leukemia. 2023;37(3):617–626. doi:10.1038/s41375-023-01829-9
43. Wright GD. Back to the future: a new “old” lead for tuberculosis. EMBO Mol Med. 2012;4(10):1029–1031. doi:10.1002/emmm.201201811
44. Ji W-J, Jiang J-Y, Hong M, Zhu B, Ren G-B, Qi M-H. Colorful prothionamide salt forms with enhancement in water solubility and dissolution behavior. Cryst Growth Des. 2023;23(8):5770–5784. doi:10.1021/acs.cgd.3c00389
45. Drost L, Finke JB, Behrje A, Rebeck D, Domes G, Schächinger H. Optimal timing of oral metyrapone intake for the suppression of cold-pressor stress-induced cortisol release. Psychoneuroendocrinology. 2023;156:106328. doi:10.1016/j.psyneuen.2023.106328
46. Nayak N, Ramprasad J, Dalimba U. New INH-pyrazole analogs: design, synthesis and evaluation of antitubercular and antibacterial activity. Bioorganic Med Chem Lett. 2015;25(23):5540–5545. doi:10.1016/j.bmcl.2015.10.057
47. Deshpande D, Pasipanodya JG, Mpagama SG, et al. Ethionamide pharmacokinetics/pharmacodynamics-derived dose, the role of MICs in clinical outcome, and the resistance arrow of time in multidrug-resistant tuberculosis. Clin Infect Dis. 2018;67(Suppl 3):S317–S326. doi:10.1093/cid/ciy609
48. Maschouf C, Robinson RW, Lebeau RJ. Evaluation of nialamide on the coagulation of blood. Blood. 1964;24(3):289–298. doi:10.1182/blood.V24.3.289.289
49. D’alessandro C, Benedetti A, Di Paolo A, Giannese D, Cupisti A. Interactions between food and drugs, and nutritional status in renal patients: a narrative review. Nutrients. 2022;14(1). doi:10.3390/nu14010212
50. Ramachandran G, Swaminathan S. Role of pharmacogenomics in the treatment of tuberculosis: a review. Pharmgenomics Pers Med. 2012;5(1):89–98. doi:10.2147/PGPM.S15454
51. Kumar D, Khare G, Beena B, et al. Novel isoniazid–amidoether derivatives: synthesis, characterization and antimycobacterial activity evaluation. Medchemcomm. 2015;6(1):131–137. doi:10.1039/C4MD00288A
52. Hearn MJ, Cynamon MH, Chen MF, et al. Preparation and antitubercular activities in vitro and in vivo of novel Schiff bases of isoniazid. Eur J Med Chem. 2009;44(10):4169–4178. doi:10.1016/j.ejmech.2009.05.009
53. Bosworth DM. Iproniazid: a brief review of its introduction and clinical use. Ann N Y Acad Sci. 2006;80(3):809–819. doi:10.1111/j.1749-6632.1959.tb49257.x
54. Crowe M, Inder M, McCall C. Experience of antidepressant use and discontinuation: a qualitative synthesis of the evidence. J Psychiatr Ment Health Nurs. 2023;30(1):21–34. doi:10.1111/jpm.12850
55. Van Der Walt M, Keddy KH. The tuberculosis-depression syndemic and evolution of pharmaceutical therapeutics: from ancient times to the future. Front Psychiatry. 2021;12:1–9. doi:10.3389/fpsyt.2021.617751
56. Nielsen JR, Pedersen KE, Dahlstrøm CG, et al. Analgetic treatment in acute myocardial infarction. a controlled clinical comparison of morphine, nicomorphine and pethidine. Acta Med Scand. 1984;215(4):349–354. doi:10.1111/j.0954-6820.1984.tb05017.x
57. Yaqoob S, Nasim N, Khanam R, et al. Synthesis of highly potent anti-inflammatory compounds (ROS inhibitors) from isonicotinic acid. Molecules. 2021;26(5):2–9. doi:10.3390/molecules26051272
58. Bustamante D, Miranda HF, Pelissier T, Paeile C. Analgesic action of clonixin, nifedipine and morphine using the formalin test. Gen Pharmacol. 1989;20(3):319–322. doi:10.1016/0306-3623(89)90266-8
59. Cremonesi G, Cavalieri L. Efficacy and safety of morniflumate for the treatment of symptoms associated with soft tissue inflammation. J Int Med Res. 2015;43(3):290–302. doi:10.1177/0300060514567212
60. Vaile JH, Davis P. Topical NSAIDs for musculoskeletal conditions. Drugs. 1998;56(5):783–799. doi:10.2165/00003495-199856050-00004
61. Sydnes OA. A clinical investigation of niflumic acid in the treatment of rheumatoid arthritis. Scand J Rheumatol. 1973;2(sup1):8–11. doi:10.3109/03009747309095202
62. Hart FD, Boardman PL. Trial of nifenazone (“Thylin”). BMJ. 1964;1(5397):1553–1554. doi:10.1136/bmj.1.5397.1553
63. Marchesi N, Govoni S, Allegri M. Non‐drug pain relievers active on non‐opioid pain mechanisms. Pain Pract. 2022;22(2):255–275. doi:10.1111/papr.13073
64. Abdel-moety EM. Simultaneous derivative spectrophotometric quantification of diethylamine salicylate and methyl nicotinate in ointments. Spectrosc Lett. 1990;23(5):669–677. doi:10.1080/00387019008054448
65. Bailly C. the potential value of amlexanox in the treatment of cancer: molecular targets and therapeutic perspectives. Biochem Pharmacol. 2022;197:114895. doi:10.1016/j.bcp.2021.114895
66. Carlson LA. Nicotinic acid and inhibition of fat mobilizing lipolysis. present status of effects on lipid metabolism. Adv Exp Med Biol. 1978;109:225–238. doi:10.1007/978-1-4684-0967-3_12
67. Carlson LA, Fröberg SO, Nye ER. Acute effects of nicotinic acid on plasma, liver, heart and muscle lipids: nicotinic acid in the rat. II. Acta Med Scand. 1966;180(5):571–580. doi:10.1111/j.0954-6820.1966.tb02872.x
68. Wang Y-T, Liu C-H, Zhu H-L. Fatty Acid Binding Protein (FABP) inhibitors: a patent review (2012-2015). Expert Opin Ther Pat. 2016;26(7):767–776. doi:10.1080/13543776.2016.1182500
69. Vella S, Floridia M. Saquinavir. Clin Pharmacokinet. 1998;34(3):189–201. doi:10.2165/00003088-199834030-00002
70. Cooper EC, Curtis N, Cranswick N, Gwee A. Pristinamycin: old drug, new tricks? J Antimicrob Chemother. 2014;69(9):2319–2325. doi:10.1093/jac/dku167
71. Nakagawa K, Koyama M, Matsui H, et al. Pharmacokinetics of cefpiramide (SM-1652) in humans. Antimicrob Agents Chemother. 1984;25(2):221–225. doi:10.1128/AAC.25.2.221
72. Crumplin GC, Smith JT. Nalidixic acid: an antibacterial paradox. Antimicrob Agents Chemother. 1975;8(3):251–261. doi:10.1128/AAC.8.3.251
73. Arayne S, Sultana N, Haroon U, Mesaik MA. Synthesis, characterization, antibacterial and anti-inflammatory activities of enoxacin metal complexes. Bioinorg Chem Appl. 2009;2009:1–6. doi:10.1155/2009/914105
74. Feng L, Lv K, Liu M, et al. Synthesis and in vitro antibacterial activity of gemifloxacin derivatives containing a substituted benzyloxime moiety. Eur J Med Chem. 2012;55:125–136. doi:10.1016/j.ejmech.2012.07.010
75. Harada K, Irie S, Ohnishi M, Kataoka Y. Assessment of the usefulness of cefapirin and cefalonium disks for susceptibility testing of staphylococcus aureus isolates from bovine mastitis. Antibiotics. 2020;9(4):197. doi:10.3390/antibiotics9040197
76. Neu HC, Scully BE. Activity of cefsulodin and other agents against pseudomonas aeruginosa. Clin Infect Dis. 1984;6(Supplement_3):S667–S677. doi:10.1093/clinids/6.Supplement_3.S667
77. Peloquin CA, James GT, Craig LD, et al. Pharmacokinetic evaluation of aconiazide, a potentially less toxic isoniazid prodrug. Pharmacother J Hum Pharmacol Drug Ther. 1994;14(4):415–423. doi:10.1002/j.1875-9114.1994.tb02831.x
78. Fox HH. Synthetic tuberculostats. I. pyridine carboxylic acid derivatives. J Org Chem. 1952;17(4):542–546. doi:10.1021/jo01138a005
79. Herbert Fox H. Synthetic tuberculostats. II. amino- and hydroxy-pyridine carboxylic acid derivatives. J Org Chem. 1952;17(4):547–554. doi:10.1021/jo01138a006
80. Boltz D, Peng X, Muzzio M, Dash P, Thomas PG, Margitich V. Activity of enisamium, an isonicotinic acid derivative, against influenza viruses in differentiated normal human bronchial epithelial cells. Antivir Chem Chemother. 2018;26:204020661881141. doi:10.1177/2040206618811416
81. Owada A, Suda S, Hata T. Antiproteinuric effect of niceritrol, a nicotinic acid derivative, in chronic renal disease with hyperlipidemia: a randomized trial. Am J Med. 2003;114(5):347–353. doi:10.1016/S0002-9343(02)01567-X
82. O’Connor P, Feely J, Shepherd J. Lipid lowering drugs. BMJ. 1990;300(6725):667–672. doi:10.1136/bmj.300.6725.667
83. Squires H, Simpson E, Meng Y, et al. A systematic review and economic evaluation of cilostazol, naftidrofuryl oxalate, pentoxifylline and inositol nicotinate for the treatment of intermittent claudication in people with peripheral arterial disease. Clin Gov An Int J. 2012;17(2). doi:10.1108/cgij.2012.24817baa.004
84. O’Hara J, Jolly PN, Nicol CG. The therapeutic efficacy of inositol nicotinate (hexopal®) in intermittent claudication: a controlled trial. Int J Clin Pract. 1988;42(9):377–383. doi:10.1111/j.1742-1241.1988.tb08609.x
85. Sposito AC, Mansur AP, Maranhão RC, Rodrigues-Sobrinho CRM, Coelho OR, Ramires JAF. Etofibrate but not controlled-release niacin decreases LDL cholesterol and lipoprotein (a) in type iib dyslipidemic subjects. Braz J Med Biol Res. 2001;34(2):177–182. doi:10.1590/S0100-879X2001000200004
86. Tarkin JM, Kaski JC. Vasodilator therapy: nitrates and nicorandil. Cardiovasc Drugs Ther. 2016;30(4):367–378. doi:10.1007/s10557-016-6668-z
87. Pearce L, Carr RD, Yellon DM, Davidson SM. Nicorandil — an effective multitarget drug for cardioprotection? Cardiovasc Drugs Ther. 2023;37(1):5–8. doi:10.1007/s10557-022-07397-x
88. Velasco M, Gilbert CA, Rutledge CO, McNay JL, McNay JL. Antihypertensive effect of a dopamine beta hydroxylase inhibitor, bupicomide: a comparison with hydralazine. Clin Pharmacol Ther. 1975;18(2):145–153. doi:10.1002/cpt1975182145
89. Chrysant SG, Adamopoulos P, Tsuchiya M, Frohlich ED. Systemic and renal hemodynamic effects of bupicomide: a new vasodilator. Am Heart J. 1976;92(3):335–339. doi:10.1016/S0002-8703(76)80114-7
90. Hawkes N. Merck Ends trial of potential alzheimer’s drug verubecestat. BMJ. 2017;356:j845. doi:10.1136/bmj.j845
91. Aygören-Pürsün E, Magerl M, Graff J, et al. Prophylaxis of hereditary angioedema attacks: a randomized trial of oral plasma kallikrein inhibition with avoralstat. J Allergy Clin Immunol. 2016;138(3):934–936.e5. doi:10.1016/j.jaci.2016.03.043
92. Giembycz MA. Can the anti-inflammatory potential of PDE4 inhibitors be realized: guarded optimism or wishful thinking? Br J Pharmacol. 2008;155(3):288–290. doi:10.1038/bjp.2008.297
93. Coe D, Fox D. Recent disclosures of clinical candidates. Drugs Future. 2010;35(4):349. doi:10.1358/dof.2010.35.4.1487337
94. Wu C-P, Murakami M, Wu Y-S, et al. The multi-targeted tyrosine kinase inhibitor SKLB610 resensitizes ABCG2-overexpressing multidrug-resistant cancer cells to chemotherapeutic drugs. Biomed Pharmacother. 2022;149:112922. doi:10.1016/j.biopha.2022.112922
95. Rouillé T, Barbosa S, Steinhoff A, et al. LB1777 a novel ex vivo model of human hair follicle immune privilege collapse reveals the potential of farudodstat, a DHODH inhibitor, as a therapeutic for alopecia areata treatment. J Invest Dermatol. 2023;143(9):B31. doi:10.1016/j.jid.2023.06.160
96. Wang Y-J, Kathawala RJ, Zhang Y-K, et al. Motesanib (AMG706), a potent multikinase inhibitor, antagonizes multidrug resistance by inhibiting the efflux activity of the ABCB1. Biochem Pharmacol. 2014;90(4):367–378. doi:10.1016/j.bcp.2014.06.006
97. Goeldner C, Kishnani PS, Skotko BG, et al. A randomized, double-blind, placebo-controlled Phase II trial to explore the effects of a GABAA-Α5 NAM (basmisanil) on intellectual disability associated with down syndrome. J Neurodev Disord. 2022;14(1):10. doi:10.1186/s11689-022-09418-0
98. Roviello G, Ravelli A, Polom K, et al. Apatinib: a novel receptor tyrosine kinase inhibitor for the treatment of gastric cancer. Cancer Lett. 2016;372(2):187–191. doi:10.1016/j.canlet.2016.01.014
99. Hundertmark M, Siu AG, Matthews V, et al. A Phase 2a trial investigating ninerafaxstat – a novel cardiac mitotrope for the treatment of diabetic Cardiomyopathy (IMPROVE-DiCE). Eur Heart J. 2022;43:
100. Ciofalo VB, Latranyi MB, Patel JB, Taber RI. Flunixin meglumine: a non-narcotic analgesic. J Pharmacol Exp Ther. 1977;200(3):501–507.
101. Medhurst AD, Atkins AR, Beresford IJ, et al. GSK189254, a novel H 3 receptor antagonist that binds to histamine H 3 receptors in alzheimer’s disease brain and improves cognitive performance in preclinical models. J Pharmacol Exp Ther. 2007;321(3):1032–1045. doi:10.1124/jpet.107.120311
102. Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the P38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(1):63–72. doi:10.1016/S2213-2600(13)70200-5
103. DelMonte AJ, Fan Y, Girard KP, et al. Kilogram synthesis of a second-generation LFA-1/ICAM inhibitor. Org Process Res Dev. 2011;15(1):64–72. doi:10.1021/op100225g
104. Hatch SB, Yapp C, Montenegro RC, et al. Assessing histone demethylase inhibitors in cells: lessons learned. Epigenet Chromatin. 2017;10(1):9. doi:10.1186/s13072-017-0116-6
105. Johansson C, Velupillai S, Tumber A, et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat Chem Biol. 2016;12(7):539–545. doi:10.1038/nchembio.2087
106. Ajani JA, Javle M, Eng C, et al. Phase I study of DFP-11207, a novel oral fluoropyrimidine with reasonable AUC and low cmax and improved tolerability, in patients with solid tumors. Invest New Drugs. 2020;38(6):1763–1773. doi:10.1007/s10637-020-00939-w
107. Pennington LD, Moustakas DT. The necessary nitrogen atom: a versatile high-impact design element for multiparameter optimization. J Med Chem. 2017;60(9):3552–3579. doi:10.1021/acs.jmedchem.6b01807
108. Ravindra M, Wallace-Povirk A, Karim MA, et al. Tumor targeting with novel pyridyl 6-substituted pyrrolo[2,3- d]pyrimidine antifolates via cellular uptake by folate receptor α and the proton-coupled folate transporter and inhibition of de novo purine nucleotide biosynthesis. J Med Chem. 2018;61(5):2027–2040. doi:10.1021/acs.jmedchem.7b01708
109. Li JJ, Nahra J, Johnson AR, et al. Quinazolinones and pyrido[3,4- d]pyrimidin-4-ones as orally active and specific matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. J Med Chem. 2008;51(4):835–841. doi:10.1021/jm701274v
110. Khan KM, Gillani SS, Saleem F. Role of Pyridines as enzyme inhibitors in medicinal chemistry. In: Recent Developments in the Synthesis and Applications of Pyridines. Elsevier; 2023:207–252. doi:10.1016/B978-0-323-91221-1.00010-5
111. Ali B, Khan KM, Hussain S, et al. Synthetic nicotinic/isonicotinic thiosemicarbazides: in vitro urease inhibitory activities and molecular docking studies. Bioorg Chem. 2018;79:34–45. doi:10.1016/j.bioorg.2018.04.004
112. Ijuin R, Umezawa N, Higuchi T. Design, synthesis, and evaluation of new type of l-amino acids containing pyridine moiety as nitric oxide synthase inhibitor. Bioorg Med Chem. 2006;14(10):3563–3570. doi:10.1016/j.bmc.2006.01.020
113. Lin Y-S, Chen S-H, Huang W-J, et al. Effects of nicotinic acid derivatives on tyrosinase inhibitory and antioxidant activities. Food Chem. 2012;132(4):2074–2080. doi:10.1016/j.foodchem.2011.12.052
114. Santos DC, Henriques RR, Junior MAAL, et al. Acylhydrazones as isoniazid derivatives with multi-target profiles for the treatment of alzheimer’s disease: radical scavenging, myeloperoxidase/acetylcholinesterase inhibition and biometal chelation. Bioorg Med Chem. 2020;28(10):115470. doi:10.1016/j.bmc.2020.115470
115. Lu X, Zhang H, Li X, et al. Design, synthesis and biological evaluation of pyridine acyl sulfonamide derivatives as novel COX-2 inhibitors. Bioorg Med Chem. 2011;19:6827–6832. doi:10.1016/j.bmc.2011.09.034
116. Di bello E, Sian V, Bontempi G, et al. Novel pyridine-containing histone deacetylase inhibitors strongly arrest proliferation, induce apoptosis and modulate MiRNAs in cancer cells. Eur J Med Chem. 2023;247:115022. doi:10.1016/j.ejmech.2022.115022
117. Baker MD, Campbell M, Diallo H, et al. Demethylase enzymes inhibitors. WO 2013/143597 Al. 2013.
118 Shirasaki Y, Miyashita H, Yamaguchi M. Exploration of orally available calpain inhibitors. part 3: dipeptidyl α-ketoamide derivatives containing pyridine moiety. Bioorg Med Chem. 2006;14(16):5691–5698. doi:10.1016/j.bmc.2006.04.013
119. Desogus A, Schenone S, Brullo C, Tintori C, Musumeci F. Bcr-Abl tyrosine kinase inhibitors: a patent review. Expert Opin Ther Patents. 2015;397–412. doi:10.1517/13543776.2015.1012155
120. Zhu W, Wang W, Xu S, et al. Bioorganic & medicinal chemistry synthesis, and docking studies of phenylpyrimidine-carboxamide derivatives bearing 1 h -pyrrolo [2, 3- b] pyridine moiety as c-met inhibitors. Bioorg Med Chem. 2016;24(8):1749–1756. doi:10.1016/j.bmc.2016.02.046
121. Zhu W, Wang W, Xu S, et al. Bioorganic & medicinal chemistry design, synthesis, and docking studies of phenylpicolinamide derivatives bearing 1 h -pyrrolo [2, 3- b] pyridine moiety as c-met inhibitors. Bioorg Med Chem. 2016;24(4):812–819. doi:10.1016/j.bmc.2016.01.001
122. Tang Q, Wang L, Tu Y, et al. Bioorganic & medicinal chemistry letters discovery of novel pyrrolo [2, 3-b] pyridine derivatives bearing 1, 2, 3-triazole moiety as c-met kinase inhibitors. Bioorg Med Chem Lett. 2016;26(7):1680–1684. doi:10.1016/j.bmcl.2016.02.059
123. Copeland RA, Harpel MR, Tummino PJ. Targeting enzyme inhibitors in drug discovery. Expert Opin Ther Targets. 2007;11(7):967–978. doi:10.1517/14728222.11.7.967
124. Cusack KP, Hoemann MZ, Kinsman DA, et al. Preparation of nicotinamide derivatives containing pyrazole and related heterocycles as RIPK1 inhibitors for the treatment of diseases. WO2023018643. 2023.
125. Ulrich B, Wengner AM, Holger S, et al. Combinations of inhibitors of IRAK4 with inhibitors of BTK. WO 2016/174183 A1. 2016.
126. Rowbottom MW. Apoptosis signal-regulating kinase 1 (ASK1) inhibitor compounds. WO 2019/099703 A1. 2019.
127. Bouyssou, Gottschling T, Heine D, et al. Pyridine carbonyl derivatives and therapeutic uses thereof as TRPC6 inhibitors. WO2019081637, 2019.
128. Gogliotti RD, Stauffer SR, Jeon K, et al. WDR5 - MLL1 inhibitors and modulators. US 2020/0055824 A1. 2020.
129. Zhang Z, Zhou X, Arend MP, Kjaergaard CH. Compounds, Composition and methods for histone demethylase inhibition. 2022.
130. Salas Solana J, Carceller Gonzalez E, Ortega Munoz A. 2-(Bicyclo-heteroaryl)-isonicotinic derivatives as histone demethylase inhibitors. WO 2018/149986 Al. 2018.
131. Labelle M, Boesen T, Mehrotra M, Khan Q, Ullah F. Inhibitors of histone demethylases. WO 2014/053491 Al. 2014.
132. Kanouni T, Stafford JA, Veal JM, Wallace MB. Histone demethylase inhibitors. WO 2014/100463 Al. 2014.
133. Chen YK, Wallace MB. Histone demethylase inhibitors. WO 2015/200709 Al. 2015.
134. Tripathi S, Narayan R, Abhijith Biji RSR. Antiviral applications of picolinic acid and its derivatives. 2022.
135. Ian B, Sylvie C, pascal F, et al. Solid forms of bicyclic heterocyclic derivatives as PDGF receptor mediators. WO 2014/132220 Al. 2014.
136. Sarah Elizabeth S, Sharanjeet Kaur B, Nigel Alan S, Kiyoyuki O, Mark David A. N-Acylpiperidine Ether Tropomyosin- Related Kinase Inhibitors. WO 2015/092610 Al. 2015.
137. Bachovchin WW, Sen Lai H, O’Connell DP, Wu W, Kiritsy CP. Niacin mimetics and methods of use thereof. US 8,937,063 B2. 2015.
138. Al-Horani RA, Desai UR. Factor xia inhibitors: a review of the patent literature. Expert Opin Ther Pat. 2016;26(3):323–345. doi:10.1517/13543776.2016.1154045
139. Anand Venkataramana S, Iain David R, Andrew R. Novel polymorphic forms of a TGFB inhibitors. WO 2020/128850 A1. 2020.
140. Ashok A, Ian MB, Michael JB, et al. 2-Amino-N-hetero aryl -nicotinamides as NAV1.8 inhibitors. WO 2020/092667 A1. 2020.
141. Laria JCCP, Sola ME, Toribio MEL, Romero EN. Amino nicotinic and isonicotinic acid derivatives as DHODH inhibitors. US 8,691,852 B2, 2014.
142. Song JU, Tae Kim G, Pil Choi S, et al. Compounds effective as xanthine oxidase inhibitors, method for preparing the same, and pharmaceutical composition containing the same. US 8,729,273 B2. 2014.
143. Luo G, Chen L, Dubowchik GM, et al. GSK-3 Inhibitors. WO 2015/069594 Al. 2015.
144. Andrew MSQ. Therapeutic inhibitory compounds. WO 2016/011209 Al. 2016.
145. Dotiwala F, Salvino JM. IsPH inhibitors, and methods of making and using same. 2021.
146. Braithwaite SP, Minami SS, Nikolich K. Methods and composition for treating aging-associated impairment using CCR-3 inhibitors. 2022.
147. Sintim HO, Laroque E, Naganna N. Alkynyl nicotinamide compounds as kinase inhibitors. WO2020/053812A1. 2020.
148. Sougato B, Shimiao W, Thomas M, Julie W, Jie S, Michael R. Substituted nicotinamide derivatives as kinase inhibitors. WO 2015/089220 Al. 2015.
149. Bai Y-R, Yang W-G, Hou X-H, et al. The Recent advance of interleukin-1 receptor associated kinase 4 inhibitors for the treatment of inflammation and related diseases. Eur J Med Chem. 2023;258:115606. doi:10.1016/j.ejmech.2023.115606
150. Bhide RS, Keon A, Weigelt C, et al. Discovery and structure-based design of 4,6-diaminonicotinamides as potent and selective IRAK4 inhibitors. Bioorg Med Chem Lett. 2017;27(21):4908–4913. doi:10.1016/j.bmcl.2017.09.029
151. McElroy WT. Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) inhibitors: an updated patent review (2016-2018). Expert Opin Ther Pat. 2019;29(4):243–259. doi:10.1080/13543776.2019.1597850
152. Bryan MC, Drobnick J, Gobbi A, et al. Development of potent and selective pyrazolopyrimidine IRAK4 inhibitors. J Med Chem. 2019;62(13):6223–6240. doi:10.1021/acs.jmedchem.9b00439
153. Zhai W, Lu Y, Zhu Y, et al. Discovery and optimization of a potent and selective indazolamine series of IRAK4 inhibitors. Bioorg Med Chem Lett. 2021;31:127686. doi:10.1016/j.bmcl.2020.127686
154. Cumming IA, Degorce SL, Aagaard A, et al. Identification and optimisation of a pyrimidopyridone series of IRAK4 inhibitors. Bioorg Med Chem. 2022;63:116729. doi:10.1016/j.bmc.2022.116729
155. Dudhgaonkar S, Ranade S, Nagar J, et al. Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. J Immunol. 2017;198(3):1308–1319. doi:10.4049/jimmunol.1600583
156. Xin Z, Himmelbauer MK, Jones JH, et al. Discovery of CNS-penetrant Apoptosis Signal-Regulating Kinase 1 (ASK1) inhibitors. ACS Med Chem Lett. 2020;11(4):485–490. doi:10.1021/acsmedchemlett.9b00611
157. Qin J, Cao M, Hu X, et al. Dual inhibitors of ASK1 and PDK1 kinases: design, synthesis, molecular docking and mechanism studies of N-benzyl pyridine-2-one containing derivatives as anti-fibrotic agents. Eur J Med Chem. 2023;247:115057. doi:10.1016/j.ejmech.2022.115057
158. Ogier JM, Nayagam BA, Lockhart PJ. ASK1 inhibition: a therapeutic strategy with multi-system benefits. J Mol Med. 2020;98(3):335–348. doi:10.1007/S00109-020-01878-Y
159. Obsilova V, Honzejkova K, Obsil T. Structural insights support targeting ASK1 kinase for therapeutic interventions. Int J Mol Sci. 2021;22(24):13395. doi:10.3390/IJMS222413395
160. Okamoto M, Saito N, Kojima H, et al. Identification of novel ASK1 inhibitors using virtual screening. Bioorg Med Chem. 2011;19(1):486–489. doi:10.1016/j.bmc.2010.11.004
161. Papaconstantinou J. The Role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells. 2019;8(11):1383. doi:10.3390/cells8111383
162. Tesch GH, Ma FY, Nikolic-Paterson DJ. ASK1: a new therapeutic target for kidney disease. Am J Physiol. 2016;311(2):F373–F381. doi:10.1152/AJPRENAL.00208.2016/ASSET/IMAGES/LARGE/ZH20111679670003.JPEG
163. Clapham DE, Runnels LW, Strübing C. The trp ion channel family. Nat Rev Neurosci. 2001;2(6):387–396. doi:10.1038/35077544
164. Pan Z, Yang H, Reinach PS. Transient Receptor Potential (TRP) gene superfamily encoding cation channels. Hum Genomics. 2011;5(2):108. doi:10.1186/1479-7364-5-2-108
165. Prikhodko V, Chernyuk D, Sysoev Y, Zernov N, Okovityi S, Popugaeva E. Potential drug candidates to treat TRPC6 channel deficiencies in the pathophysiology of alzheimer’s disease and brain ischemia. Cells. 2020;9(11):2351. doi:10.3390/cells9112351
166. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76(1):387–417. doi:10.1146/annurev.biochem.75.103004.142819
167. Häfner S, Burg F, Kannler M, et al. A (+)-Larixol congener with high affinity and subtype selectivity toward TRPC6. ChemMedChem. 2018;13(10):1028–1035. doi:10.1002/cmdc.201800021
168. Preti D, Szallasi A, Patacchini R. TRP Channels as therapeutic targets in airway disorders: a patent review. Expert Opin Ther Pat. 2012;22(6):663–695. doi:10.1517/13543776.2012.696099
169. Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev. 2014;66(3):676–814. doi:10.1124/pr.113.008268
170. Häfner S, Urban N, Schaefer M. Discovery and characterization of a positive allosteric modulator of Transient Receptor Potential Canonical 6 (TRPC6) channels. Cell Calcium. 2019;78:26–34. doi:10.1016/j.ceca.2018.12.009
171. Ye X, Zhang R, Lian F, et al. The identification of novel small-molecule inhibitors targeting WDR5-MLL1 interaction through fluorescence polarization based high-throughput screening. Bioorg Med Chem Lett. 2019;29(4):638–645. doi:10.1016/j.bmcl.2018.12.035
172. Bolshan Y, Getlik M, Kuznetsova E, et al. Optimization, and evaluation of novel small molecules as antagonists of WDR5-MLL interaction. ACS Med Chem Lett. 2013;4(3):353–357. doi:10.1021/ml300467n
173. Dharmarajan V, Lee J-H, Patel A, Skalnik DG, Cosgrove MS. Structural basis for WDR5 interaction (win) motif recognition in human SET1 family histone methyltransferases. J Biol Chem. 2012;287(33):27275–27289. doi:10.1074/jbc.M112.364125
174. Getlik M, Smil D, Zepeda-Velázquez C, et al. Structure-based optimization of a small molecule antagonist of the interaction between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1). J Med Chem. 2016;59(6):2478–2496. doi:10.1021/acs.jmedchem.5b01630
175. Mitchell K, Sprowls SA, Arora S, et al. WDR5 represents a therapeutically exploitable target for cancer stem cells in glioblastoma. Genes Dev. 2023;37(3–4):86–102. doi:10.1101/gad.349803.122
176. Chen W, Chen X, Li D, et al. Discovery of a potent MLL1 and WDR5 protein-protein interaction inhibitor with in vivo antitumor activity. Eur J Med Chem. 2021;223:113677. doi:10.1016/j.ejmech.2021.113677
177. Karatas H, Townsend EC, Bernard D, Dou Y, Wang S. Analysis of the Binding of Mixed Lineage Leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1−WDR5 interaction. J Med Chem. 2010;53(14):5179–5185. doi:10.1021/jm100139b
178. Britten O, Ragusa D, Tosi S, Mostafa Kamel Y. MLL-rearranged acute leukemia with t(4;11)(Q21;Q23)—current treatment options. Is there a role for CAR-T cell therapy? Cells. 2019;8(11):1341. doi:10.3390/cells8111341
179. Małecki PH, Rüger N, Roatsch M, et al. Front cover: structure‐based screening of tetrazolylhydrazide inhibitors versus KDM4 histone demethylases (ChemMedChem 21/2019). ChemMedChem. 2019;14(21):1803. doi:10.1002/cmdc.201900598
180. He X, Zhang H, Zhang Y, et al. Drug discovery of Histone Lysine Demethylases (KDMs) inhibitors (progress from 2018 to present). Eur J Med Chem. 2022;231:114143. doi:10.1016/j.ejmech.2022.114143
181. Nie Z, Shi L, Lai C, et al. Structure-based design and discovery of potent and selective KDM5 inhibitors. Bioorg Med Chem Lett. 2018;28(9):1490–1494. doi:10.1016/j.bmcl.2018.03.083
182. Chen YK, Bonaldi T, Cuomo A, et al. Design of KDM4 inhibitors with antiproliferative effects in cancer models. ACS Med Chem Lett. 2017;8(8):869–874. doi:10.1021/acsmedchemlett.7b00220
183. Morera L, Roatsch M, Fürst MCD, et al. 4-biphenylalanine- and 3-phenyltyrosine-derived hydroxamic acids as inhibitors of the JumonjiC-domain-containing histone demethylase KDM4A. ChemMedChem. 2016;11(18):2063–2083. doi:10.1002/cmdc.201600218
184. Małecki PH, Rüger N, Roatsch M, et al. Structure‐based screening of tetrazolylhydrazide inhibitors versus KDM4 histone demethylases. ChemMedChem. 2019;14(21):1828–1839. doi:10.1002/cmdc.201900441
185. Wu Q, Young B, Wang Y, Davidoff AM, Rankovic Z, Yang J. Recent advances with KDM4 inhibitors and potential applications. J Med Chem. 2022;65(14):9564–9579. doi:10.1021/acs.jmedchem.2c00680
186. Bavetsias V, Lanigan RM, Ruda GF, et al. 8-substituted pyrido[3,4- d]pyrimidin-4(3 h)-one derivatives as potent, cell permeable, KDM4 (JMJD2) and KDM5 (JARID1) histone lysine demethylase inhibitors. J Med Chem. 2016;59(4):1388–1409. doi:10.1021/acs.jmedchem.5b01635
187. Lee DH, Kim GW, Jeon YH, Yoo J, Lee SW, Kwon SH. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J. 2020;34(3):3461–3484. doi:10.1096/fj.201902584R
188. Fu Y-D, Huang M-J, Guo J-W, et al. Targeting histone demethylase KDM5B for cancer treatment. Eur J Med Chem. 2020;208:112760. doi:10.1016/j.ejmech.2020.112760
189. Labbé RM, Holowatyj A, Yang ZQ. Histone lysine demethylase (Kdm) subfamily 4: structures, functions and therapeutic potential. Am J Transl Res. 2014; 2014:1–15.
190. Chin YW, Han SY. KDM4 histone demethylase inhibitors for anti-cancer agents: a patent review. Expert Opin Ther Patents. 2015;135–144. doi:10.1517/13543776.2014.991310
191. Aguayo E, Appleby T, Birkus G, et al. Methods of treating hepatitis B virus. US 2018/0042905 A1. 2018.
192. Mohebbi A, Lorestani N, Tahamtan A, Kargar NL, Tabarraei A. An overview of hepatitis B virus surface antigen secretion inhibitors. Front Microbiol. 2018;9:1–9. doi:10.3389/fmicb.2018.00662
193. Hu Z, Hu J, Ren F, et al. Nobiletin, a novel inhibitor, inhibits HBsAg production and hepatitis B virus replication. Biochem Biophys Res Commun. 2020;523(3):802–808. doi:10.1016/j.bbrc.2019.12.099
194. Dai Y. Platelet-derived growth factor receptor tyrosine kinase inhibitors: a review of the recent patent literature. Expert Opin Ther Pat. 2010;20(7):885–897. doi:10.1517/13543776.2010.493559
195. Liu G, Campbell BT, Holladay MW, et al. Discovery of AC710, a globally selective inhibitor of platelet-derived growth factor receptor-family kinases. ACS Med Chem Lett. 2012;3(12):997–1002. doi:10.1021/ml300214g
196. Norman P. Evaluation of WO-2014132220, selective PDGFR inhibitors for the treatment of pulmonary arterial hypertension. Expert Opin Ther Pat. 2015;25(4):493–499. doi:10.1517/13543776.2015.1007042
197. Medarametla V, Festin S, Sugarragchaa C, et al. PK10453, a nonselective platelet‐derived growth factor receptor inhibitor, prevents the progression of pulmonary arterial hypertension. Pulm Circ. 2014;4(1):82–102. doi:10.1086/674881
198. Shaw DE, Baig F, Bruce I, et al. Optimization of Platelet-Derived Growth Factor Receptor (PDGFR) inhibitors for duration of action, as an inhaled therapy for lung remodeling in pulmonary arterial hypertension. J Med Chem. 2016;59(17):7901–7914. doi:10.1021/acs.jmedchem.6b00703
199. Qiu L, Levine K, Gajiwala KS, et al. Small molecule inhibitors reveal PTK6 kinase is not an oncogenic driver in breast cancers. PLoS One. 2018;13(6):e0198374. doi:10.1371/journal.pone.0198374
200. Bagal SK, Andrews M, Bechle BM, et al. Discovery of potent, selective, and peripherally restricted pan-trk kinase inhibitors for the treatment of pain. J Med Chem. 2018;61(15):6779–6800. doi:10.1021/acs.jmedchem.8b00633
201. Bailey JJ, Schirrmacher R, Farrell K, Bernard-Gauthier V. Tropomyosin receptor kinase inhibitors: an updated patent review for 2010-2016–part II. Expert Opin Ther Patents. 2017;831–849. doi:10.1080/13543776.2017.1297797
202. Guyton JR, Bays HE. Safety considerations with niacin therapy. Am J Cardiol. 2007;99(6 SUPPL. 1). doi:10.1016/j.amjcard.2006.11.018
203. Dawood DH, Srour AM, Saleh DO, Huff KJ, Greco F, Osborn HMI. New pyridine and chromene scaffolds as potent vasorelaxant and anticancer agents. RSC Adv. 2021;11(47):29441–29452. doi:10.1039/D1RA04758B
204. Sinthupoom N, Prachayasittikul V, Prachayasittikul S, Ruchirawat S, Prachayasittikul V. Nicotinic acid and derivatives as multifunctional pharmacophores for medical applications. Eur Food Res Technol. 2014;240(1):1–17. doi:10.1007/s00217-014-2354-1
205. Lazarova TI, Jin L, Rynkiewicz M, et al. Synthesis and in vitro biological evaluation of aryl boronic acids as potential inhibitors of factor XIa. Bioorganic Med Chem Lett. 2006;16(19):5022–5027. doi:10.1016/j.bmcl.2006.07.043
206. Al-Horani RA. Factor XI(a) inhibitors for thrombosis: an updated patent review (2016-present). Expert Opin Ther Patents. 2020;30:39–55. doi:10.1080/13543776.2020.1705783
207. Abdel-Magid AF. Inhibitors of transforming growth factor beta receptor 1 (TGFβr1) may enhance the efficacy of several monoclonal antibodies as cancer therapy. ACS Med Chem Lett. 2022;13(9):1405–1407. doi:10.1021/acsmedchemlett.2c00356
208. Pujala B, Ramachandran SA, Sonawane M, et al. Discovery of MDV6058 (PF-06952229), a selective and potent TGFβR1 inhibitor: design, synthesis and optimization. Bioorg Med Chem Lett. 2022;75:128979. doi:10.1016/j.bmcl.2022.128979
209. Vyas KV, Ghate M. Recent developments in the medicinal chemistry and therapeutic potential of Dihydroorotate Dehydrogenase (DHODH) inhibitors. Mini-Reviews Med Chem. 2011;11(12):1039–1055. doi:10.2174/138955711797247707
210. Zhou Y, Tao L, Zhou X, et al. DHODH and Cancer: promising prospects to be explored. Cancer Metab. 2021;9(1):1–25. doi:10.1186/S40170-021-00250-Z
211. DeRatt LG, Christine Pietsch E, Tanner A, et al. A carboxylic acid isostere screen of the DHODH inhibitor brequinar. Bioorg Med Chem Lett. 2020;30(22):127589. doi:10.1016/j.bmcl.2020.127589
212. Madak JT, Bankhead A, Cuthbertson CR, Showalter HD, Neamati N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol Ther. 2019;195:111–131. doi:10.1016/j.pharmthera.2018.10.012
213. Vyas VK, Ghate M. 2D and 3D QSAR study on amino nicotinic acid and isonicotinic acid derivatives as potential inhibitors of Dihydroorotate Dehydrogenase (DHODH). Med Chem Res. 2012;21(10):3021–3034. doi:10.1007/s00044-011-9837-4
214. Luna G, Dolzhenko AV, Mancera RL. Inhibitors of xanthine oxidase: scaffold diversity and structure-based drug design. ChemMedChem. 2019;14(7):714–743. doi:10.1002/CMDC.201900034
215. Kaur G, Singh A, Arora G, et al. Synthetic heterocyclic derivatives as promising xanthine oxidase inhibitors: an overview. Chem Biol Drug Des. 2022;100(3):443–468. doi:10.1111/cbdd.14109
216. Luna G, Dolzhenko AV, Mancera RL. Inhibitors of xanthine oxidase: scaffold diversity and structure‐based drug design. ChemMedChem. 2019;14(7):714–743. doi:10.1002/cmdc.201900034
217. Borges F, Fernandes E, Roleira F. Progress towards the discovery of xanthine oxidase inhibitors. Curr Med Chem. 2002;9(2):195–217. doi:10.2174/0929867023371229
218. Pacher P, Nivorozhkin A, Szabó C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58(1):87–114. doi:10.1124/pr.58.1.6
219. Ojha R, Singh J, Ojha A, Singh H, Sharma S, Nepali K. An Updated Patent Review: Xanthine Oxidase Inhibitors for the Treatment of Hyperuricemia and Gout (2011-2015). In: Expert Opinion on Therapeutic Patents. Taylor & Francis; 2017:311–345. doi:10.1080/13543776.2017.1261111
220. Arciniegas Ruiz SM, Eldar-Finkelman H. Glycogen synthase kinase-3 inhibitors: preclinical and clinical focus on CNS-A decade onward. Front Mol Neurosci. 2022;14:792364. doi:10.3389/fnmol.2021.792364
221. Srivani G, Sharvirala R, Veerareddy PR, Pal D, Kiran G. GSK-3 inhibitors as new leads to treat type-II diabetes. Curr Drug Targets. 2021;22(13):1555–1567. doi:10.2174/1389450122666210120144428
222. Kandar CC, Sen D, Maity A. Anti-inflammatory potential of GSK-3 inhibitors. Curr Drug Targets. 2021;22(13):1464–1476. doi:10.2174/1389450122666210118150313
223. Morales-García JA, Susín C, Alonso-Gil S, et al. Glycogen synthase kinase-3 inhibitors as potent therapeutic agents for the treatment of Parkinson disease. ACS Chem Neurosci. 2013;4(2):350–360. doi:10.1021/cn300182g
224. Pal D, Mukherjee S, Song I-H, Nimse SB. GSK-3 inhibitors: a new class of drugs for alzheimer’s disease treatment. Curr Drug Targets. 2021;22(15):1725–1737. doi:10.2174/1389450122666210114095307
225. Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev. 2004;3(6):479–487. doi:10.1038/nrd1415
226. Xie Z, Li Z, Shao Y, Liao C. Discovery and development of plasma kallikrein inhibitors for multiple diseases. Eur J Med Chem. 2020;190:112137. doi:10.1016/j.ejmech.2020.112137
227. Li Z, Partridge J, Silva-Garcia A, et al. Structure-guided design of novel, potent, and selective macrocyclic plasma kallikrein inhibitors. ACS Med Chem Lett. 2017;8(2):185–190. doi:10.1021/acsmedchemlett.6b00384
228. Hussain Z, Mahmood A, Shah Q, et al. Synthesis and evaluation of amide and thiourea derivatives as Carbonic Anhydrase (CA) inhibitors. ACS Omega. 2022;7(50):47251–47264. doi:10.1021/acsomega.2c06513
229. Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett. 2010;20(12):3467–3474. doi:10.1016/j.bmcl.2010.05.009
230. Wiggins SV, Schreiner R, Ferreira J, Marmorstein AD, Levin LR, Buck J. Carbonic anhydrase inhibitor modulation of intraocular pressure is independent of soluble adenylyl cyclase. J Ocul Pharmacol Ther. 2023;39(5):317–323. doi:10.1089/jop.2022.0180
231. Ciccone L, Cerri C, Nencetti S, Orlandini E. Carbonic anhydrase inhibitors and epilepsy: state of the art and future perspectives. Molecules. 2021;26(21):6380. doi:10.3390/molecules26216380
232. Bulut N, Kocyigit UM, Gecibesler IH, et al. Synthesis of some novel pyridine compounds containing Bis-1,2,4-triazole/thiosemicarbazide moiety and investigation of their antioxidant properties, carbonic anhydrase, and acetylcholinesterase enzymes inhibition profiles. J Biochem Mol Toxicol. 2018;32(1):1–10. doi:10.1002/jbt.22006
233. Menabuoni L, Scozzafava A, Mincione F, Briganti F, Mincione G, Supuran CT. Carbonic anhydrase inhibitors. water-soluble, topically effective intraocular pressure lowering agents derived from isonicotinic acid and aromatic/heterocyclic sulfonamides: is the tail more important than the ring? J Enzyme Inhib. 1999;14(6):457–474. doi:10.3109/14756369909030336
234. Mohammad HK, Alzweiri MH, Khanfar MA, Al-Hiari YM. 6-substituted nicotinic acid analogues, potent inhibitors of caiii, used as therapeutic candidates in hyperlipidemia and cancer. Med Chem Res. 2017;26(7):1397–1404. doi:10.1007/s00044-017-1825-x
235. Carlos S, Minami SS, Carlos S. Methods and compositions for treating aging-associated impairments using CCR3-Inhibitors. US11382907B2, 2022.
236. Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477(7362):90–96. doi:10.1038/nature10357
237. Nawaz M, Taha M, Qureshi F, et al. Structural elucidation, molecular docking, α-amylase and α-glucosidase inhibition studies of 5-amino-nicotinic acid derivatives. BMC Chem. 2020;14(1):1–11. doi:10.1186/s13065-020-00695-1
238. Luo QL, Li JY, Liu ZY, et al. Inhibitors of type I MetAPs containing pyridine-2-carboxylic acid thiazol-2-ylamide. Part 1: SAR studies on the determination of the key scaffold. Bioorganic Med Chem Lett. 2005;15(3):635–638. doi:10.1016/j.bmcl.2004.11.034
239. Grant RS, Coggan SE, Smythe GA. The physiological action of picolinic acid in the human brain. Int J Tryptophan Res. 2009;2(1):71–79. doi:10.4137/ijtr.s2469
240. Amaral M, Levy C, Heyes DJ, et al. Structural basis of kynurenine 3-monooxygenase inhibition. Nature. 2013;496(7445):382–385. doi:10.1038/nature12039
241. Hughes TD, Güner OF, Iradukunda EC, Phillips RS, Bowen JP. The kynurenine pathway and kynurenine 3-monooxygenase inhibitors. Molecules. 2022;27(1):1–26. doi:10.3390/molecules27010273
242. Shave S, McGuire K, Pham NT, Mole DJ, Webster SP, Auer M. Diclofenac identified as a kynurenine 3-monooxygenase binder and inhibitor by molecular similarity techniques. ACS Omega. 2018;3(3):2564–2568. doi:10.1021/acsomega.7b02091
243. El-Dash Y, Khalil NA, Ahmed EM, Hassan MSA. Synthesis and biological evaluation of new nicotinate derivatives as potential anti-inflammatory agents targeting COX-2 enzyme. Bioorg Chem. 2021;107:104610. doi:10.1016/j.bioorg.2020.104610
244. O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. doi:10.1016/j.ccr.2009.09.028
245. Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome–positive leukemias. N Engl J Med. 2012;367(22):2075–2088. doi:10.1056/NEJMoa1205127
246. Larocque E, Chu EFY, Naganna N, Sintim HO. Nicotinamide–ponatinib analogues as potent Anti-CML and Anti-AML compounds. ACS Omega. 2020;5(6):2690–2698. doi:10.1021/acsomega.9b03223
247. He C, Medley SC, Hu T, et al. PDGFRβ signalling Regulates Local Inflammation And Synergizes With Hypercholesterolaemia To Promote Atherosclerosis. Nat Commun. 2015;6:1–14. doi:10.1038/ncomms8770
248. Jo N, Mailhos C, Ju M, et al. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol. 2006;168(6):2036–2053. doi:10.2353/ajpath.2006.050588
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.