")
Back to Journals » International Journal of Nanomedicine » Volume 20
Mitochondrial Reactive Oxygen Species (mROS) Generation and Cancer: Emerging Nanoparticle Therapeutic Approaches
Received 7 December 2024
Accepted for publication 24 April 2025
Published 13 May 2025 Volume 2025:20 Pages 6085—6119
DOI https://doi.org/10.2147/IJN.S510972
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Farooq A. Shiekh
Xinyao Wang,1,2 Xiangyang Xiong1,3
1The MOE Basic Research and Innovation Center for the Targeted Therapeutics of Solid Tumors, School of Basic Medical Sciences, Jiangxi Medical College, Nanchang University, Nanchang, People’s Republic of China; 2Queen Mary School of Nanchang University, Nanchang, People’s Republic of China; 3Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Nanchang University, Nanchang, People’s Republic of China
Correspondence: Xiangyang Xiong, School of Basic Medical Sciences, Nanchang University, Nanchang, Jiangxi, People’s Republic of China, Email [email protected]
Abstract: Mitochondrial reactive oxygen species (mROS) are generated as byproducts of mitochondrial oxidative phosphorylation. Changes in mROS levels are involved in tumorigenesis through their effects on cancer genome instability, sustained cancer cell survival, metabolic reprogramming, and tumor metastasis. Recent advances in nanotechnology offer a promising approach for precise regulation of mROS by either enhancing or depleting mROS generation. This review examines the association between dysregulated mROS levels and key cancer hallmarks. We also discuss the potential applications of mROS-targeted nanoparticles that artificially manipulate ROS levels in the mitochondria to achieve precise delivery of antitumor drugs.
Keywords: mitochondria reactive oxygen species, nanoparticle, drug delivery, cancer therapy
Graphical Abstract:

Introduction
Mitochondria, one of the first organelles discovered during endosymbiosis, is known as “the powerhouse of the cell”.1 More than 98% of the oxygen is consumed to produce energy in the form of adenosine phosphate (ATP) by oxidative phosphorylation. In contrast, 1–2% of the electrons leak to oxygen to form reactive oxygen species (ROS).2,3 The major forms of ROS include free oxygen radicals, such as superoxide and hydroxyl radicals, and non-radical biomolecules, such as hydrogen peroxide.4 These different ROS can interconvert to maintain the redox balance in the cell. For example, superoxide anions are produced when electrons leak and react with oxygen in complexes I and III in mitochondria. Manganese superoxide dismutase (MnSOD), a mitochondrial superoxide scavenging enzyme, converts these superoxide anions into hydrogen peroxide, which is less reactive but can still cause oxidative stress if not managed.5 The role of mROS in cancer can be summarized in two aspects: First, increased mROS levels have been implicated in many tumor types by altering many biological mechanisms.6–9 Therefore, the novelty of our review is that we combined mROS alterations with related cancer hallmarks that explain the general process of neoplastic progression, as summarized by Hanahan and Weinberg, providing an approach to explore abnormal mROS levels with highly conserved principles in tumor progression.10 Second, alteration of mROS can also be used as a targeted therapy for cancer, either by an increase in ROS production in the mitochondria to induce cancer cell death or a reduction in mROS production to limit cancer cell proliferation.11 Significantly, the modulation of mROS can serve as a strategy to tackle multidrug resistance, a primary factor contributing to tumor relapse following conventional chemotherapy.12 For example, a mROS-targeted nanoparticle was evaluated for its antitumor efficacy in a multidrug-resistant ovarian cancer cell line. The generated ROS reduced the proton concentration gradient across the inner mitochondrial membrane, thereby inhibiting ATP-dependent efflux pumps in cancer cells. These pumps are a major reason why many antitumor drugs cannot be retained within cancer cells.13
In recent years, nanoparticles have emerged as a novel drug delivery system that utilizes 1–100 nm biomaterials to deliver therapeutic payloads into pathological tissues.14 Pioneer works have reported that targeting mitochondria shows great therapeutic potential to disrupt abnormal energy production,15 induces cellular apoptosis16 and overcomes drug resistance in tumorigenesis.17 In contrast to untargeted ROS inducer/suppressor, specifically modulation of ROS in mitochondria demonstrated improved antitumor activities and reduced side effects.18,19 However, guiding nanoparticles to reach mitochondria in clinical use still faces significant challenges compared to other emerging cancer therapies, such as immunotherapy and gene therapy. Recently, several nanoparticle formulations, including those based on gold, polymers, liposomes, and upconversion nanoparticles, have been developed to enhance mitochondrial targeting.20–23 Also, functional modifications, such as mitochondrial-targeting peptides or ligands, enable nanoparticles to cross the mitochondrial membrane and accumulate specifically within the mitochondria.24 Nanoparticles can be engineered to target mitochondria with remarkable precision, delivering mROS-inducing agents to induce cell death through apoptosis, necroptosis, ferroptosis, pyroptosis, and cuproptosis or by disrupting antioxidant defenses specifically within the mitochondria of cancer cells. By harnessing the unique role of mROS in cancer biology, nanoparticle-based therapies hold promise for advancing cancer treatment, offering a novel and selective approach for tumor eradication. This review discusses the role of mROS in the normal cellular environment and the effect of mROS alterations in promoting cancer cell growth, genome instability, metabolic reprogramming, and cancer cell metastasis. In addition, we summarize mROS-targeted nanoparticles that induce cell death via five cell death mechanisms or deplete ROS generation in mitochondria via a series of “Mito” products, providing a promising future for targeted cancer therapies.
mROS Generation in Normal Homeostatic Environment
In normal physiological environments, ROS are produced from three major sources: membrane-associated NADPH oxidases (NOXs), peroxisomes, and the mitochondria.25 NOXs were first recognized by Iyer et al in 1961 as a source of ROS generation in phagocytes.26 ROS are directly generated by enzymes in phagocytes to kill microorganisms and are essential for protecting cells and preventing inflammation. Conventionally, ROS-mediated oxidant signaling activated NOXs.27–29 However, in the last two decades, many researchers have found that mitochondrial ROS also plays a role in many cellular processes, such as proliferation,9 stem cell generation,30 cellular plasticity,31 and cell cycle control.32
Mitochondria produce ROS as byproducts of oxidative phosphorylation. As the final step of cellular respiration, the mitochondrial electron transport chain converts more than 90% of oxygen into water, which requires intricate interplay with the tricarboxylic acid (TCA) cycle.33,34 The TCA cycle generates a series of biomolecules including citric acid, succinyl-CoA succinate, fumarate, and oxaloacetate. It also produces building blocks that enter the ETC chain, such as nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD). NAD+ and FAD are reduced into NADH and FADH2 by “giving up” electrons from complex I to complex IV. However, approximately 1–2% of the electrons leak out in the ETC chain, which transfers electrons to oxygen, thus creating a highly reactive oxygen radical superoxide (12). At the ultrastructural level, complex I (ubiquinone oxidoreductase) and complex II (Succinate dehydrogenase) in the ETC produce O2·− in the mitochondrial matrix, whereas complex III (cytochrome c oxidoreductase) creates O2·− in both the mitochondrial matrix and the intermembrane space (Figure 1).35,36 The exact site of ROS generation has not yet been determined, but the employment of specific site inhibitors has elucidated site IQ, which contributes to most ROS generation in mitochondria.37,38
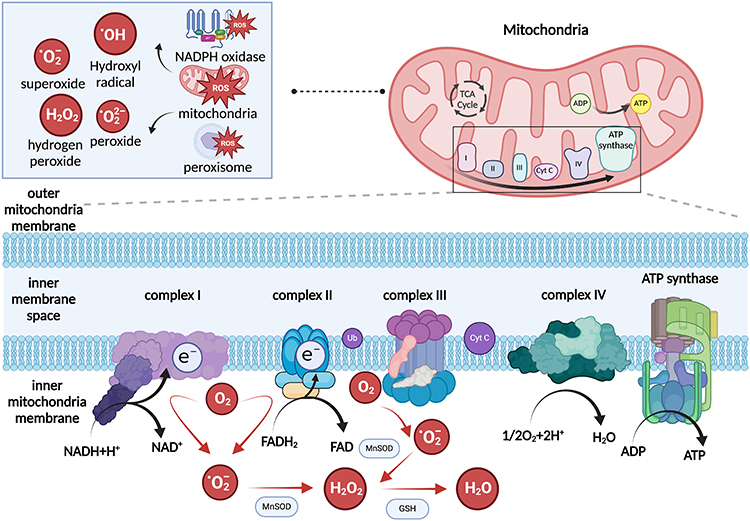 |
Figure 1 Schematic representation of ROS generation in mitochondria. ROS generation has three major sources: mitochondria, NADPH oxidase, and peroxisome. In mitochondria, mROS is generated as a byproduct of the electron transport chain. Complex I and III in the electron transport chain generate superoxide from oxygen. Manganese superoxide dismutase converts superoxide and hydrogen peroxide, and glutathione converts hydrogen peroxide to water. Created in BioRender. Wang, X. (2025) https://BioRender.com/t14f418. |
These highly reactive oxygen species include oxygen free radicals such as superoxide (O2·−) and hydroxyl (OH), and non-free radicals such as hydrogen peroxide (H2O2). The transport of one electron to O2 produces O2·−, which has a relatively weak oxidant property.39 Additional electron donation converts O2·− to H2O2 via MnSOD. H2O2 can also be converted to hydroxyl radicals with transition metals, such as Cu+ and Fe2+.


These free radicals play a central role in signaling cascades by acting as intermediates. The increase in mROS levels directly recapitulates the level of one cyclin-dependent kinase (CDK2), thereby promoting cell proliferation.25 mROS levels also correlate with cellular differentiation in hair follicle cells. Ablation of mitochondrial transcription factor A (TFAM) interrupts mROS generation, thus inhibiting downstream Notch/β-catenin–dependent transcription.40 ROS not only promote an analytical cycle of other biomolecules but also induce the ROS regenerative cycle in mitochondria, named ROS-Induced ROS Release (RIRR).41 Previously produced ROS triggered the induction of permeability transition pore (PTP) in mitochondria. Subsequently, PTP activation forms a positive feedback loop that results in an elevation in ROS levels in both the mitochondria and cytosol. In the latter condition, cytosolic ROS further activates ROS production in the neighboring mitochondria. This adaptive change is efficient in removing damaged cells and inducing apoptosis under pathological conditions.42 Apart from the functions discussed above, other roles of mROS in normal cellular environments have been summarized by Dunn et al.43
Production of mROS in Cancer Progression
The production of mROS is thought to play a pivotal role in cancer progression. Therefore, the molecular mechanisms underlying the appropriate control of mROS concentrations have been a focus of research. Increased mROS levels, accompanied by damaged antioxidant defense systems, enable tumors to acquire hallmark capabilities including genome instability, sustained cancer cell survival, metabolic reprogramming, and cancer metastasis (Figure 2). The following sections explain the relationship between mROS generation and these key regulatory processes.
 |
Figure 2 The role of mitochondrial ROS in cancer progression. From left to right: increased mROS generation could affect genome instability, maintain sustained cell survival, mediate metabolic reprogramming and keep tumor metastasis ability. Details are mentioned in each part. Created in BioRender. Wang, X. (2025) https://BioRender.com/1673kb1. |
mROS Generation and Genome Instability
Genome instability, including chromosomal instability (CIN), microsatellite instability (MSI), epigenetic-mediated instability, and instability of the DNA repair system, is recognized as one of the hallmarks of cancer in the next generation (Figure 2).44,45 With a series of gene mutations, tumorigenesis is a multistep progression that acquires successive ‘cancer driver mutations’ and finally develops into cancer cell lines.10
High levels of mROS production in cancer cells promote mutations in the nuclear DNA, amplifying the genomic instability that drives cancer progression. For example, the P53 gene is the most well-known tumor suppressor gene that maintains stability in the human genome. Mutations in the P53 gene and expression of tumor protein 53 (TP53) have been observed in more than 50% of cancer types.46,47 Whole-genome sequencing of 48 ovarian cancer (OVCA) revealed a novel p53 mutation, the TP53I3 p.S252X variant (rs145078765, MAF = 0.0016), which may decrease ROS production and dampen mitophagy in apoptosis, thus affecting the efficacy of several cytotoxic agents that target mROS production to induce apoptosis of tumor cells (33). In contrast, P53 gene also affects mROS generation, which contributes to cancer cell proliferation, migration, and differentiation.48 The wide-type P53 gene maintains genomic instability of mitochondrial DNA by supporting DNA repair functions and interacting with mitochondrial polymerase γ to reduce mitochondrial DNA mutations. In P53+/+ cells, knockout of P53 by 100 nM and 500 nM rotenone reagent increased O₂⁻ levels to 150% and 210% of control levels, respectively, suggesting loss of P53 gene leads to increased mROS production and loss of mitochondrial respiration.49
Apart from control mutant genotypes in cancer, mROS also affects CIN by disrupting micronuclei integrity.50 CIN is the most common type of genomic instability, including changes in the number or structure of chromosomes.10,51,52 Mis-segregation of nuclear chromosomes forms micronuclei, which is a feature of CIN.53 Micronuclei are precarious structures with fragile nuclear envelope. Rupture of micronuclei exposes chromatin and induces complex chromatin rearrangement, named chromothripsis.54 A pan-cancer genome-wide association study identified 2,658 tumors from 38 cancer types related to chromothripsis, with mutations such as oncogene activation, tumor suppressor science, and mismatch repair mutation.55 Recently, Martin et al discovered that mROS plays a crucial role in promoting micronuclei collapse, thus pointing to mitochondrial oxidative damage and genome instability (Figure 3).50
 |
Figure 3 Molecular mechanisms of mROS drive micronuclei and chromosome instability. (A) CIN refers to an increased rate of changes in chromosome structure or number within cancer cells. The formation and collapse of micronuclei is a highly conserved activity of CIN. (B) Normal micronuclear envelope integrity is maintained by coordination between ESCRT-III complex (endosomal sorting complex required for transport III), CHMP7 (ESCRT-II/ESCRT-III hybrid protein Cmp7p) and LEMD2 (LEM (Lap2-Emerin-Man1) Domain Nuclear Envelope Protein 2). Created in BioRender. Wang, X. (2025) https://BioRender.com/l1hwbvr. |
ESCRT-III plays a role in microautophagy, a process where lysosomes directly engulf parts of the cytosol without the need for an autophagosome, which are responsible for the delivery of damaged cellular material. Mitochondria ROS disrupt micronuclei integrity by inducing oligomerization of CHMP7, aberrant binding of CHMP7 to LEMD2, and autophagy-related protein P62 aggregation.
The presence of mitochondria and micronuclei showed significant colocalization. Subsequently, micronuclei close to the mitochondria had a higher rupture rate. It was found that mROS interferes with the regular activity of the ESCRT-III complex and the autophagy-related protein p62, which are essential for maintaining micronuclei integrity.56 Most tumor cells accumulate more mROS under hypoxic conditions. Therefore, this study also tested the link between hypoxia and micronuclei catastrophe. Over 10% enhanced micronuclei collapse was observed after HeLa cells were cultured in a hypoxic environment. Typically, the tumor core is the most hypoxic in the tumor microenvironment, with insufficient oxygen supply.57 This study tested micronuclei rupture in tumor cores in human high-grade serous ovarian cancer (HGSOC) and human papillomavirus-induced (HPV+) head and neck squamous cell carcinoma (HNSCC) and found higher micronuclei rupture, supporting this notion in many cancer types.
mROS and Sustained Cell Survival
Self-sufficiency of cell growth factors is a fundamental hallmark of cancer progression (Figure 2). Although mROS production is primarily recognized as detrimental to cell survival, elevation of ROS generation also has a mitogenic effect on cancer cells.58 Immortalized murine embryonic fibroblasts (MEFs) demonstrate a decreased cell proliferation rate after treatment with mitochondria-targeted antioxidants, suggesting the role of mROS in promoting cell growth. This effect is mediated by ERK-MAPK signaling, consistent with previous studies showing that the activation of ERK-MAPK promotes carcinoma development.59 Mitochondrial nitric oxide (MCP and MCTPO) can abolish mROS by acting as a superoxide dismutase mimetic and scavenger. Phosphorylation of ERK isoforms 1 and 2 was enhanced after treatment with mitochondria-targeted MCP and MCTPO.60 Therefore, mROS dampens ERK-MAPK phosphorylation and increases the mitogenic ability of tumor cells.
Similarly, mROS drives hepatocellular carcinoma (HCC) growth and metastasis. This promoting effect is mediated by the activation of transcription elongation factor of mitochondria (TEFM), a nucleus-encoded transcription factor for mitochondrial elongation.61 More than 83% of HCCs were observed with TEFM overexpression, leading to lower overall survival (OS) and recurrence-free survival (RFS) in patients with higher TEFM.62 Analysis of mROS levels in both TEFM-overexpressing and TEFM-knockdown HCC cell lines revealed that mROS levels were positively associated with tumor cell proliferation in HCC cell lines. Furthermore, treatment with ROS scavengers significantly reduced ERK signaling in HCC cell lines. Collectively, these results suggest that the mROS/ERK signaling pathway promotes HCC cell growth and metastasis.63
In addition to activating proliferation signaling cascades, mROS promote cancer cell survival via mitochondria transfer.9 Mitochondria transfer is a preventive strategy in cancer cells to receive “mitochondria donation” from the tumor microenvironment, allowing cancer cells to restore oxidative phosphorylation to generate ATP.64 For example, bone marrow stromal cells (BMSC) transport mitochondria to acute myeloid leukemia (AML) cells to improve AML cell survival.65 Several cellular structures are linked to the donor and recipient cells, such as extracellular vesicles,66 tunneling nanotubes (TNTs),67 and gap junctions.68
Intriguingly, some transferred mitochondria are dysfunctional with high levels of mROS production 9. These dysfunctional mitochondria accumulate high amounts of mROS and significantly increase the proliferation of tumor cells. Subsequent analysis revealed that mROS accumulation leads to downstream ERK signaling, suggesting activation of downstream proliferation signaling in an mROS-ERK-mediated manner.69 Cancer-associated fibroblasts (CAFs) in the tumor microenvironment are essential for cancer cell survival. In lung cancer, primary tumor cells transfer damaged mitochondria to normal fibroblasts (NFs) via extracellular vesicles. The presence of damaged mitochondria triggers the production of mROS in NFs, driving the transformation of NFs into CAFs.66 Other roles of mROS in transferred mitochondria are summarized in Table 1.
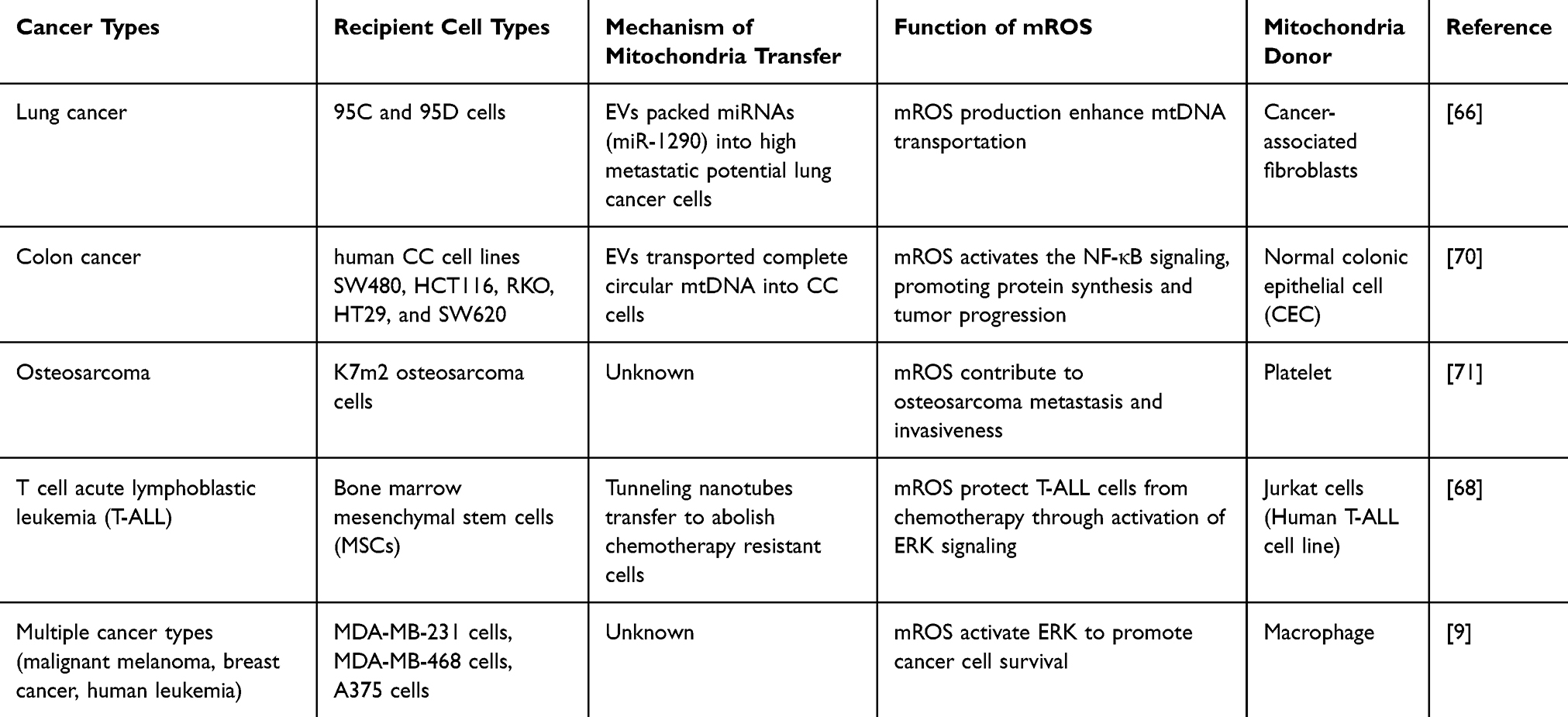 |
Table 1 Mitochondria Transfer in Tumorigenesis and the Roles of mROS in Mitochondria Transfer |
However, most studies of mROS signaling focus on its apoptotic effects in mediating cancer cell death, but there is a lack of studies to demonstrate the role of mROS in cancer cell survival.7,72–75 In contrast, the mitogenic effect mediated by mROS has been reported in normal cell proliferation, with its activity activating cyclin family members. For example, mROS prompts cardiomyocyte proliferation in mice by controlling the downstream phosphorylation of Akt, GSK-3β, β-catenin, and cyclin, thus activating cell proliferation signaling cascades.76 mROS can be recruited to neural precursor cells (NPCs) for lineage specification and activate downstream signaling for proliferation in cells with higher ROS levels, such as cyclin E1 (CCNE1), cyclin A2 (CCNA2), and CDK2. In particular, mROS targets cell cycle progression during proliferation and is coupled with CDK2.25,77 These findings collectively indicate that mROS may play a role in mediating ordinary cell multiplication through various signaling cascades. However, different tumor cells in distinct tumor types have dramatically different mitochondrial ROS levels, leading to diverse oxidative stress responses. For example, some cancer subpopulations exhibit proliferative ability with lower mitochondrial ROS levels. Compared to other tumorigenic progeny, maintaining low ROS levels is essential for self-renewal in cancer stem cells in cancer types such as glioblastoma and breast cancer cells.78,79 Therefore, whether mROS production promotes cancer cell division or how to determine the level of mROS that mediates cell death and cell survival is a question to be addressed in future studies.
mROS and Metabolic Reprogramming
Cancer cells are characterized by uncontrolled proliferation that requires large amounts of oxygen and nutrients to adapt to rapid cell division. Therefore, redox systems forficate homeostatic systems to counteract the metabolic effects of oncogene activation, tumor suppressor gene loss, and other stressors, known as metabolic reprogramming.10 The Warburg effect is a classic example of a reprogrammed metabolic pathway in cancer, in which cancer cells transform glucose to lactate regardless of oxygen availability.80 The consequent low oxygen supply, also known as hypoxia, is one of the hallmarks of solid tumors and tumor metastasis in the tumor microenvironment (Figure 2).81,82 The Warburg effect is often promoted under low-oxygen conditions (hypoxia) and regulated by factors such as hypoxia-inducible factors (HIFs).83
Chronic hypoxia has also been reported to increase mROS generation in the electron transport chain.84 Specifically, mROS stabilizes the transcriptional activity of HIF-1α. HIFs are formed by two subunits, oxygen-regulated HIF-1α and constitutively active HIF-β. The coordination of HIF-1α and HIF-β acts as a complex to control the transcription of many enzymes, including those involved in cell metabolism.85 Under normoxic conditions, HIF-1 levels are strictly controlled by the degradation of the HIF-1α subunit. Hydroxylation of proline 462 and proline 564 of HIF-1α by the enzyme prolyl hydroxylase (PHD) leads to the formation of a covalent bond between HIF-1α and von Hippel-Lindau tumor suppressor protein (pVHL), thus resulting in the proteolytic degradation of HIF-1α.86,87 As HIF-1α is constitutively degraded in the cytosol, HIF cannot translocate to the nucleus and induce a downstream transcriptional response. However, the accumulation of oncometabolites, including mROS, nitric oxide, and succinate, can inhibit the catalytic activity of PHD in hypoxic tumor microenvironments. Therefore, it enables accumulation of the HIF-1α subunit and HIF complex formation.88 The stabilization of HIF-1α and HIF-β isoforms has been reported in many cancer types, including gastric cancer,89 lung cancer90 and melanoma.91 Notably, the combination of HIF-1α and HIF-β regulates hypoxia at the transcriptional level by binding to the hypoxia response element (HRE) in the cancer cell nucleus and activating genes involved in glycolysis to support cancer cell survival in oxygen-poor environments92 (Figure 4).
 |
Figure 4 Cellular responses to oxygen levels, comparing conditions of normoxia (normal oxygen levels) and hypoxia (low oxygen levels). In normoxia, abundant oxygen allows the enzyme PHD to hydroxylate HIF-α. The hydroxylated HIF-α is recognized by pVHL, which binds to it and targets it for degradation. Without HIF-α, there is no binding to HIF-β and no activation of HRE, preventing the transcription of HIF-targeted genes. Hypoxia can lead to increased production of mROS. As a result, HIF-α is not marked for degradation. HIF-α accumulates and binds to HIF-β, forming a complex. This HIF-α/HIF-β complex binds to HRE on DNA, initiating the transcription of HIF-targeted genes. Created in BioRender. Wang, X. (2025) https://BioRender.com/8v6165z. |
Furthermore, many studies have identified multiple upstream signaling of mROS/ HIF-1α in different tumor microenvironments. In murine lung cancer carcinoma, downregulation of the tumor suppressor genes PDZ and LIM domain 2 (PDLIM2) constitutively activates succinate dehydrogenase (SDH) in the mitochondrial respiratory chain. This enables mitochondrial dysfunction, including accumulation of mROS and succinate. Additionally, mROS accumulation stabilizes HIF-1α, facilitating the translocation of HIF into the nucleus.93 In 5-fluorouracil (5-FU)-resistant colorectal cancer cells, damaged mitochondria induce metabolic reprogramming by shifting oxidative phosphorylation to alternative aerobic glycolysis, thereby increasing mROS generation and lactate metabolism. mROS further upregulates HIF-1α, driving further changes in glycolytic enzymes.94 OMA1 is an ATP‐independent zinc metalloprotease that cleaves ubiquinol-cytochrome c reductase complex assembly factor 3 (UQCC3) in the mitochondrial complex III. OMA1 is downregulated in hypoxic hepatocellular carcinoma, thereby increasing UQCC3 expression in cancer cells. In turn, UQCC3 forms a positive feedback loop with mROS to initiate hypoxic signaling with stabilization of HIF-1α and high glucose uptake in cancer cells.95
While interactions between HIF-1α and mROS help cancer progression, the overproduction of mROS leads to cell apoptosis. Under these conditions, HIF-1α acts as a negative feedback regulator of the mROS levels. This property has been exploited in antitumor therapies. Cardamonin is an antitumor drug used to treat Triple-negative breast cancer (TNBC). Cardamonin treatment reduced the production of HIF-1α, thus enhancing mROS production and inducing apoptosis in triple-negative breast cancer cell lines.96
mROS and Cancer Metastasis/Invasion
Metastasis is the seeding of tumor cells away from primary sites to other parts of the body, and is the primary cause of mortality in more than 90% of cancer patients (Figure 2).97 The interaction between cancer cells and mROS in the tumor microenvironment allows the cancer cells to precede the dissemination process. Epithelial-mesenchymal transition (EMT) is the initial stage of cancer metastasis, in which epithelial cells at the primary tumor site acquire motility and transform into a mesenchymal phenotype. Complex I in the respiratory chain is one of the main sites for mitochondrial ROS production. Complex I was downregulated in CMS4 human colorectal cell lines compared with that in CMS1 human colorectal cell lines. Clinically, CMS4 subtypes have poor prognosis and prominent EMT activation. However, the contribution of the signaling pathways between the two CMS subtypes remains unclear. It has been reported that CMS4 cell lines have significantly higher expression of ROS than CMS1 cell lines, while the expression of the mROS-scavenging enzyme SOD2 was much lower than that in CMS1 cells. We assessed the correlation between mROS expression and EMT. Surprisingly, both inhibition and activation of mROS production can inhibit CMS4 CRC cell migration, indicating that CMS4 cells finely orchestrate mROS levels to a favorable level that facilitates tumor migration.98
Mechanistically, EMT involves reconstruction of the cytoskeleton, and epithelial tissues contain basement membranes that communicate with the extracellular matrix (ECM).99 Cancer cells must break the link between the basement membrane and ECM to evade the primary site. Matrix metalloproteinases (MMPs) been implicated in tumor progression and metastasis. Breaking down ECM barriers enables cancer cells to invade nearby tissues and to spread to distant organs.100 A TGFβ-derived protein, HIC-5, has been implicated in EMT and invadopodia formation during breast cancer metastasis. When HIC-5 -/- MDA‐MB‐231 breast cancer cells were implanted into mice, HIC-silenced cells formed more lung metastasis nexus than the control groups. HIC-5 expression is sensitive to redox changes in mROS.100,101 When breast cancer cells were treated with mitochondria-targeted antioxidants to abolish mROS generation, the enhancement of MMP9 expression was diminished. Altogether, these results indicate that HIC-5 controls EMT through the mROS-mediated modulation of MMP9. Other MMPs also induce mesenchymal changes in breast cancer; for example, MMP-3 triggers EMT via the activation of the Rho GTPase family, Rac1b. In mouse mammary epithelial cells, either expression of Rac1b or treatment with excessive MMP-3 enhances ROS production in mitochondria.102
Depletion of mROS by Antioxidant Systems
Antioxidants are chemical compounds that protect cells from ROS-induced damage. In both normal and cancer cells, the mitochondria maintain a dynamic redox balance between ROS generation and ROS depletion by antioxidants. The antioxidative defense system for mROS involves both endogenous antioxidants, naturally synthesized in the body to protect mitochondrial function, and exogenous antioxidants, obtained through diet and supplements, to support mitochondrial antioxidant defenses.103,104
Endogenous Antioxidant
Manganese Superoxide Dismutase (SOD2)
As the most well-known ROS scavenger, Superoxide Dismutases exerts its effects by converting O2•− to H2O2 and O2. There are three types of SOD distributed in cells: SOD1/ Cu–Zn-SOD that localizes in the cytosol, SOD2/ Mn-SOD present specifically in the mitochondria, and SOD3/ EC-SOD as an extracellular enzyme.105 Evidence has suggested that increased oxidative stress due to lower SOD2 levels can trigger signaling pathways (such as NF-κB and HIF-1) that promote cancer cell growth; therefore, many cancers such as lung cancer, colorectal cancer, esophageal cancer, and leukemia cells have low or undetectable SOD2 levels.106
While low levels of SOD2 enhance mROS generation at a sublethal high level, high levels of SOD2 play a role in suppressing cancer progression.107 High CD44 expression is a marker of cancer stem cells (CSC) in head and oral cancers. The EMT process, which transforms the CD44L epithelial phenotype to the CD44H mesenchymal phenotype, is related to the upregulation of SOD2. Additionally, the knockdown of SOD2 altered the distribution of CD44H mesenchymal cells in the cell culture media of CD44H and CD44L cells. SOD2 knockdown also decreased EMT efficiency in CD44L cells compared to that in the control groups with regular SOD2 expression.
Further analysis of mROS expression in the whole CSC culture found that mROS reached the highest level in cell subpopulation with mediate CD44 expression between CD44L and CD44H cells, which was suspected as ‘transition cells’ in response to SOD2 changes.108 According on the transformation of cancer cells in response to SOD2 change, the role of SOD2 in cellular motility was further investigated. In ovarian cancer cells, tumor invasion is accompanied by spheroid formation with cytoskeletal anchorage. SOD-2 knockdown in ES-2 cells (ovarian cancer cell line) reduced spheroid body size. H2O2 levels increased by 50% in SOD2 knockdown groups compared to control groups, while cancer cell migration was significantly slowed, indicating the role of SOD2 in abrogating mitochondrial H2O2 to control tumor invasion.109 Reduced SOD2 levels are accompanied by low levels of anchorage-independent spheroid outgrowth in ovarian cancer cells, potentially by shifting steady-state H2O2 levels in the mitochondria.109 Another study further elucidated the mechanism between SOD2 increase and anchorage independence during metastasis: SOD2-dependent H2O2, production triggers the expression of MMP family members MMP2 and MMP9, and EMT-related proteins such as E-cadherin in lung adenocarcinoma, all of which are related to tumor progression and migration.110
Peroxiredoxin III (PRDX III)
Peroxiredoxins (PRDXs) are another kind of antioxidants that facilitate the conversion of H2O2 to H2O, and hydroperoxides to alcohols. There are six isoforms of PRDXs characterized by their distribution in subcellular structures: PRDX V distributed throughout the cell; PRDX I, II, and VI in the cytoplasm; PRDX IV in the ER; and PRDX functions explicitly in mitochondria. PRDX and its mitochondrial electron donors, Trx and Trx reductase (TrxR), could serve as critical defense mechanisms against H2O2 generated by the mitochondrial respiratory chain. In the presence of H2O2, PRDX catalyzes the conversion of a peroxidative cysteine to a sulfenic acid (–SOH) intermediate. Another cysteine residue in the adjacent site forms a disulfide bond with oxidized cysteine. In a typical physiological environment, PRDX is reactivated by Thioredoxin 2 (TRX2) to exert its antioxidant function.111 Overexpression of PRDX III in the cytosol has been observed in many carcinomas, including breast,112 gastric,113 and lung cancer.111 In contrast, the production of mitochondrial oxidants (90% H2O2) was significantly reduced in human malignant mesothelioma cells (HM) after PRDX III knockout, indicating a therapeutic option for inhibiting PRDX III function to induce cancer cell death.
Glutathione Peroxidase 1 (GPX1)
Glutathione peroxidase 1 (GPx1) are a selenoprotein ubiquitously expressed in the cytoplasm and mitochondria, converting H2O2 and other lipid hydroperoxides into nontoxic products.114 GPx1 plays a complex role in cancer because of its dual effects on oxidative stress and cell survival. Elevated GPx1 levels have been observed in certain cancers, where it enables cells to avoid apoptosis (programmed cell death) by reducing ROS, allowing cancer cells to proliferate even under stressful conditions (see115 for review). Mechanistically, Gpx1 interacts with tumor necrosis factor receptor-associated factors (TRAF2 and TRAF6) to interfere with their binding to apoptosis signal-regulating kinase 1 (ASK1).116 Under normal conditions, ASK1 remains inactive and binds to redox-active thioredoxin 1. However, ROS generation can dissociate ASK1 and induce subsequent apoptosis.117 GPx1 maintains cellular redox homeostasis by controlling ROS, which suppresses the activation of ROS-mediated apoptotic pathways.
Exogenous Antioxidant
Exogenous antioxidants are compounds obtained from external sources, such as diet or supplements, which help neutralize free radicals and reduce oxidative stress in the body. Unlike endogenous antioxidants, which are naturally produced within cells, exogenous antioxidants are ingested and distributed to various tissues to maintain cellular health and protect against damage from mROS. Vitamin C (Ascorbic Acid) is a water-soluble antioxidant that neutralizes free radicals in the aqueous environment of cells and the blood. Vitamin C functions as an antioxidant at low levels but becomes a pro-oxidant at higher concentrations, which increases the complexity of its use to inhibit tumor growth.118 One study tested the effect of antioxidant supplementation (β-carotene, vitamin C, and vitamin E) on skin cancer risk in a large cohort of French adults and found that women in the antioxidant group had a higher incidence of skin cancer, with a hazard ratio of 1.68 (95% confidence interval: 1.02–2.77), indicating a 68% increased risk of developing skin cancer among women taking antioxidants.119 Another type of antioxidant is mineral antioxidant, which contributes to an exogenous antioxidant defense system by acting as a cofactor for antioxidant enzymes or stabilizing cell structures to prevent oxidative damage. Some minerals, such as selenium, copper, and manganese, are cofactors for endogenous antioxidants that reduce hydrogen peroxide and organic hydroperoxides, thus protecting cells from oxidative stress.120–122 Cerium, particularly in the form of cerium oxide (CeO2), is known for its antioxidant properties. Cerium ions (Ce3+ and Ce4+) can alternate between oxidation states, continuously neutralize ROS, and regenerate active sites, thereby making them a potential application in treating diseases with higher than normal ROS levels, such as cancer, ischemia-reperfusion injury (IRI), and neurodegenerative diseases.123–125
The Design of Nanoparticle Carriers to Target ROS in Mitochondria
Selectively targeting mitochondria and altering their redox signaling during the generation of mROS is one of the most effective pathways for delivering drugs to mitochondria. Due to the negative potential in the inner membrane of mitochondria, the mitochondrial uptake of drugs covalently linked to positive anions is high. Three major types of biomolecules have been used to assemble encapsulated nanoparticles targeting mROS (Figure 5).
 |
Figure 5 Nanoparticles can be classified into three main types: artificial, biomimetic, and hybrid mito-targeted nanoparticles. Artificial nanoparticles are synthetically engineered particles, designed for specific functions, often using materials like metals, polymers, or silica. Biomimetic nanoparticles are inspired by biological structures and aim to mimic natural properties, such as cell membranes, to improve biocompatibility and targeting. Hybrid nanoparticles combine elements of both artificial and biomimetic particles, integrating synthetic materials with biological components, to enhance functionality and versatility in applications like drug delivery, diagnostics, and imaging. Created in BioRender. Wang, X. (2025) https://BioRender.com/f1f98qo. |
First, artificial mito-targeted nanoparticles utilize synthetic molecules to precisely manipulate drug design. However, these drugs are at risk of immune clearance and have the lowest biocompatibility. Secondly, biological mito-targeted nanoparticles directly use cell membranes derived from red blood cells, platelets, or cancer cells to evade immune recognition. These drugs possess the highest biocompatibility but lack the ability to target mitochondria precisely. Third, hybrid metal-organic nanoparticles that contain metal ions and multiple organic linkers, for example, a series of developments in Metal-organic frameworks, provide a novel strategy for targeting mROS for antitumor therapy.
Artificial Nanoparticle
Mitochondria-targeted Triphenylphosphonium (TPP) is the most commonly used artificial nanoparticle carrier. The accumulation of drugs depends on the structure of the mitochondria, which are the only animal organelles that contain both inner and outer membranes. In oxidative phosphorylation reactions, the transportation of electrons drives the accumulation of protons in the mitochondrial matrix, thus producing a positively charged matrix and negatively charged inner membrane.126 As a lipophilic cation, TPP can localize to the mitochondria in two stages. First, TPP passes through a negatively charged (30mv-50mv) inner cell membrane to enter the cytoplasm; second, a stronger mitochondrial membrane potential of 150–180 mV (negatively charged inner membrane) pulls them into the mitochondria, completing the second enrichment phase. TPP uptake concentration depends on the Nernst equation:

At standard temperatures (298 K or 25°C), the concentrations of TPP inside and outside the membrane were determined by the potential on the membrane. According to the inner cell and inner mitochondrial membranes, TPP concentration increases by 3~5 folds in the cytoplasm and 100~1000 in the mitochondria, making TPP an ideal compound to target mitochondria.127
Biological Nanoparticle
Nanoparticles must camouflage themselves to prevent phagocytosis by the immune system. Recent studies have utilized biomimetic membranes, including cancer cells, red blood cells, and platelet membranes, for immune evasion.128,129
As the most abundant cell type in circulation, erythrocytes have become ideal drug carriers. Moreover, membrane proteins on red blood cells (RBCs) such as CD47, membrane cofactor protein, and C8 binding protein (C8bp) release “don’t eat me” signals to phagocytic cells, thus preventing the decomposition of drugs.130 Huang et al designed RBC-coated nanoparticle with the chemotherapeutic agent doxorubicin (Dox), which was linked to the RBC membrane through glutaraldehyde (Glu), to treat uterine sarcoma, named Dox-gluRDV. Because the RBC-encapsulated membrane does not have specificity, this study used an indirect pathway to induce mROS release through the lysosomal-mitochondrial axis.131 In multidrug-resistant human uterine sarcoma cells, Dox-gluRDVs exhibited a half-maximal inhibitory concentration (ICso) of approximately 0.5 µM. In contrast, free Dox had an ICso exceeding 10 µM, indicating a significantly enhanced cytotoxicity effect of targeted therapy. While previous studies have found that calcium release from lysosomes can stimulate mROS production, the treatment also increased the intracellular calcium concentration released from lysosomes to mitochondria.
Moreover, Inhibition of key enzymes in the mitochondrial ETC chain, such as 2-oxoglutarate dehydrogenase (OGDH), significantly decreased the cytotoxic effect of Dox, indicating that Dox induces cytotoxicity via mROS production.131 The same strategy used in another study achieved a similar apoptotic effect of Dox-gluRDVin in three other cancer cell lines via mROS production. Critical regulators of cellular apoptotic processes, such as ERK 1/2 and caspase 3, are activated, whereas in free Dox treatment, activation of these molecules is weaker.132 The lipophilic property of RBC membranes of another nanoparticle, Cy5-labeled Ang-MNPs@(Reg/Cy5), also facilitates drug delivery across the blood-brain barrier (BBB) in some brain tumors, such as glioblastoma. After 24 hours of Cy5-labeled Ang-MNPs@(Reg/Cy5), these targeted nanoparticles achieved a cumulative transport ratio of 20.4%, which was 1.6x higher than that of non-targeted drugs.133
Platelets are another important component of the circulation that can be used to design mROS-targeted nanoparticles. Mai et al developed platelet membrane-packed multifunctional nanoparticle for breast cancer treatment. The study effectively killed tumor cells using two methods: IR780, as a photodynamic agent, produces a toxic level of ROS to kill the cells. Meanwhile, the introduction of metformin (Met) reduces the function of the mitochondrial respiration chain, subsequently leading to a hypoxic TME and mROS generation.134
The cancer cell membrane is another source of nanoparticle that ensures efficient endocytosis and ROS accumulation. B16F10@CaCO3–CU@MnO2 was formed by dual ions of Mn2+ and Ca2+ bound to the B16F10 cancer cell membrane. As mentioned above, an intracellular increase in Ca2+ stimulates the mitochondria to generate more ROS and execute immunogenic cell death, which results in about 60% cancer cell death at 50 μg/mL CM NP concentration. Similarly, Mn2+ promotes hypoxia in the TME, further inducing mROS generation and cancer cell death.135
Apart from the use of natural membranes derived from cells, recent studies have combined cellular membranes with subcellular organelle membranes to achieve higher accuracy in targeting specific organelles such as the ER and mitochondria. Gboxin is an oxidative phosphorylation inhibitor that disrupts mitochondrial function by compromising oxygen consumption and inhibiting ATP synthase activity, leading to reduced energy production in GBM cells. Moreover, the mechanism of Gboxin involves targeting of the mitochondrial ETC, which is linked to the production of ROS as a byproduct of ATP production. Mitochondrial PTP can mitigate mROS, suggesting that the effects of Gboxin may also influence mROS levels in GBM cells. Gboxin increases mROS production by disrupting mitochondrial function, contributing to its cytotoxic effects on cancer cells.136 However, it has a half-life of less than 5 min and poor BBB penetration. Recently, a study adopted a ‘Trojan horse strategy’ to pack Gboxin in nanoparticle camouflaged by both cancer cells and mitochondrial membranes.22 By fusing cancer cells and mitochondrial membranes, Gboxin (HM-NPs@G) can target both cancer cells and mitochondria in cancer cells. Moreover, the drug core included one ROS-responsive drug, PEG-PHB, which releases Gboxin from nanoparticles in the presence of high levels of ROS (86.6% at 1 mM H2O2, 50.2% at 0.1 mM H2O2 after 24h incubation), leading to the collapse of ETC and cell apoptosis.
The drug complex Ca@GOx is another nanoparticle that integrates cancer cells and mitochondrial membranes to deliver drugs that are composed of calcium phosphate and glucose oxidase. The target of mROS is achieved by incorporating agents that induce calcium overload in the mitochondria, which increases mROS through the fine regulation of ETC enzymes.137 Upregulation of Ca2+ in the mitochondria can be achieved in nanoparticles with other chemotherapeutic agents to exert a synergistic effect in generating mROS and mitochondrial dysfunction. The nanoplatform generates ROS through a Fenton reaction by tannic acid (TA) in the acidic environment of the tumor, which disrupts mitochondrial calcium buffering capacity. Subsequently, near-infrared (NIR) irradiation facilitates the formation of the IP3R-Grp75-VDAC1 channel to transport Ca2+ from the ER directly into the mitochondria. This synergistic effect further enhances mitochondrial dysfunction and mROS release during cell death.138
Hybrid Nanoparticle
Biological ions such as Mn2+, Ca2+, Cu+, and Fe2+ play key roles in normal physiological environments.139 Ion overloading may be an effective treatment option for inducing cell death in the tumor environment. Metal-organic frameworks (MOFs) are designed with metal ions or clusters connected by organic ligands, which can lead to mitochondrial damage, mROS production, and cell death. FMUP nanoagents comprise a core MOF shell and upconversion nanoparticles (UCNPs).140 The shell is created using a metal ion called Fe3+ (iron), which connects with other molecules called carboxyl groups from substances like BTC (1,3,5-benzenetricarboxylic acid) and folic acid. Folic acid helps nanoagents target cancer cells more effectively. When the FMUP nanoagents are exposed to NIR light of 808nm, they release H+, which makes the TME more acidic in releasing excess Ca2+ in the mitochondria, and Fe3+ helps generate large amounts of ROS in the proximity of the mitochondria. This dual effect of Fe3+ and Ca2+ reinforces mROS production and cell death in HeLa cells. Under 808 nm laser irradiation, UCMT-treated cells exhibited a marked reduction in viability, dropping to 27.0%, 1.9 times lower than the viability observed in UCM-treated cells. Another MOF compound, UCMTs, is a stepwise nanoplatform that triggers the production of mROS. Nanoparticle combine with TPP, which is used to target mitochondria, and lanthanide-doped upconversion nanoparticles (UCNPs), which can activate ROS generation in mitochondria with NIR light. In mouse breast cancer cells, UCMTs demonstrated significant reactive oxygen species (ROS) in the mitochondria upon 808 nm NIR laser irradiation.20 However, most of these nanoparticles are still in progress based on preclinical trials, and some have shown conflicting results in animal models, warranting further investigation of their efficacy in tumors.
mROS-Targeted Therapy with Nanoparticles in Cancer Therapy
Previous studies have summarized mROS as a ‘double-edged sword’ for cancer cell survival and apoptosis. While we have summarized above that a moderate increase in mROS is beneficial to tumor progression, the artificial elevation of mROS levels to kill cancer cells or inhibition of mROS generation to inhibit tumor growth are both current therapeutic approaches in the targeted therapy of mROS. The advent of nanoparticles has enabled selective production of ROS in mitochondria, contributing to precision-targeted therapy in cancer treatment (Figure 6).
 |
Figure 6 Schematic representation of mROS-targeted nanoparticles as therapeutic strategies in cancer. The figure is divided into two panels: mROS enhancing nanoparticles (left) and mROS scavenging nanoparticles (right). mROS-enhancing nanoparticles induce oxidative stress, leading to various regulated cell death pathways including apoptosis, necroptosis, ferroptosis, pyroptosis, and cuproptosis. mROS-scavenging nanoparticles, such as triphenylphosphonium-based core-shell nanoparticles loaded with agents like Mito-E, Mito-Q, and cerium oxide, scavenge excess mROS to inhibit tumor growth via anti-angiogenesis, anti-proliferation, and anti-hypoxia effects. Created in BioRender. Wang, X. (2025) https://BioRender.com/14ki86j. |
Nanoparticles to Increase ROS Production in Mitochondria
Many nanoparticles are designed to increase mROS levels inside cancer cells, which can lead to oxidative stress, DNA damage, and ultimately, cell death (Figure 7).
Figure 7 Continued. Figure 7 Role of mROS-targeted nanoparticle in inducing cell death (A–E): mROS-targeted nanoparticle that induces apoptosis, necroptosis, ferroptosis, pyroptosi,s and cuproptosis. (F) Schematic representation of nanoparticles used to increase mROS and cell death. Created in BioRender. Wang, X. (2025) https://BioRender.com/d34lt7f.

Increased mROS to Enhance Apoptosis
Apoptosis is a type of programmed cell death that occurs in both normal and cancerous cells (Figure 7A). Apoptosis is initiated by two main pathways: (1) the extrinsic pathway (also known as the death receptor pathway), which relies on the interaction between proapoptotic death receptors in the cell membrane, such as Fas, TNFR1, TNFR2, and TRAIL receptors, and their ligands FasL, TNF, and TRAIL, to form a death-inducing signaling complex (DISC).141 (2) the intrinsic pathway (also known as the mitochondrial pathway), which includes mitochondrial participation, and Proapoptotic Bcl-2 proteins (Bax and Bak) are central regulators of intrinsic apoptosis and act as anti-apoptotic protein, helping cells to avoid undergoing apoptosis in response to various stress signals. Upon cellular stress stimulation, a process controlled by the Bax and Bak family proteins generates mitochondria outer membrane permeabilization (MOMP). This conformational change in the mitochondria allows the release of proapoptotic factors (cytochrome c) from the inner membrane of the mitochondria into the cytosol. Subsequently, cytochrome c forms a complex called the apoptosome with apoptotic peptidase activator 1 (APAF1), recruiting caspase 9 as an initiator for mitochondria-directed apoptosis.142,143 Caspases are initially generated as inactive enzyme precursors, known as procaspases, which require dimerization or oligomerization to become active. During activation, the protease effector domains of these procaspases are split into large and small subunits, which combine to form complexes that perform enzymatic functions.144,145
Chemotherapeutic agents are frequently used in cancer therapy to induce apoptosis by directly damaging the DNA or inhibiting cell division. Agents, such as alkylating agents, topoisomerase inhibitors, and antimetabolites, cause DNA damage and activate intrinsic apoptotic pathways. Drugs such as paclitaxel and vincristine inhibit mitosis to apoptosis via mitotic checkpoint failure.146–148 However, apoptosis induced by chemotherapy is often a byproduct of stress or damage to cancer cells rather than the precise modulation of apoptotic pathways, which makes them lack the specificity to target a single apoptosis pathway. The application of mitochondria-targeted chemotherapeutic nanoparticles facilitated mROS production and disrupted the intrinsic cell apoptosis pathway, with high biocompatibility (Table 2).
 |
Table 2 Chemotherapy-Based Nanoparticles to Elevate mROS Production in Apoptosis |
Different from chemotherapeutic nanoenzymes which are primarily focused on improving the delivery and efficacy of existing cancer drugs, radiotherapy nanosensitizers are also inducers for mROS generation in cancer cell apoptosis, thereby achieving a. Studies have shown that AuNPs sensitize cancer cells to radiation via strong attenuation of photons by high-Z atoms.158 Additionally, AuNPs exhibit effects similar to glucose oxidase (GOx), which oxidizes glucose into H2O2 and gluconic acid.159,160 One study constructs a novel TPP-conjugated AuNPs to enhance both X-ray radiation and photodynamic therapy: 4 Gy radiation treated human colon adenocarcinoma HCT 116 cells have equivalent controlled growth compared to 12 Gy radiation without AuNPs, which is caused by enhanced production of mROS and disrupting MPP. Treatment of TPP-conjugated AuNPs results in a substantial increase in the apoptotic cell population (from 5.3 ± 1.8% to 37.4 ± 4.7%), whereas nanoparticles alone or X-rays alone induce a much lower apoptosis rate of 8.9 ± 2.9% and 7.8 ± 3.5%, respectively. Moreover, mice treated with AuNPs experienced less weight loss after irradiation. Together, these results indicate the synergistic effects of combining AuNPs with.161 dAuNP-TPP is another mitochondria-targeted AuNP with 5.22-fold higher mitochondrial accumulation than free AuNP-TPP. After treating dAuNP-TPP for 24h in 4T1 cells, morphological analysis demonstrated that dAuNP-TPP-treated cells had dysfunctional mitochondria with membrane fragmentation, and apoptosis-regulated factors, Bax and Bcl-2, were also expressed at higher levels than those in control cells. Finally, in vivo, photoacoustic (PA) imaging was used to assess radiotherapeutic enhancement by dAuNP-TPP in 4T1 tumor-bearing mice, and dAuNP-TPP treated mice demonstrated the strongest PA signals compared to other untreated groups.162
Increased mROS to Enhance Necroptosis
Necroptosis is a caspase-independent type of cell death that triggers multiple organelle dysfunction, cell swelling, and membrane rupture, ultimately inducing cell death (Figure 7B). It acts as a backup for cells that lack apoptosis-related factors: the production of TNFα can initiate the activation of various cellular receptors, including death receptors (Fas) and toll-like receptors (TLRs).163 The binding of TNFα to its receptor TNFR1 in cell membrane promotes the formation of TNFR1 signaling complex–complex I, which is composed of multiple adapter proteins such as FAS-associated death domain protein (FADD), TNFR1-associated death domain (TRADD) and receptor-interacting protein [RIP] kinase 1 (RIPK1), named as complex I. At this point, the central regulator of cell death is RIPK1, which is deubiquitinated by other proteins to limit the cell survival and NF-κB pathways. Complex II (necrosome) is formed by RIPK1, RIPK3, and caspase-8, and recruits MLKL to induce cell rupture.164,165 RIPK3 activation during necroptosis is related to the production of mROS via activation of pyruvate dehydrogenase (PDH), thereby enhancing aerobic production and mROS generation.166 In contrast, mROS production could also induce the RIPK1/RIPK3/MLKL signaling pathway to promote necroptosis, providing a potential for nanotechnology to induce cell death.167 CPT/MT-NG is a redox-sensitive nanoparticle that resulted in the elevation of mitochondria oxidative stress to enhance synergistic cancer cell death. Encapsulating NG with mitochondria-targeting monomer (MT monomer) can lead to the accumulation and release of the anticancer drug camptothecin (CPT) to induce mitochondrial ROS generation. In MDA-MB-231 human breast cancer cells, CPT/MT-NGs reduced cell viability to approximately 34%, compared to 58% observed with mono-therapeutic CPT-NGs, highlighting a synergistic enhancement in anticancer activity. Notably, treatment with either an apoptosis inhibitor or necroptosis inhibitor (Nectastatin-1) could significantly reduce the population of dead cells, confirming that CPT/MT-NG efficiently activates both cell death pathways and enhances antitumor therapy.168
Overproduction of ROS in the mitochondria also causes oxidative DNA damage in cancer cells, subsequently activating the DNA damage response (DDR) via ataxia-telangiectasia mutated/checkpoint kinase 2 (ATM/Chk2), which leads to programmed cell death.169 One study developed a small nanosphere TPA-N-n with superior antiproliferative performance, while another developed a small nanosphere TPA-N-n with superior antiproliferative performance: First, according to Pearson’s correlation coefficient (PCC) values with MitoTracker Green (0.818 and 0.805), two kinds of TPA-N-n nanoparticles (TPA-N-4, TPA-N-8) exhibit strong mitochondrial targeting ability. Then Aggregation Emission (AIE) molecules localize in mitochondria and begin to promote mROS generation. In addition, Chk2 phosphorylation was upregulated after treatment, with an elevation in RIP3 expression, suggesting the presence of mROS-mediated necroptosis.170
Increased mROS to Enhance Ferroptosis
Ferroptosis is a type of iron-dependent cell death characterized by dysfunctional mitochondria that are smaller than the average size, increased mitochondrial membrane density, accumulation of mitochondrial ROS, and lipid peroxidation in the cytoplasm (Figure 7C).171 In tumor cells, accelerated metabolic rates enhance the production of mROS, making them intrinsically susceptible to ferroptosis.172 Therefore, the development of nanoparticles that enhance mROS production is a potential therapeutic option for inducing tumor ferroptosis. A multifunctional mitochondrion-targeted liposomal nanoparticle, Mito@Lip/R162/IR780 (MLipRIR NPs), is able to disrupt glutaminolysis pathway in mitochondria to induce redox imbalance and ferroptosis in 4T1 breast cancer cell lines: after TPP-based nanoparticle accumulate in mitochondria, first a glutamate dehydrogenase 1 (GDH1) inhibitor, R162, are released into cytoplasm to inhibit endogenous antioxidant enzyme (GPx) activity to trigger ferroptosis via increase in lipid peroxidation.173 Furthermore, IR780-mediated ultrasound (US) exposure for 1 minute leads to ROS enrichment in mitochondria, which in turn depletes glutathione and aggravates ferroptosis.174 Following IR irradiation, cell viability significantly decreased to 16.4% in MLipRIR nanoparticle-treated groups. By contrast, tumor cell eradication by MLipRIR without US only causes moderate cytotoxicity with more than 75% cell viability, indicating synergistic effects of loaded R162 and IR780. As mentioned above, mROS accumulation is related to the hypoxic tumor microenvironment. Previous studies have reported that HIF-1α in a hypoxic environment aggravates ferroptotic cell death by upregulating transferrin receptor 1 in non-small cell lung cancer and glioblastoma cell lines, providing a novel method to manipulate hypoxia and induce ferroptosis in tumor.175–177 Based on this principle, Tian H et al reported mitochondria-targeted HL/MOS@M780 and LOD nanoparticles to activate ferroptosis in 4T1 tumor-xenografted mice; once the nanoparticles reached the tumor area, mROS generated by mitochondria-targeted IR780 (M780) and lactate depletion by lactate oxidase (LOD) played dual roles in depleting oxygen and further upregulating transferrin to activate ferroptosis. Altogether, the overexpression of mROS heralds a new path for designing nanoparticles to activate ferroptosis in tumor cell death.178
Increase mROS and Pyroptosis
Increased ROS production may trigger pyroptosis, an inflammatory type of programmed cell death (Figure 7D). Pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), together with nucleotide-binding oligomerization domain-like receptors (NLRs) assemble inflammasomes to activate downstream caspase 1. Caspase 1 further cleaves the pivotal gasdermin D (GSDMD) protein into the N and C domains, resulting in cell pore formation and swelling of the cell, which eventually leads to cell rupture and the release of cellular contents.16,179 Yang et al constructed a tumor-targeting nanoparticle (CS-HAP@ATO NPs) to target mouse colorectal cancer cell lines via the mitochondria-mediated pyroptosis pathway; once the nanoparticle was internalized into tumor cells, atorvastatin (ATO) was released to hamper mitochondrial membrane potential and ATP production, while the other two components in the nanoparticle, chondroitin sulfate (CS) and hydroxyapatite (HAP), caused calcium overload in the mitochondria. These synergistic effects trigger mROS production and fragmentation of mitochondrial DNA, promoting aggregation of the NLRP3 inflammasome and recruitment of caspase-1, eventually leading to pyroptosis. In the following tumor volume analysis, CS-HAP@ATO nanoparticles administration showed a 93% inhibition rate, and tumor weight measurements on day 15 indicated an 85% reduction in the nanoparticle group compared to the control.180 Similar results were also observed in NIR-II Z1 nanoparticles and DPITQ nanoparticles, combining photodynamic therapy to induce mROS generation and further activate caspase-1 to induce both apoptosis and pyroptosis to kill tumors effectively.16,179 In the analysis of immunogenic cell death (ICD) in response to mROS generation triggered by Photocatalytic CDs, the study also found immune preventive effects caused by nanoparticle-induced pyroptotic cancer cells (PCIPs). After injecting PCIPs into mice with breast cancer, the number of T helper cells (memory T cells) was significantly increased in the lymph nodes and spleen, suggesting the potential to develop photocatalytic CDs for cancer vaccine.181 Recently, a mitochondria-dependent non-canonical pathway was found to be associated with mitochondrial dysfunction. Mitochondrial abnormalities further induce MOMP and the overproduction of mROS, leading to the activation of caspase 3 and GSDMD cleavage. Peng et al developed a mitochondria-targeted nanoparticle (Th-M) that induces pyroptosis in tongue squamous cell carcinoma. Fluorescence intensity measurements demonstrated that cells treated with Th-M for 5 minutes have markedly increase in fluorescence, with cells in the control group only showed minimal fluorescence, suggesting Th-M specifically induced robust mROS generation. Active caspase 3 and protein levels of GSDME-N were increased in tongue squamous cell carcinoma, whereas full-length GSDMD was reduced under light irradiation, demonstrating the potential of Th-M to initiate pyroptosis.182
Increased mROS and Cuproptosis
In 2022, Tsvetkov et al identified copper-dependent cell death and apoptosis as new forms of cell death that are highly related to the mitochondrial respiratory pathway (Figure 7E).183 Unlike ferroptosis, which induces cell death by increasing lipid peroxidation, cuproptosis is triggered by direct accumulation of excessive copper or copper ionophores.184 Overloading Cu subsequently alters a series of enzymes in the TCA cycle through lipoylation (a post-transcriptional process), such as dihydrolipoamide S-acetyltransferase (DLAT) and iron-sulfur (Fe–S) cluster proteins, resulting in mitochondrial dysfunction and cell death.185 Therefore, the development of novel nanoparticles that induce both mROS generation and Cu overload in tumor cells could be beneficial for inducing cancer cell death. D@HCC-CuT is a Ca/Cu bimetallic nanoplatform that combines calcium and copper overload that leads to dual effects in two cancer cell lines (4T1 and MCF-7): First, calcium carbonate (CaCO3) triggers endogenous stress and induces Ca2+ outflow from the ER to mitochondria, leading to the production and release of apoptosis-related factors. Meanwhile, the copper coordination polymer (CuT) shell releases Cu2+ provokes DLAT aggregation and loss of the Fe-S cluster in the mitochondria. Furthermore, Cu2+ significantly exacerbates mROS production via Fenton-like reactions that convert H2O2 to •OH. D@HCC-CuT caused a tremendous decrease in ATP levels, reducing it to 21.4% of the control level, which is related to a significant impairment of mitochondrial energy metabolism (Figure 8).186 In tumor cells, cuproptosis is often restricted because of insufficient endogenous copper levels and overexpression of the antioxidant glutathione (GSH).187 To address the gap in Cu levels, another study designed an Fe/Cu bimetallic nanoplatform (DSF3@HMCIS2-PEG-FA) to achieve ferroptosis-cuproptosis for potent antitumor therapy. Robust ferroptosis not only produces large amounts of H2O2 in the mitochondria but also disrupts cellular antioxidant GPX production, promoting copper overload and cuproptosis.188
 |
Figure 8 mROS-targeted nanoparticle (D@HCC-CuT) to induce cuproptosis in two cancer cell lines, (A) schematic representation of D@HCC-CuT construction and delivery. Calcium carbonate (CaCO3) induces cellular stress by causing Ca²⁺ release from the ER to mitochondria, triggering apoptosis. Simultaneously, the CuT shell releases Cu2+, which promotes DLAT aggregation, disrupts mitochondrial Fe-S clusters, and enhances mROS production. (B) Ca2+ overload and ROS production in mitochondria after D@HCC-CuT administration. (C) D@HCC-CuT injection leads to mitochondrial dysfunction in cancer cells. (D) anti-tumor efficacy of D@HCC-CuT demonstrated as changes in tumor volume. Reprinted (adapted) with permission from Xu, Weijun et al. Hollow Calcium/Copper Bimetallic Amplifier for Cuproptosis/Paraptosis/Apoptosis Cancer Therapy via Cascade Reinforcement of Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. ACS nano. vol. 18,43 (2024): 30,053–30068. based on cc By License, copyright 2024 American Chemical Society.186 |
Nanoparticles to Depletes ROS Production in Mitochondria
Increased mROS levels have been implicated in many tumor types. Therefore, it is reasonable to postulate that mROS reduction inhibits cancer cell growth. Many preclinical and clinical studies have suggested that reducing mROS production may have clinical benefits in preventing cancer incidence (Table 3). Despite growing evidence supporting the anticancer activity of reducing mROS, the controversy surrounding its role in cancer may arise from its redox properties and the dynamic interconversion of these drugs to inhibit cancer cell proliferation and promote tumor progression. For example, several randomized clinical trials have revealed that treatment with NAC promotes tumorigenesis in various types of cancer, including lung cancer,189,190 melanoma,191 colorectal cancer192 and hepatocellular carcinoma.193 One possible reason for the lack of efficacy is that these general antioxidants suffer from poor localization in the mitochondria, which limits their ability to effectively combat oxidative stress. Conventional antioxidants, such as green tea and vitamins C and E, are widely dispersed throughout the body, and only a small percentage of drugs reach the mitochondria to function.194 Therefore, mitochondria-targeted antioxidants can overcome this limitation by specifically accumulating in the mitochondria, providing more potent protection against oxidative damage.
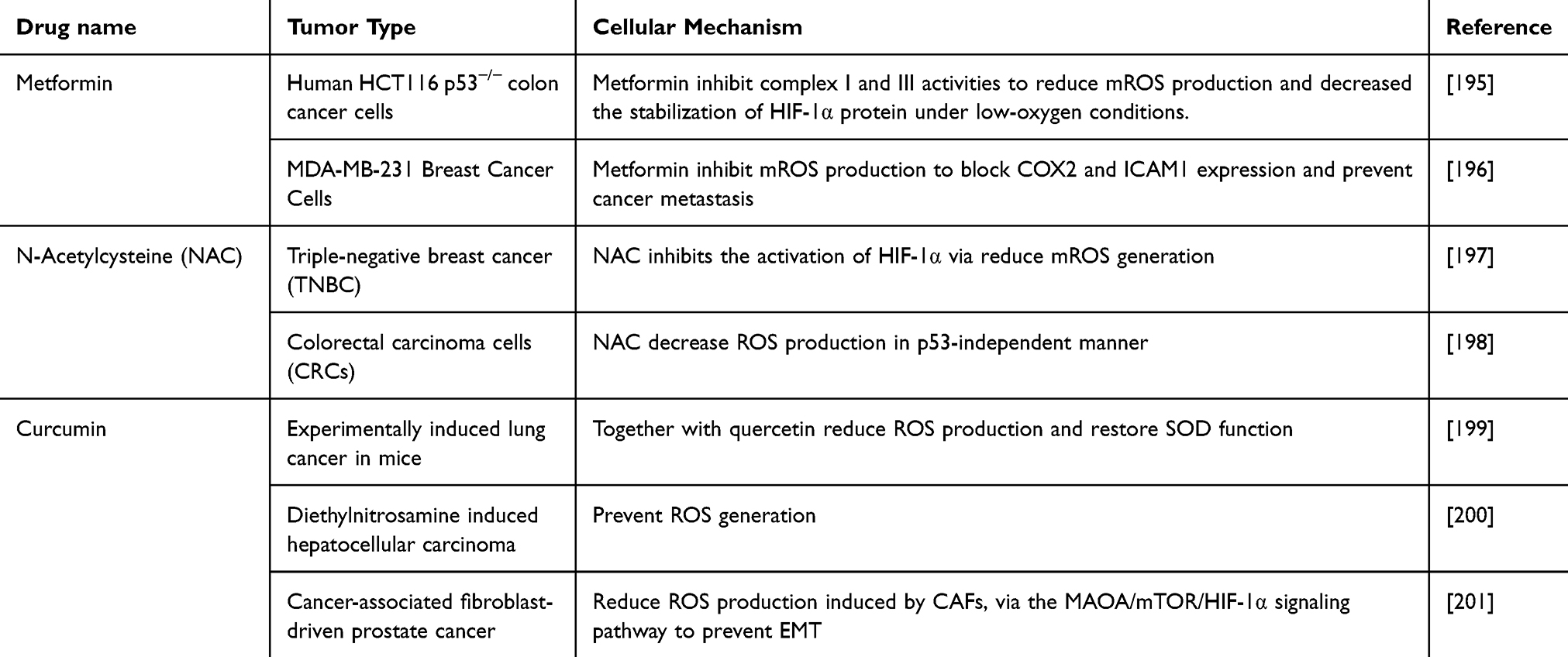 |
Table 3 Antioxidants Used in Preclinical and Clinical Trials to Combat ROS Generation and Cancer Progression |
Several antioxidants exist in the form of nanoparticles that dampen mROS production and are used in anticancer therapy. Mito E and Mito Q is two mitochondrial-targeted antioxidants have been used in many animal models and clinical trials. Mito E2 was the first mitochondria-targeted antioxidant (vitamin E) developed by Smith in 1999.202 This study increased the level of antioxidant vitamin E (α-tocopherol) more than 80 folds in the mitochondria of human osteosarcoma 143 B cells. However, in this study, vitamin E was found in the lipid bilayer of the mitochondrial inner membrane, whereas TPP entered the mitochondrial matrix, thereby, encapsulation of Mito E in TPP-based nanoparticles might have influenced the efficacy of vitamin E.
In addition, MitoQ10 extends the accuracy of drugs via target to ubiquinol portion of coenzyme Q in the electron transport chain. Compared to untargeted ubiquinone analogs, MitoQ10 demonstrates a superior potential to accumulate in the mitochondria up to 1000 folds.203 In animal models, MitoQ10 has been proven not only to decrease mROS production but also to inhibit cancer cell proliferation. In aggressive triple-negative and HER2-positive human breast cancer cell lines, MitoQ10 treatment has cytostatic effects on human breast cancer cells, reducing MDA-MB-231 cell numbers. Researchers have explored the use of nanoparticles that encapsulate MitoQ (nMitoQ) in treating prenatal hypoxia. As a result, nMitoQ treatment increased fetal blood space area under hypoxic conditions in the female placenta, suggesting the therapeutic potential of nanoparticle-encapsulated MitoQ to combat ROS.204
Recently, researchers developed an antioxidant (Cerium oxide) based nanoparticle (Nanoceria) for melanoma management. In vitro experiments demonstrated that Nanoceria exhibits unique redox properties that enable them to scavenge excess ROS production, which is related to a dose-dependent decrease in the expression level of HIF-1α and vascular endothelial growth factor (VEGF), contributing to anti-angiogenic cancer treatments.205 Moreover, the scavenging effect of Nanoceria is related to nanoparticle size, treatment duration, and specific cancer cell type. Larger nanoceria particles (94 nm) showed a greater capacity to scavenge ROS than smaller particles (7 nm), and the results of cell internalization were distinct in two colon cancer cell lines.206
Nanoparticles Targeted mROS Production as Enhanced Photodynamic Therapy
In the past two decades, photodynamic therapy (PDT) has emerged as a promising antitumor therapy. It relies on the generation of ROS by photosensitizers to exert cytotoxic effects and to induce cell death. Conventional photodynamic therapies involve two stages: First, the Photosensitizers induce the generation of detrimental free radicals after absorbing light. Two major photosensitizers can either react with biomolecules to release ROS (type I), or directly interact with oxygen to generate ROS (type II)207 (Figure 9). Second, near-infrared (NIR) lasers release the appropriate wavelengths to activate photosensitizers. Normally, light with a higher wavelength has better tissue penetration ability, referred to as a “near-infrared window”.208,209
 |
Figure 9 Type I and type II photodynamic therapy mechanism to induce robust mROS generation by nanoparticles. In the type I process, PDT-based nanoparticles produce mROS from biomolecules; in the type II process, PDT-based nanoparticles generate singlet oxygen from existing oxygen. Created in BioRender. Wang, X. (2025) https://BioRender.com/knynrm3. |
PDT selectivity of PDT is originally achieved by the location of light irradiation at the tumor sites. However, PDT has a limited tissue penetration (5 mm). This restricts its use to surface or shallow tumors, such as skin or oral cancers, making it less effective for deep-seated cancers.210,211 In addition, a lack of selectivity can cause prolonged sensitivity to light, leading to photosensitivity reactions that may last for days or even weeks. ROS release may activate many cytokines including IL-1β, IL-2, IL-6, and IL-8, which induce pain, inflammation, and allergic reactions.212 Moreover, common sensitizers, such as phthalocyanine, phthalocyanine, and chlorine, are only slightly soluble in water, prolonging the retention of drugs localized in cell membranes and limiting their efficacy.208,213
Hence, when PDT is combined with mROS-targeted nanotechnology, its effectiveness is amplified by specifically disrupting mitochondrial function and elevating mROS levels, leading to enhanced oxidative stress within cancer cells. LinTT1-HFtn is a mitochondria-targeted nanoparticle encapsulated in photosensitizers with ROS-generating abilities. When compared with free PDT-treated groups, LinTT1-HFtn demonstrated the highest mitochondrial ROS level and prominent release of cytochrome c from the mitochondria, demonstrating better therapeutic effects after direct ROS generation in the mitochondria. Fluorescence analysis showed that 64.4% of HepG2 cells underwent apoptosis after treatment with LinTT1-HFtn-AIE nanoparticles, which is higher than other control groups.214 Compared to free photosensitizers, photosensitizers with an nanoparticle design demonstrated a slight decay after 30 minutes of irradiation.215 As mentioned above, limited penetration is one of the biggest challenges for phototherapy applications in deep tumors such as liver, pancreatic, and gallbladder cancers. Several studies have reported sustained NIR-emitting luminescence using photosensitizer-incorporated nanoparticles. Juengpanich S et al developed tumor-targeted photodynamic nanoparticles (STPNs) the treatment of gallbladder cancer. To prolong the luminescence of photosensitizers, the study used up-conversion nanoparticles are a class of luminescent nanomaterials that can absorb low-energy photons, typically in the near-infrared (NIR) range, and convert them into high-energy emissions in the visible or ultraviolet (UV) range. This study identified exceptional antitumor activities in gallbladder cancer cell lines after treatment, demonstrating higher ROS generation and significant activation of the apoptotic pathway in tumor cells.216 For brain tumors, the presence of the blood-brain barrier is another important issue that influences many photosensitizers. For example, Indocyanine green (ICG) serves as a water-soluble photosensitizer, which limits its use in brain tumors. Recently, Liang et al designed a biomimetic nanocarrier containing ICG to target glioblastoma. The mitochondria-targeted nanoparticle surface was loaded with low-density lipoprotein receptor-related protein-1 (LRP1), a member of transferrin (TF) that can pass through the BBB. After dissociation, ICG was released into the mitochondria and significantly increased ROS production.217
Evidence has shown that mitochondria-targeted photodynamic therapy can induce innate immunity via activation of cyclic GMP-AMP synthase (cGAS)-stimulator of interferon gene (STING) pathway.218,219 Therefore, mitochondria-targeted photodynamic therapy which generates mROS to induce cell death, and immunotherapy, which induces ICD, play a synergistic role in cancer treatment. ALA&Dz@ZIF-PEG, designed by Zhao et al, is a mitochondria-targeted nanoparticle that exhibits the dual effects of photodynamics and immunotherapy. Once the nanoparticle accumulates in the mitochondria of cancer cells, 5-aminolevulinic acid (ALA) is released to induce the photodynamic effect of mROS generation and mitochondrial dysfunction. These changes subsequently drive the release of double-stranded mitochondrial DNA into the cytoplasm, initiating activation of the cGAS-STING pathway and activation of immune cells such as T natural killer (NK) cells, dendritic cells (DCs), and T lymphocytes (CTLs).220
mROS Targeted Nanoparticles vs Other Targeted Cancer Therapies
mROS Targeted Nanoparticles vs Immunotherapy
Among all other targeted treatment options, immunotherapy is currently the most innovative method that eliminates tumors via the host immune system. Significant progress has been made in preventing tumor immune evasion. FDA has approved many immune checkpoint inhibitors for future clinical trials, yet mROS-targeted nanoparticles have seen slower clinical translation.221 In the past decade, although numerous nanoparticles that promote ROS generation in mitochondria have been investigated, only one type of reactive oxygen species (ROS)-generating nanoparticle, ferumoxytol, has received FDA approval for treating iron deficiency anemia.222 Immunotherapy also faces significant challenges, including a very low response rate and the potential for severe side effects; under this circumstance, the combination of mROS-targeted nanoparticles and immunotherapy may achieve a “1+1>2” effect in tumor treatment. For example, one multifunctional nanoparticle contains curcumin (CU), a Ca2+ enhancer, co-loaded with CaCO3 and MnO2 to promote mROS generation that induces immunogenic cell death (ICD). Moreover, When combined with anti-PD-1 (immune checkpoint inhibition) therapy, the nanoparticle significantly enhances antitumor efficacy, suggesting its potential as a combinational strategy in immunotherapy.135
mROS Targeted Nanoparticles vs Gene Targeted Therapy
Cancer gene therapy has revolutionized cancer treatment over the past few decades by replacing tumor suppressor genes, silencing oncogenes, and introducing silencing suicide genes.223 In recent years, chimeric antigen receptor (CAR)-T cell therapy combines cancer immunotherapy and gene engineering of T cells to restore the host immune system and to attack cancer cells. Markedly, the success of CAR-T therapy has been observed in many types of cancers, especially hematological cancers such as non-Hodgkin lymphoma and leukemia.224,225 For example, enhanced CAR-T cells were effective in patients with relapsed/refractory aggressive B cell non-Hodgkin lymphoma with an 87.5% remission rate and negligible adverse events.224 By contrast, mROS-targeted nanoparticles remain in the “pre-clinical” stage. Although most studies reported selective cancer cell death and excellent biosafety in animal models, there are no documented ongoing clinical trials, and translating mROS-targeted nanoparticle therapies into clinical use requires further research to establish their safety and efficacy.
mROS Targeted Nanoparticles vs Untargeted ROS Inducers/Suppressors
Tumor heterogeneity significantly influences how mROS affect tumor growth within or across tumor types. On the one hand, variation of mROS in cancer cells across tumors can lead to vastly different outcomes: elevated mtROS is often observed in some solid tumors such as colon cancer and breast cancer, enhancing cell proliferation and tumor growth.60 However, hematologic cancers generally have lower baseline mitochondrial ROS, which depends on mitochondrial oxidative phosphorylation to prevent ROS-mediated apoptosis.226 On the other hand, tumor heterogeneity also results in variations of mitochondrial ROS within the same tumor types. Researchers discovered that glioblastoma CSCs maintain lower mROS levels than bulk tumor cells through the action of prohibitin (PHB), while knockout of the PHB gene could suppress CSCs’ self-renewal. Also, CSCs of acute myeloid leukemia have an inverse correlation between mROS levels and stemness potential.227
The dynamism of mROS between and within tumors may change therapeutic sensitivity and bring unpredictability to tumor responses when treated with untargeted mROS inducers/suppressors. For example, a potent ROS inducer, elesclomol, leads to CSCs copper dependent cell death in CSCs of glioblastoma, suggesting that elevation in mROS of CSCs could potentially lead to tumor eradication.228 However, elevated levels of mROS in CSCs contribute to metastasis formation in 4 different tumor cell lines via activation of MAPK signaling and subsequent EMT transformation.229 Likewise, the use of antioxidants was initially recognized to inhibit ROS generation that potentially inhibits cancer cell proliferation, but antioxidants such as vitamin C and vitamin E could paradoxically trigger tumor progression in patients with stage III/IV tumors.230
Fine-tune mROS levels within specific tumor compartments precisely destroy cancer cell mitochondria by altering redox homeostasis. A “Trojan horse” nanoparticle system has been developed that can initiate or halt mROS production in a controlled manner by adjusting the NIR wavelengths, which helps to limit damage to healthy cells while maximizing cancer cell death. The nanoregulator PIHR adjusts the mROS levels by responding to different NIR laser wavelengths. At 808 nm, it produces ROS to kill cancer cells, whereas at 1064 nm, it releases hydrogen (H2) to eliminate excess ROS to protect normal cells.231 mROS-targeted nanoparticles become increasingly favored in modern approaches to address the potential adverse effects of untargeted mROS modulations. For example, compared to single elesclomol to induce intracellular ROS, these nanoparticles encapsulate with elesclomol to generate a higher level of ROS and induce more antitumor activities.19 Additionally, nanoparticles that reduce mROS production by encapsulating antioxidants derived from coenzyme Q10 (Mito Q) suppress breast cancer and glioma cell proliferation, which suggested opposite results compared to free antioxidants.18 One explanation for this difference may be attributed to the poor mitochondrial accumulation of general antioxidants that limit their ability to alleviate oxidative stress, which is critical in limiting cell proliferation.
Challenges of mROS-Targeted Nanoparticles in Clinical Translation
FDA-regulated new drugs must “be safe and effective”, with confirmed benefits exceeding associated risks.232 Despite the growing number of nanoparticle-based therapies entering clinical trials and the increasing number of FDA-approved nanomedicines for cancer treatment, there are currently no clinical trials specifically focused on mROS-targeted nanoparticles in cancer therapy. Therefore, we searched for nanoparticles approved by the FDA in the recent decade and analyzed their significant clinical benefits and potential risks (Table 4) (http://www.clinicaltrials.gov). While some nanoparticles such as Onivyde™ and Vyxeos® show manageable toxicity profiles in cancer patients, other formulations like Apretude® and Aristada Initio® exhibit adverse events, including injection site reactions, stroke, or ischemic attacks.233–236 These examples underscore the unresolved challenges in nanoparticle clinical translation. Herein, we analyze three major challenges behind the clinical translation of mROS-targeted nanoparticles.
 |
Table 4 FDA-Approved Nanoparticles |
Biosafety and Toxicity
The biosafety and toxicity of mROS-targeted nanoparticles remain a major concern to clinical translation in cancer therapy. Many mROS-targeted nanoparticles incorporate mineral components to facilitate ROS generation; however, metal-based nanoparticles, such as those containing gold or silver, can be cytotoxic at higher doses, necessitating biocompatible coatings or alternative materials that reduce toxicity. As the primary detoxifying organ, nanoparticles are more likely to accumulate in the liver and induce various adverse events.238 One mROS-targeted nanoparticle, CPD, exhibits an increase in alkaline phosphatase (ALP) with aspartate aminotransferase (AST), which are markers of potential liver injury.149 The injection of Ag-NPs markedly increased liver inflammatory factors such as TNF-α and IL-6 and decreased GSH levels in rats, suggesting the induction of inflammation and ROS-induced damage.239 Additionally, the accumulation of ROS-responsive nanoparticles modulates immune activities by shifting macrophage polarization from the M1 state to the M2 state and inhibiting pro-inflammatory factors, including IL-1β, IL-6, and TNF-α in the injured liver, suggesting a potential interaction between mROS-targeted nanoparticles and immune system.240,241
Although numerous nanoparticles that promote ROS generation in mitochondria have exhibited excellent biocompatibility and imperceptible systemic injury in vitro and in vivo studies, the administration of nanoparticles may have long-term toxicities to organs. While commonly assumed to be non-toxic in the short-term, selenium nanoparticle administration in the long term raised atherosclerosis and caused liver and kidney damage in murine models.242 Yet, there is a critical need to define toxicity thresholds based on mROS-targeted nanoparticle dose and size to ensure future clinical application.243,244 For future studies to optimize mROS-targeted nanoparticles therapeutic windows and prevent toxicological side effects, several strategies that precisely control the characterization of nanoparticles and target accuracy are key to extend selectivity: Han and colleagues prepared three sizes of ROS- scavenging nanoparticles with PC-1 (~575 nm), PC-2 (~729 nm), and PC-3 (~1071 nm) to examine the influence of nanoparticle to therapeutic efficacy. PC-2 achieved the highest lung-to-liver fluorescence ratio in mice with hyperoxia-induced acute lung injury, demonstrating the lowest liver deposition rate.245 Another X-ray responsive nanoparticle (DMSNs@AO) incorporates diselenide bonds into silica nanoparticles. Diselenide bonds are cleavable in the presence of both oxidative stress and X-ray conditions, thereby ensuring the target release of nanoparticles to tumor tissues. Mice that administered DMSNs@AO showed less dose-dependent weight loss compared to free AO groups, indicating low systemic toxicity.246
Biodistribution Variation
Biodistribution directly influences the spread, release, and clearance of mROS-targeted nanoparticles throughout the body after administration. Yet, among all the studies mentioned above, only a few discussed organ biodistribution of mROS-targeted nanoparticles due to the lack of in vivo studies. Moreover, the lack of measurement techniques to measure how nanoparticles reach mitochondria in specific organs or tissues also contributes to a poor understanding of distribution profiles.247 The degree of nanoparticle distribution varies significantly depending on the kind of animal models and the type of tumor.248,249 A comparative study using different murine models for breast cancer showed varying spatial biodistribution of nanoparticles, which were related to varied tumor stromal cells in the tumor microenvironment, highlighting a poor understanding of the complex interplay of nanoparticle and host-dependent differences.250 Apart from the difference in biodistribution in animal models, in vitro models could also affect the extent of nanoparticle distribution. In a study using ceria-based nanoparticles to induce ROS-mediated antitumor activities in melanoma spheroids, significant differences in mROS generation were observed between 2D and 3D models, pointing out the current limitations of current in vitro 2D cell culture studies.251
Off-Target Obstacles
For nanoparticles to target cancer cells or even subcellular organelles such as mitochondria, research has mainly focused on either enhanced permeability and retention (EPR) effect to target cancer cell mitochondria passively or modulate nanoplatform surface to make them “recognizable” for cancer cells.252 Although several designs of nanoparticle carriers, such as TPP-based nanocarriers and biomimetic design, were utilized to ensure the drug reaches the mitochondria, there is still undesirable deposition of nanoparticles in major organs apart from tumor sites. HM-NPs@G is a mROS-targeted nanoparticle that hybridizes cancer cell membrane and mitochondria membrane to promote higher deposition of ROS-responsive drugs. Compared to the free drug group with a 0.47-hour half-life, HM-NPs@G exhibited a prolonged circulation half-life of approximately 4.9 hours. Moreover, it could effectively cross the BBB, which remains to be a major limitation in the treatment of brain carcinoma. However, the accumulation of HM-NPs@G was also observed in the spleen and liver, which are two organs that are primarily involved in NP clearance, indicating a potential risk of organ injuries.22
Discussion
The knowledge of ROS generated from mitochondria and the related mechanisms in cancer has increased in recent years. Elevated levels of mROS can activate signaling pathways that promote cancer cell proliferation, survival, and metastasis. As ROS scavengers, antioxidants prevent the oxidation of other molecules by neutralizing ROS and other free radicals. In the context of cancer, while low or undetectable levels of antioxidants could promote tumorigenesis, higher than normal levels of antioxidants may encourage cancer metastasis and prevent cancer cell apoptosis.253,254 Importantly, mROS in cancer serves as a potential target for cancer therapies. Nanoparticles with mROS-regulating properties have been exploited as ROS modulators in mitochondria to suppress cancer progression. Unlike untargeted ROS modulators, which may produce unpredictable effects due to tumor heterogeneity and cancer stem cell plasticity, mROS-targeted nanoparticles potentially minimize off-target toxicity and enhance therapeutic efficacy.19 There are two major strategies to modulate mROS with nanoparticles: (1) Increase ROS production to lethal levels that trigger cell death via several mechanisms such as apoptosis, necroptosis, ferroptosis, pyroptosis, and cuproptosis. (2) Inhibit mROS production to inhibit cancer progression. In addition, photodynamic therapy-based nanoparticles to mitochondria is a therapeutic strategy to produce ROS in the mitochondria artificially. Unfortunately, the role of mROS in inducing cell death and promoting tumorigenesis remains poorly understood. Some studies have reported that a “moderate level” of ROS could promote tumorigenesis and that “excessive” ROS exerts an antitumor effect.255 However, the boundaries between these two levels still require clarification. Compared to other emerging therapeutic therapies like immunotherapy and gene-targeted therapies, mROS-targeted nanoparticles remain in the early stages of development, with no clinical trials currently ongoing/under recruitment (https://clinicaltrials.gov/). Several barriers must be addressed, including biosafety and long-term toxicity, variation in biodistribution and clearance, and challenges in achieving mitochondrial specificity in heterogeneous tumor environments.
In conclusion, the regulation of mitochondrial ROS by nanoparticles in cancer still heralds precise drug delivery approaches, and future studies are required to pave the way for more antitumor strategies (Figure 10).
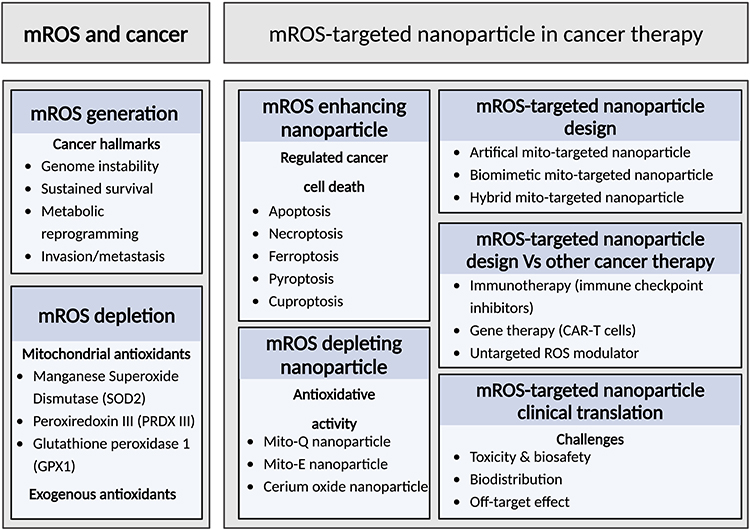 |
Figure 10 Overview of mitochondrial ROS (mROS) and mROS-targeted nanoparticle strategies in cancer therapy. This schematic summarizes the dual roles of mROS in cancer progression and therapeutic intervention. The left panel outlines the generation of mROS and its association with cancer hallmarks, as well as the role of endogenous and exogenous antioxidants in mROS depletion. The right panel illustrates current strategies for leveraging mROS-targeted nanoparticles in cancer therapy. The figure also highlights key design approaches for mitochondria-targeted nanoparticles, comparisons with other cancer therapies and major translational challenges. Created in BioRender. Wang, X. (2025) https://BioRender.com/z7e7ibg. |
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC) (Grant No. 82260531 and 81760509) and Natural Science Foundation of Jiangxi Province of China (Grant No. 20224BAB206057 and 20181BAB205043). The graphical abstract was Created in BioRender. Wang, X. (2025) https://BioRender.com/z4utvor.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Ma C, Xia F, Kelley SO. Mitochondrial targeting of probes and therapeutics to the powerhouse of the cell. Bioconjug Chem. 2020;31(12):2650–2667. doi:10.1021/acs.bioconjchem.0c00470
2. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling. Int J Mol Med. 2019;44(1):3–15. doi:10.3892/ijmm.2019.4188
3. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res off J Am Assoc Cancer Res. 2018;24(11):2482–2490. doi:10.1158/1078-0432.CCR-17-3070
4. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–947. doi:10.1038/nrd4002
5. Keele BB, McCord JM, Fridovich I. Superoxide dismutase from Escherichia coli B. A new manganese-containing enzyme. J Biol Chem. 1970;245(22):6176–6181.
6. Deng R, Zhang HL, Huang JH, et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy. 2021;17(10):3011–3029. doi:10.1080/15548627.2020.1850609
7. Iskandar K, Foo J, Liew AQX, et al. A novel MTORC2-AKT-ROS axis triggers mitofission and mitophagy-associated execution of colorectal cancer cells upon drug-induced activation of mutant KRAS. Autophagy. 2024;20(6):1418–1441. doi:10.1080/15548627.2024.2307224
8. Owari T, Tanaka N, Nakai Y, et al. 5-aminolevulinic acid overcomes hypoxia-induced radiation resistance by enhancing mitochondrial reactive oxygen species production in prostate cancer cells. Br J Cancer. 2022;127(2):350–363. doi:10.1038/s41416-022-01789-4
9. Kidwell CU, Casalini JR, Pradeep S, et al. Transferred mitochondria accumulate reactive oxygen species, promoting proliferation. eLife. 2023:
10. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
11. Missiroli S, Perrone M, Genovese I, Pinton P, Giorgi C. Cancer metabolism and mitochondria: finding novel mechanisms to fight tumours. EBioMedicine. 2020;59:102943. doi:10.1016/j.ebiom.2020.102943
12. Cui Q, Wang JQ, Assaraf YG, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1–25. doi:10.1016/j.drup.2018.11.001
13. Wang H, Gao Z, Liu X, et al. Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat Commun. 2018;9:562. doi:10.1038/s41467-018-02915-8
14. Khan I, Saeed K, Khan I. Nanoparticles: properties, applications and toxicities. Arab J Chem. 2019;12(7):908–931. doi:10.1016/j.arabjc.2017.05.011
15. Cui L, Gouw AM, LaGory EL, et al. Mitochondrial copper depletion suppresses triple-negative breast cancer in mice. Nat Biotechnol. 2021;39(3):357–367. doi:10.1038/s41587-020-0707-9
16. Wang B, Zhou H, Chen L, et al. A mitochondria-targeted photosensitizer for combined pyroptosis and apoptosis with NIR-II imaging/photoacoustic imaging-guided phototherapy. Angew Chem Int Ed Engl. 2024;63(39):e202408874. doi:10.1002/anie.202408874
17. Wang H, Shi W, Zeng D, et al. pH-activated, mitochondria-targeted, and redox-responsive delivery of paclitaxel nanomicelles to overcome drug resistance and suppress metastasis in lung cancer. J Nanobiotechnology. 2021;19(1):152. doi:10.1186/s12951-021-00895-4
18. Cheng G, Karoui H, Hardy M, Kalyanaraman B. Redox-crippled MitoQ potently inhibits breast cancer and glioma cell proliferation: a negative control for verifying the antioxidant mechanism of MitoQ in cancer and other oxidative pathologies. Free Radic Biol Med. 2023;205. doi:10.1016/j.freeradbiomed.2023.06.009
19. Wu H, Zhang Z, Cao Y, et al. A self‐amplifying ROS‐responsive nanoplatform for simultaneous cuproptosis and cancer immunotherapy. Adv Sci. 2024;11(23):2401047. doi:10.1002/advs.202401047
20. Liu C, Liu B, Zhao J, et al. Nd3+-sensitized upconversion metal–organic frameworks for mitochondria-targeted amplified photodynamic therapy. Angew Chem Int Ed. 2020;59(7):2634–2638. doi:10.1002/anie.201911508
21. Zhang J, Gao B, Ye B, et al. Mitochondrial-targeted delivery of polyphenol-mediated antioxidases complexes against pyroptosis and inflammatory diseases. Adv Mater Deerfield Beach Fla. 2023;35(11):e2208571. doi:10.1002/adma.202208571
22. Zou Y, Sun Y, Wang Y, et al. Cancer cell-mitochondria hybrid membrane coated Gboxin loaded nanomedicines for glioblastoma treatment. Nat Commun. 2023;14(1):4557. doi:10.1038/s41467-023-40280-3
23. Zhou Z, Li C, Li C, et al. Mitochondria-targeted nanoadjuvants induced multi-functional immune-microenvironment remodeling to sensitize tumor radio-immunotherapy. Adv Sci Weinh Baden-Wurtt Ger. 2024;11(26):e2400297. doi:10.1002/advs.202400297
24. Lyu M, Chen M, Liu L, et al. A platelet-mimicking theranostic platform for cancer interstitial brachytherapy. Theranostics. 2021;11(15):7589. doi:10.7150/thno.61259
25. Kirova DG, Judasova K, Vorhauser J, et al. A ROS-dependent mechanism promotes CDK2 phosphorylation to drive progression through S phase. Dev Cell. 2022;57(14):1712–1727.e9. doi:10.1016/j.devcel.2022.06.008
26. Iyer GYN, Islam MF, Quastel JH. Biochemical aspects of phagocytosis. Nature. 1961;192:4802):535–541. doi:10.1038/192535a0
27. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–421. doi:10.1038/nrm3801
28. Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43(3):332–347. doi:10.1016/j.freeradbiomed.2007.03.027
29. Pecchillo Cimmino T, Ammendola R, Cattaneo F, Esposito G. NOX dependent ROS generation and cell metabolism. Int J Mol Sci. 2023;24(3):2086. doi:10.3390/ijms24032086
30. Khacho M, Clark A, Svoboda DS, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19(2):232–247. doi:10.1016/j.stem.2016.04.015
31. Miao CC, Hwang W, Chu LY, et al. LC3A-mediated autophagy regulates lung cancer cell plasticity. Autophagy. 202218(4):921–934. doi:10.1080/15548627.2021.1964224
32. Ashwaq AAS, Al-Qubaisi MS, Rasedee A, Abdul AB, Taufiq-Yap YH, Yeap SK. Inducing G2/M cell cycle arrest and apoptosis through generation reactive oxygen species (ROS)-mediated mitochondria pathway in HT-29 cells by dentatin (DEN) and dentatin incorporated in hydroxypropyl-β-cyclodextrin (DEN-HPβCD). Int J Mol Sci. 2016;17(10):1653. doi:10.3390/ijms17101653
33. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11(1):102. doi:10.1038/s41467-019-13668-3
34. Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. 1977;180(2):248–257. doi:10.1016/0003-9861(77)90035-2
35. Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane*. J Biol Chem. 2004;279(47):49064–49073. doi:10.1074/jbc.M407715200
36. Diebold L, Chandel NS. Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med. 2016;100:86–93. doi:10.1016/j.freeradbiomed.2016.04.198
37. Orr AL, Ashok D, Sarantos MR, Shi T, Hughes RE, Brand MD. Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic Biol Med. 2013;65:1047–1059. doi:10.1016/j.freeradbiomed.2013.08.170
38. Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1(1):304–312. doi:10.1016/j.redox.2013.04.005
39. Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi:10.1146/annurev.micro.57.030502.090938
40. Hamanaka RB, Glasauer A, Hoover P, et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci Signal. 2013;6(261):ra8. doi:10.1126/scisignal.2003638
41. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi:10.1152/physrev.00026.2013
42. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–1014. doi:10.1084/jem.192.7.1001
43. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485. doi:10.1016/j.redox.2015.09.005
44. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:6625):623–627. doi:10.1038/386623a0
45. Yoshioka KI, Kusumoto-Matsuo R, Matsuno Y. Genomic instability and cancer risk associated with erroneous DNA repair. Int J Mol Sci. 2021;22(22):12254. doi:10.3390/ijms222212254
46. Deininger P. Genetic instability in cancer: caretaker and gatekeeper genes. Ochsner J. 1999;1(4):206–209.
47. Chaudhry SR, Lopes J, Levin NK, Kalpage H, Tainsky MA. Germline mutations in apoptosis pathway genes in ovarian cancer; the functional role of a TP53I3 (PIG3) variant in ROS production and DNA repair. Cell Death Discov. 2021;7:62. doi:10.1038/s41420-021-00442-y
48. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi:10.1038/nature01322
49. Achanta G, Sasaki R, Feng L, et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol γ. EMBO J. 2005;24(19):3482–3492. doi:10.1038/sj.emboj.7600819
50. Di Bona M, Chen Y, Agustinus AS, et al. Micronuclear collapse from oxidative damage. Science. 2024;385:
51. Garribba L, De Feudis G, Martis V, et al. Short-term molecular consequences of chromosome mis-segregation for genome stability. Nat Commun. 2023;14(1):1353. doi:10.1038/s41467-023-37095-7
52. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability — an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–228. doi:10.1038/nrm2858
53. Di Bona M, Bakhoum SF. Micronuclei and Cancer. Cancer Discov. 2024;14(2):214–226. doi:10.1158/2159-8290.CD-23-1073
54. Zhang CZ, Spektor A, Cornils H, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:7555):179–184. doi:10.1038/nature14493
55. Cortés-Ciriano I, Lee JJK, Xi R, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet. 2020;52(3):331–341. doi:10.1038/s41588-019-0576-7
56. Martin S, Scorzoni S, Cordone S, et al. A p62-dependent rheostat dictates micronuclei catastrophe and chromosome rearrangements. Science. 2024. doi:10.1126/science.adj7446
57. Greenwald AC, Darnell NG, Hoefflin R, et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell. 2024;187(10):2485–2501.e26. doi:10.1016/j.cell.2024.03.029
58. Kang MA, So EY, Simons AL, Spitz DR, Ouchi T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012;3(1):e249. doi:10.1038/cddis.2011.134
59. Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–327. doi:10.1016/S1470-2045(05)70168-6
60. Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107(19):8788–8793. doi:10.1073/pnas.1003428107
61. Posse V, Shahzad S, Falkenberg M, Hällberg BM, Gustafsson CM. TEFM is a potent stimulator of mitochondrial transcription elongation in vitro. Nucleic Acids Res. 2015;43(5):2615–2624. doi:10.1093/nar/gkv105
62. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia NYN. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
63. Wan L, Wang Y, Zhang Z, et al. Elevated TEFM expression promotes growth and metastasis through activation of ROS/ERK signaling in hepatocellular carcinoma. Cell Death Dis. 2021;12(4):325. doi:10.1038/s41419-021-03618-7
64. Zampieri LX, Silva-Almeida C, Rondeau JD, Sonveaux P. Mitochondrial transfer in cancer: a comprehensive review. Int J Mol Sci. 2021;22(6):3245. doi:10.3390/ijms22063245
65. Marlein CR, Zaitseva L, Piddock RE, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood. 2017;130(14):1649–1660. doi:10.1182/blood-2017-03-772939
66. Zhou Z, Qu C, Zhou P, et al. Extracellular vesicles activated cancer-associated fibroblasts promote lung cancer metastasis through mitophagy and mtDNA transfer. J Exp Clin Cancer Res CR. 2024;43(1):158. doi:10.1186/s13046-024-03077-w
67. Burt R, Dey A, Aref S, et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood. 2019;134(17):1415. doi:10.1182/blood.2019001398
68. Wang J, Liu X, Qiu Y, et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol. 2018;11:11. doi:10.1186/s13045-018-0554-z
69. Ullah R, Yin Q, Snell AH, Wan L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin Cancer Biol. 2022;85:123–154. doi:10.1016/j.semcancer.2021.05.010
70. Guan B, Liu Y, Xie B, et al. Mitochondrial genome transfer drives metabolic reprogramming in adjacent colonic epithelial cells promoting TGFβ1-mediated tumor progression. Nat Commun. 2024;15:3653. doi:10.1038/s41467-024-48100-y
71. Zhang W, Zhou H, Li H, et al. Cancer cells reprogram to metastatic state through the acquisition of platelet mitochondria. Cell Rep. 2023;42(9):113147. doi:10.1016/j.celrep.2023.113147
72. Peng C, Li X, Ao F, et al. Mitochondrial ROS driven by NOX4 upregulation promotes hepatocellular carcinoma cell survival after incomplete radiofrequency ablation by inducing of mitophagy via Nrf2/PINK1. J Transl Med. 2023;21:218. doi:10.1186/s12967-023-04067-w
73. Pi H, Xu S, Reiter RJ, et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11(7):1037–1051. doi:10.1080/15548627.2015.1052208
74. Min Y, Kim MJ, Lee S, Chun E, Lee KY. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy. 2018;14(8):1347–1358. doi:10.1080/15548627.2018.1474995
75. Gong Y, Tang N, Liu P, et al. Newcastle disease virus degrades SIRT3 via PINK1-PRKN-dependent mitophagy to reprogram energy metabolism in infected cells. Autophagy. 2022;18(7):1503–1521. doi:10.1080/15548627.2021.1990515
76. Blasco N, Beà A, Barés G, et al. Involvement of the mitochondrial nuclease EndoG in the regulation of cell proliferation through the control of reactive oxygen species. Redox Biol. 2020;37:101736. doi:10.1016/j.redox.2020.101736
77. Zhang X, Liu L, Chen C, et al. Interferon regulatory factor-1 together with reactive oxygen species promotes the acceleration of cell cycle progression by up-regulating the cyclin E and CDK2 genes during high glucose-induced proliferation of vascular smooth muscle cells. Cardiovasc Diabetol. 2013;12:147. doi:10.1186/1475-2840-12-147
78. Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:7239):780–783. doi:10.1038/nature07733
79. Huang H, Zhang S, Li Y, et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat Commun. 2021;12:3720. doi:10.1038/s41467-021-24108-6
80. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215). https://pubmed.ncbi.nlm.nih.gov/13351639/?dopt=Abstract.
81. Jing X, Yang F, Shao C, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157. doi:10.1186/s12943-019-1089-9
82. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. 2020;21(5):268–283. doi:10.1038/s41580-020-0227-y
83. Dabral S, Muecke C, Valasarajan C, et al. A RASSF1A-HIF1α loop drives Warburg effect in cancer and pulmonary hypertension. Nat Commun. 2019;10(1):2130. doi:10.1038/s41467-019-10044-z
84. Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):401–408. doi:10.1016/j.cmet.2005.05.001
85. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–3671. doi:10.1172/JCI67230
86. Maxwell PH, Eckardt KU. HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat Rev Nephrol. 2016;12(3):157–168. doi:10.1038/nrneph.2015.193
87. Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi:10.1016/j.ccr.2004.11.022
88. Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. doi:10.1016/j.cell.2004.08.025
89. Yang Q, Lei X, He J, et al. N4-acetylcytidine drives glycolysis addiction in gastric cancer via NAT10/SEPT9/HIF-1α positive feedback loop. Adv Sci Weinh Baden-Wurtt Ger. 2023;10(23):e2300898. doi:10.1002/advs.202300898
90. Yang Y, Cheng C, He B, et al. Cigarette smoking, by accelerating the cell cycle, promotes the progression of non-small cell lung cancer through an HIF-1α-METTL3-m6A/CDK2AP2 axis. J Hazard Mater. 2023;455:131556. doi:10.1016/j.jhazmat.2023.131556
91. Vara-Pérez M, Rossi M, Van den Haute C, et al. BNIP3 promotes HIF-1α-driven melanoma growth by curbing intracellular iron homeostasis. EMBO J. 2021;40(10):e106214. doi:10.15252/embj.2020106214
92. Dachs GU, Patterson AV, Firth JD, et al. Targeting gene expression to hypoxic tumor cells. Nat Med. 1997;3(5):515–520. doi:10.1038/nm0597-515
93. Yang JX, Chuang YC, Tseng JC, et al. Tumor promoting effect of PDLIM2 downregulation involves mitochondrial ROS, oncometabolite accumulations and HIF-1α activation. J Exp Clin Cancer Res CR. 2024;43:169. doi:10.1186/s13046-024-03094-9
94. Dong S, Liang S, Cheng Z, et al. ROS/PI3K/Akt and Wnt/β-catenin signalings activate HIF-1α-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J Exp Clin Cancer Res CR. 2022;41:15. doi:10.1186/s13046-021-02229-6
95. Yang Y, Zhang G, Guo F, et al. Mitochondrial UQCC3 modulates hypoxia adaptation by orchestrating OXPHOS and glycolysis in hepatocellular carcinoma. Cell Rep. 2020;33(5). doi:10.1016/j.celrep.2020.108340
96. Jin J, Qiu S, Wang P, et al. Cardamonin inhibits breast cancer growth by repressing HIF-1α-dependent metabolic reprogramming. J Exp Clin Cancer Res CR. 2019;38:377. doi:10.1186/s13046-019-1351-4
97. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi:10.1038/nm1469
98. Bastin J, Sroussi M, Nemazanyy I, Laurent-Puig P, Mouillet-Richard S, Djouadi F. Downregulation of mitochondrial complex I induces ROS production in colorectal cancer subtypes that differently controls migration. J Transl Med. 2023;21:522. doi:10.1186/s12967-023-04341-x
99. Horejs CM. Basement membrane fragments in the context of the epithelial-to-mesenchymal transition. Eur J Cell Biol. 2016;95(11):427–440. doi:10.1016/j.ejcb.2016.06.002
100. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi:10.1016/j.cell.2010.03.015
101. Shibanuma M, Kim-Kaneyama JR, Ishino K, et al. Hic-5 communicates between focal adhesions and the nucleus through oxidant-sensitive nuclear export signal. Mol Biol Cell. 2003;14(3):1158–1171. doi:10.1091/mbc.02-06-0099
102. Dc R, Dd L, Le L, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436(7047). doi:10.1038/nature03688
103. Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem. 2015;97:55–74. doi:10.1016/j.ejmech.2015.04.040
104. Pisoschi AM, Pop A, Iordache F, Stanca L, Predoi G, Serban AI. Oxidative stress mitigation by antioxidants - An overview on their chemistry and influences on health status. Eur J Med Chem. 2021;209:112891. doi:10.1016/j.ejmech.2020.112891
105. Liu M, Sun X, Chen B, Dai R, Xi Z, Xu H. Insights into manganese superoxide dismutase and human diseases. Int J Mol Sci. 2022;23(24):15893. doi:10.3390/ijms232415893
106. Dhar SK, St.Clair DK. Manganese superoxide dismutase regulation and cancer. Free Radic Biol Med. 2012;52(11):2209–2222. doi:10.1016/j.freeradbiomed.2012.03.009
107. Miar A, Hevia D, Muñoz-Cimadevilla H, et al. Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radic Biol Med. 2015:85. DOI:10.1016/j.freeradbiomed.2015.04.001
108. Kinugasa H, Whelan KA, Tanaka K, et al. Mitochondrial SOD2 regulates epithelial-mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene. 2015;34(41):5229. doi:10.1038/onc.2014.449
109. Hemachandra LPMP, Shin DH, Dier U, et al. Mitochondrial superoxide dismutase has a protumorigenic role in ovarian clear cell carcinoma. Cancer Res. 2015;75(22):4973–4984. doi:10.1158/0008-5472.CAN-14-3799
110. Yi L, Shen H, Zhao M, et al. Inflammation-mediated SOD-2 upregulation contributes to epithelial-mesenchymal transition and migration of tumor cells in aflatoxin G1-induced lung adenocarcinoma. Sci Rep. 2017;7:7953. doi:10.1038/s41598-017-08537-2
111. Cunniff B, Wozniak AN, Sweeney P, DeCosta K, Heintz NH. Peroxiredoxin 3 levels regulate a mitochondrial redox setpoint in malignant mesothelioma cells. Redox Biol. 2014;3:79–87. doi:10.1016/j.redox.2014.11.003
112. Chua PJ, Ow SH, Ng CT, et al. Peroxiredoxin 3 regulates breast cancer progression via ERK-mediated MMP-1 expression. Cancer Cell Int. 2024;24(1):59. doi:10.1186/s12935-024-03248-x
113. Yan H, Cai X, Fu S, Zhang X, Zhang J. PRDX3 promotes resistance to cisplatin in gastric cancer cells. J Cancer Res Ther. 2022;18(7):1994. doi:10.4103/jcrt.jcrt_970_22
114. Sunde RA, Raines AM. Selenium regulation of the selenoprotein and nonselenoprotein transcriptomes in rodents. Adv Nutr Bethesda Md. 2011;2(2):138–150. doi:10.3945/an.110.000240
115. Handy DE, Loscalzo J. The role of glutathione peroxidase-1 in health and disease. Free Radic Biol Med. 2022;188:146. doi:10.1016/j.freeradbiomed.2022.06.004
116. Lee S, Lee EK, Kang DH, et al. Glutathione peroxidase-1 regulates ASK1-dependent apoptosis via interaction with TRAF2 in RIPK3-negative cancer cells. Exp Mol Med. 2021;53(6):1080. doi:10.1038/s12276-021-00642-7
117. Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596–2606. doi:10.1093/emboj/17.9.2596
118. Kaźmierczak-Barańska J, Boguszewska K, Adamus-Grabicka A, Karwowski BT. Two faces of vitamin C—antioxidative and pro-oxidative agent. Nutrients. 2020;12(5):1501. doi:10.3390/nu12051501
119. Hercberg S, Ezzedine K, Guinot C, et al. Antioxidant supplementation increases the risk of skin cancers in women but not in men1. J Nutr. 2007;137(9):2098–2105. doi:10.1093/jn/137.9.2098
120. Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167(12):1600–1619. doi:10.1164/rccm.200212-1479SO
121. Guo Y, Xu Y, Bao Q, et al. Endogenous copper for nanocatalytic oxidative damage and self-protection pathway breakage of cancer. ACS Nano. 2021;15(10):16286–16297. doi:10.1021/acsnano.1c05451
122. Carlisle AE, Lee N, Matthew-Onabanjo AN, et al. Selenium detoxification is required for cancer-cell survival. Nat Metab. 2020;2(7):603–611. doi:10.1038/s42255-020-0224-7
123. Zhang H, Zhang W, Hu B, et al. Precise pancreatic cancer therapy through targeted degradation of mutant p53 protein by cerium oxide nanoparticles. J Nanobiotechnology. 2023;21(1):117. doi:10.1186/s12951-023-01867-6
124. Ni D, Wei H, Chen W, et al. Ceria nanoparticles meet hepatic ischemia-reperfusion injury: the perfect imperfection. Adv Mater Deerfield Beach Fla. 2019;31(40):e1902956. doi:10.1002/adma.201902956
125. Dowding JM, Song W, Bossy K, et al. Cerium oxide nanoparticles protect against Aβ-induced mitochondrial fragmentation and neuronal cell death. Cell Death Differ. 2014;21(10):1622–1632. doi:10.1038/cdd.2014.72
126. Behl T, Makkar R, Anwer M, et al. Mitochondrial dysfunction: a cellular and molecular hub in pathology of metabolic diseases and infection. J Clin Med. 2023;12(8):2882. doi:10.3390/jcm12082882
127. Murphy MP, Smith RAJ. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi:10.1146/annurev.pharmtox.47.120505.105110
128. Sun M, Hu N, Gao Y, et al. Platelet membrane-encapsulated nanocomplexes based on profundity scavenging ROS strategy for myocardial infarction therapy. Adv Healthc Mater. 2024;13(8):e2303101. doi:10.1002/adhm.202303101
129. Chen Y, Zhi S, Ou J, et al. Cancer cell membrane-coated nanoparticle co-loaded with photosensitizer and toll-like receptor 7 agonist for the enhancement of combined tumor immunotherapy. ACS Nano. 2023;17(17):16620–16632. doi:10.1021/acsnano.3c02724
130. Xia Q, Zhang Y, Li Z, Hou X, Feng N. Red blood cell membrane-camouflaged nanoparticles: a novel drug delivery system for antitumor application. Acta Pharm Sin B. 2019;9(4):675–689. doi:10.1016/j.apsb.2019.01.011
131. Lin CP, Wu SH, Lin TY, et al. Lysosomal-targeted doxorubicin delivery using RBC-derived vesicles to overcome drug-resistant cancer through mitochondrial-dependent cell death. Pharmacol Res. 2023;197:106945. doi:10.1016/j.phrs.2023.106945
132. Wu SH, Hsieh CC, Hsu SC, et al. RBC-derived vesicles as a systemic delivery system of doxorubicin for lysosomal-mitochondrial axis-improved cancer therapy. J Adv Res. 2021;30:185–196. doi:10.1016/j.jare.2020.11.009
133. Ismail M, Yang W, Li Y, et al. Biomimetic Dp44mT-nanoparticles selectively induce apoptosis in Cu-loaded glioblastoma resulting in potent growth inhibition. Biomaterials. 2022;289:121760. doi:10.1016/j.biomaterials.2022.121760
134. Mai X, Zhang Y, Fan H, et al. Integration of immunogenic activation and immunosuppressive reversion using mitochondrial-respiration-inhibited platelet-mimicking nanoparticles. Biomaterials. 2020:232. DOI:10.1016/j.biomaterials.2019.119699
135. Luo G, Li X, Lin J, et al. Multifunctional calcium–manganese nanomodulator provides antitumor treatment and improved immunotherapy via reprogramming of the tumor microenvironment. ACS Nano. 2023;17(16):15449–15465. doi:10.1021/acsnano.3c01215
136. Shi Y, Lim SK, Liang Q, et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature. 2019;567(7748):341–346. doi:10.1038/s41586-019-0993-x
137. Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. 2018;122(10):1460–1478. doi:10.1161/CIRCRESAHA.118.310082
138. Shao F, Han J, Tian Z, Wang Z, Liu S, Wu Y. Synergistic ROS generation and directional overloading of endogenous calcium induce mitochondrial dysfunction in living cells. Biomaterials. 2023;301:122284. doi:10.1016/j.biomaterials.2023.122284
139. Serra M, Columbano A, Ammarah U, Mazzone M, Menga A. Understanding metal dynamics between cancer cells and macrophages: competition or synergism? Front Oncol. 2020;10:646. doi:10.3389/fonc.2020.00646
140. Bao W, Liu M, Meng J, et al. MOFs-based nanoagent enables dual mitochondrial damage in synergistic antitumor therapy via oxidative stress and calcium overload. Nat Commun. 2021;12:6399. doi:10.1038/s41467-021-26655-4
141. Tian X, Srinivasan PR, Tajiknia V, et al. Targeting apoptotic pathways for cancer therapy. J Clin Invest. 2024;134(14):e179570. doi:10.1172/JCI179570
142. Zhou Z, Arroum T, Luo X, et al. Diverse functions of cytochrome c in cell death and disease. Cell Death Differ. 2024;31(4):387–404. doi:10.1038/s41418-024-01284-8
143. Green DR, Llambi F. Cell death signaling. Cold Spring Harb Perspect Biol. 2015;7(12):a006080. doi:10.1101/cshperspect.a006080
144. Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11(7):725–730. doi:10.1038/nm1263
145. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and gasdermin-induced pyroptosis. Annu Rev Immunol. 2020;38:567–595. doi:10.1146/annurev-immunol-073119-095439
146. Lajous H, Lelièvre B, Vauléon E, Lecomte P, Garcion E. Rethinking alkylating(-like) agents for solid tumor management. Trends Pharmacol Sci. 2019;40(5):342–357. doi:10.1016/j.tips.2019.03.003
147. Smith ER, Wang JQ, Yang DH, Xu XX. Paclitaxel resistance related to nuclear envelope structural sturdiness. Drug Resist Updat Rev Comment Antimicrob Anticancer Chemother. 2022;65:100881. doi:10.1016/j.drup.2022.100881
148. Shukla R, Singh A, Singh KK. Vincristine-based nanoformulations: a preclinical and clinical studies overview. Drug Deliv Transl Res. 2024;14(1):1–16. doi:10.1007/s13346-023-01389-6
149. Zhang J, Li C, Xue Q, et al. An efficient carbon-based drug delivery system for cancer therapy through the nucleus targeting and mitochondria mediated apoptotic pathway. Small Methods. 2021;5(12):e2100539. doi:10.1002/smtd.202100539
150. Liu Y, Zhang X, Zhou M, Nan X, Chen X, Zhang X. Mitochondrial-targeting lonidamine-doxorubicin nanoparticles for synergistic chemotherapy to conquer drug resistance. ACS Appl Mater Interfaces. 2017;9(50):43498–43507. doi:10.1021/acsami.7b14577
151. Wu L, Shi Y, Ni Z, Yu T, Chen Z. Preparation of a self-assembled rhein–doxorubicin nanogel targeting mitochondria and investigation on its antihepatoma activity. Mol Pharm. 2022;19(1):35–50. doi:10.1021/acs.molpharmaceut.1c00565
152. Ji Y, Shan S, He M, Chu CC. A novel pseudo-protein-based biodegradable nanomicellar platform for the delivery of anticancer drugs. Small Weinh Bergstr Ger. 2017;13(1). doi:10.1002/smll.201601491
153. Liu Y, Zhou Z, Lin X, et al. Enhanced reactive oxygen species generation by mitochondria targeting of anticancer drug to overcome tumor multidrug resistance. Biomacromolecules. 2019;20(10):3755–3766. doi:10.1021/acs.biomac.9b00800
154. Tao J, Diao L, Chen F, et al. pH-sensitive nanoparticles codelivering docetaxel and dihydroartemisinin effectively treat breast cancer by enhancing reactive oxidative species-mediated mitochondrial apoptosis. Mol Pharm. 2021;18(1):74–86. doi:10.1021/acs.molpharmaceut.0c00432
155. Cheng Y, Ji Y, Tong J. Triple stimuli-responsive supramolecular nanoassembly with mitochondrial targetability for chemophotothermal therapy. J Control Release. 2020;327:35–49. doi:10.1016/j.jconrel.2020.08.006
156. Xie J, Wang H, Huang Q, et al. Enhanced cytotoxicity to lung cancer cells by mitochondrial delivery of camptothecin. Eur J Pharm Sci. 2023;189:106561. doi:10.1016/j.ejps.2023.106561
157. Wang Q, Zhang C, Zhao Y, et al. Polyprodrug nanomedicine for chemiexcitation-triggered self-augmented cancer chemotherapy and gas therapy. Biomaterials. 2024;309:122606. doi:10.1016/j.biomaterials.2024.122606
158. Laprise-Pelletier M, Simão T, Fortin MA. Gold nanoparticles in radiotherapy and recent progress in nanobrachytherapy. Adv Healthc Mater. 2018;7(16):e1701460. doi:10.1002/adhm.201701460
159. Chen M, Song J, Zhu J, et al. A dual-nanozyme-catalyzed cascade reactor for enhanced photodynamic oncotherapy against tumor hypoxia. Adv Healthc Mater. 2021;10(21):e2101049. doi:10.1002/adhm.202101049
160. Comotti M, Della Pina C, Matarrese R, Rossi M. The catalytic activity of “naked” gold particles. Angew Chem Int Ed Engl. 2004;43(43):5812–5815. doi:10.1002/anie.200460446
161. Deng W, McKelvey KJ, Guller A, et al. Application of mitochondrially targeted nanoconstructs to neoadjuvant x-ray-induced photodynamic therapy for rectal cancer. ACS Cent Sci. 2020;6(5):715. doi:10.1021/acscentsci.9b01121
162. Zhao Y, Feng Y, Li J, et al. Endogenous ROS-mediated covalent immobilization of gold nanoparticles in mitochondria: a “sharp sword” in tumor radiotherapy. ACS Chem Biol. 2022;17(8):2355–2365. doi:10.1021/acschembio.2c00475
163. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106. doi:10.1038/s41423-020-00630-3
164. Yan J, Wan P, Choksi S, Liu ZG. Necroptosis and tumor progression. Trends Cancer. 2021;8(1):21. doi:10.1016/j.trecan.2021.09.003
165. Zhang T, Wang Y, Inuzuka H, Wei W. Necroptosis pathways in tumorigenesis. Semin Cancer Biol. 2022;86(Pt 3):32. doi:10.1016/j.semcancer.2022.07.007
166. Yang Z, Wang Y, Zhang Y, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol. 2018;20(2):186–197. doi:10.1038/s41556-017-0022-y
167. Shahsavari Z, Karami-Tehrani F, Salami S. Shikonin induced necroptosis via reactive oxygen species in the T-47D breast cancer cell line. Asian Pac J Cancer Prev. 2015;16(16):7261–7266. doi:10.7314/APJCP.2015.16.16.7261
168. Choi ES, Kim S, Kim D, Choi E, Ryu JH. Drug-loaded nanogel for efficient orchestration of cell death pathways by intramitochondrial disulfide polymerization. Small Weinh Bergstr Ger. 2024;20(15):e2308872. doi:10.1002/smll.202308872
169. Jangili P, Kong N, Kim JH, et al. DNA-damage-response-targeting mitochondria-activated multifunctional prodrug strategy for self-defensive tumor therapy. Angew Chem Int Ed. 2022;61(16):e202117075. doi:10.1002/anie.202117075
170. Gao WJ, Wang MM, Su Y, Yu ZH, Liu HK, Su Z. Self-assembly mitochondria-targeting donor–acceptor type theranostic nanosphere activates ROS storm for multimodal cancer therapy. ACS Appl Bio Mater. 2023;6(2):722–732. doi:10.1021/acsabm.2c00942
171. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369. doi:10.1038/cdd.2015.158
172. Yao X, Li W, Fang D, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci. 2021;8(22):2100997. doi:10.1002/advs.202100997
173. Jin L, Chun J, Pan C, et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell. 2018;69(1):87–99.e7. doi:10.1016/j.molcel.2017.11.025
174. Ren J, Zhou J, Liu H, et al. Ultrasound (US)-activated redox dyshomeostasis therapy reinforced by immunogenic cell death (ICD) through a mitochondrial targeting liposomal nanosystem. Theranostics. 2021;11(19):9470. doi:10.7150/thno.62984
175. Henning Y, Blind US, Larafa S, Matschke J, Fandrey J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022;13(7):662. doi:10.1038/s41419-022-05121-z
176. Cai S, Ding Z, Liu X, Zeng J. Trabectedin induces ferroptosis via regulation of HIF-1α/IRP1/TFR1 and Keap1/Nrf2/GPX4 axis in non-small cell lung cancer cells. Chem Biol Interact. 2023;369:110262. doi:10.1016/j.cbi.2022.110262
177. Su X, Xie Y, Zhang J, et al. HIF-α activation by the prolyl hydroxylase inhibitor roxadustat suppresses chemoresistant glioblastoma growth by inducing ferroptosis. Cell Death Dis. 2022;13(10):861. doi:10.1038/s41419-022-05304-8
178. Tian H, Zhou L, Wang Y, Nice EC, Huang C, Zhang H. A targeted nanomodulator capable of manipulating tumor microenvironment against metastasis. J Control Release. 2022;348:590–600. doi:10.1016/j.jconrel.2022.06.022
179. Zhuang J, Ma Z, Li N, et al. Molecular engineering of plasma membrane and mitochondria dual-targeted NIR-II AIE photosensitizer evoking synergetic pyroptosis and apoptosis. Adv Mater. 2024;36(5):2309488. doi:10.1002/adma.202309488
180. Yang Y, Yang J, Zhu N, et al. Tumor-targeting hydroxyapatite nanoparticles for remodeling tumor immune microenvironment (TIME) by activating mitoDNA-pyroptosis pathway in cancer. J Nanobiotechnology. 2023;21(1):470. doi:10.1186/s12951-023-02231-4
181. Cheng Q, Zhang T, Wang Q, et al. Photocatalytic carbon dots-triggered pyroptosis for whole cancer cell vaccines. Adv Mater Deerfield Beach Fla. 2024;36(39):e2408685. doi:10.1002/adma.202408685
182. Peng Y, Mo R, Yang M, et al. Mitochondria-targeting AIEgens as pyroptosis inducers for boosting type-I photodynamic therapy of tongue squamous cell carcinoma. ACS Nano. 2024;18(38):26140–26152. doi:10.1021/acsnano.4c06808
183. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
184. Mao C, Wang M, Zhuang L, Gan B. Metabolic cell death in cancer: ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 2024;15(9):642. doi:10.1093/procel/pwae003
185. Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7:378. doi:10.1038/s41392-022-01229-y
186. Xu W, Suo A, Aldai AJM, et al. Hollow calcium/copper bimetallic amplifier for cuproptosis/paraptosis/apoptosis cancer therapy via cascade reinforcement of endoplasmic reticulum stress and mitochondrial dysfunction. ACS Nano. 2024. doi:10.1021/acsnano.4c11455
187. Tumor microenvironment responsive hollow nanoplatform for triple amplification of oxidative stress to enhance cuproptosis‐based synergistic cancer therapy. doi:10.1002/adhm.202202949.
188. Huang L, Zhu J, Wu G, et al. A strategy of “adding fuel to the flames” enables a self-accelerating cycle of ferroptosis-cuproptosis for potent antitumor therapy. Biomaterials. 2024;311:122701. doi:10.1016/j.biomaterials.2024.122701
189. Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6(221):221ra15. doi:10.1126/scitranslmed.3007653
190. Wiel C, Gal KL, Ibrahim MX, et al. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell. 2019;178(2):330–345.e22. doi:10.1016/j.cell.2019.06.005
191. Le Gal K, Ibrahim MX, Wiel C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7(308):308re8. doi:10.1126/scitranslmed.aad3740
192. Kuo CL, Ponneri Babuharisankar A, Lin YC, et al. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: foe or friend? J Biomed Sci. 2022;29(1):74. doi:10.1186/s12929-022-00859-2
193. Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217(7):2291–2298. doi:10.1083/jcb.201804161
194. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012;2012(3):CD007176. doi:10.1002/14651858.CD007176.pub2
195. Wheaton WW, Weinberg SE, Hamanaka RB, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242. doi:10.7554/eLife.02242
196. Schexnayder C, Broussard K, Onuaguluchi D, et al. Metformin inhibits migration and invasion by suppressing ROS production and COX2 expression in MDA-MB-231 breast cancer cells. Int J Mol Sci. 2018;19(11):3692. doi:10.3390/ijms19113692
197. Kwon Y. Possible beneficial effects of N-acetylcysteine for treatment of triple-negative breast cancer. Antioxidants. 2021;10(2):169. doi:10.3390/antiox10020169
198. Nargi JL, Ratan RR, Griffin DE. p53-independent inhibition of proliferation and p21Waf1/Cip1-modulated induction of cell death by the antioxidants N-acetylcysteine and vitamin E. Neoplasia N Y N. 1999;1(6):544. doi:10.1038/sj.neo.7900068
199. Liu Y. Protective effects of curcumin and quercetin during benzo(a)pyrene induced lung carcinogenesis in mice.
200. Ghosh D, Choudhury ST, Ghosh S, et al. Nanocapsulated curcumin: oral chemopreventive formulation against diethylnitrosamine induced hepatocellular carcinoma in rat. Chem Biol Interact. 2012;195(3):206–214. doi:10.1016/j.cbi.2011.12.004
201. Du Y, Long Q, Zhang L, et al. Curcumin inhibits cancer-associated fibroblast-driven prostate cancer invasion through MAOA/mTOR/HIF-1α signaling. Int J Oncol. 2015;47(6):2064–2072. doi:10.3892/ijo.2015.3202
202. Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263(3):709–716. doi:10.1046/j.1432-1327.1999.00543.x
203. Sun C, Liu X, Di C, et al. MitoQ regulates autophagy by inducing a pseudo-mitochondrial membrane potential. Autophagy. 2017;13(4):730. doi:10.1080/15548627.2017.1280219
204. Ganguly E, Kirschenman R, Spaans F, et al. Nanoparticle-encapsulated antioxidant improves placental mitochondrial function in a sexually dimorphic manner in a rat model of prenatal hypoxia. FASEB J off Publ Fed Am Soc Exp Biol. 2021;35(2):e21338. doi:10.1096/fj.202002193R
205. Yong JM, Fu L, Tang F, et al. ROS-mediated anti-angiogenic activity of cerium oxide nanoparticles in melanoma cells. ACS Biomater Sci Eng. 2022;8(2):512–525. doi:10.1021/acsbiomaterials.1c01268
206. Vassie JA, Whitelock JM, Lord MS. Endocytosis of cerium oxide nanoparticles and modulation of reactive oxygen species in human ovarian and colon cancer cells. Acta Biomater. 2017;50:127–141. doi:10.1016/j.actbio.2016.12.010
207. Kwiatkowski S, Knap B, Przystupski D, et al. Photodynamic therapy – mechanisms, photosensitizers and combinations. Biomed Pharmacother. 2018;106:1098–1107. doi:10.1016/j.biopha.2018.07.049
208. Thakur NS, Patel G, Kushwah V, Jain S, Banerjee UC. Facile development of biodegradable polymer-based nanotheranostics: hydrophobic photosensitizers delivery, fluorescence imaging and photodynamic therapy. J Photochem Photobiol B. 2019;193:39–50. doi:10.1016/j.jphotobiol.2019.02.007
209. Yang S, Wu GL, Li N, et al. A mitochondria-targeted molecular phototheranostic platform for NIR-II imaging-guided synergistic photothermal/photodynamic/immune therapy. J Nanobiotechnology. 2022;20(1):475. doi:10.1186/s12951-022-01679-0
210. Heier SK, Rothman KA, Heier LM, Rosenthal WS. Photodynamic therapy for obstructing esophageal cancer: light dosimetry and randomized comparison with Nd:YAG laser therapy. Gastroenterology. 1995;109(1):63–72. doi:10.1016/0016-5085(95)90269-4
211. Itoh Y, Ninomiya Y, Henta T, Tajima S, Ishibashi A. Topical δ-aminolevulinic acid-based photodynamic therapy for Japanese actinic keratoses. J Dermatol. 2000;27(8):513–518. doi:10.1111/j.1346-8138.2000.tb02218.x
212. Brooke RCC, Sinha A, Sidhu MK, et al. Histamine is released following aminolevulinic acid-photodynamic therapy of human skin and mediates an aminolevulinic acid Dose-Related Immediate inflammatory response. J Invest Dermatol. 2006;126(10):2296–2301. doi:10.1038/sj.jid.5700449
213. Alkan M, Oktay M, Kocakerim MM, Çopur M. Solubility of chlorine in aqueous hydrochloric acid solutions. J Hazard Mater. 2005;119(1):13–18. doi:10.1016/j.jhazmat.2004.11.001
214. Wang DP, Zheng J, Jiang FY, et al. Facile and green fabrication of tumor- and mitochondria-targeted AIEgen-protein nanoparticles for imaging-guided photodynamic cancer therapy. Acta Biomater. 2023;168:551–564. doi:10.1016/j.actbio.2023.06.048
215. A mitochondria‐targeted photosensitizer for combined pyroptosis and apoptosis with NIR‐II imaging/photoacoustic imaging‐guided phototherapy. doi:10.1002/anie.202408874.
216. Juengpanich S, Li S, Yang T, et al. Pre-activated nanoparticles with persistent luminescence for deep tumor photodynamic therapy in gallbladder cancer. Nat Commun. 2023;14:5699. doi:10.1038/s41467-023-41389-1
217. Liang J, Li L, Tian H, et al. Drug repurposing-based brain-targeting self-assembly nanoplatform using enhanced ferroptosis against glioblastoma. Small. 2023;19(46):2303073. doi:10.1002/smll.202303073
218. Liu K, Liao Y, Zhou Z, et al. Photothermal-triggered immunogenic nanotherapeutics for optimizing osteosarcoma therapy by synergizing innate and adaptive immunity. Biomaterials. 2022;282:121383. doi:10.1016/j.biomaterials.2022.121383
219. Cui M, Tang D, Wang B, Zhang H, Liang G, Xiao H. Bioorthogonal guided activation of cGAS-STING by AIE photosensitizer nanoparticles for targeted tumor therapy and imaging. Adv Mater. 2023;35(52):2305668. doi:10.1002/adma.202305668
220. Zhao X, Cheng H, Wang Q, et al. Regulating photosensitizer metabolism with DNAzyme-loaded nanoparticles for amplified mitochondria-targeting photodynamic immunotherapy. ACS Nano. 2023;17(14):13746–13759. doi:10.1021/acsnano.3c03308
221. Holder AM, Dedeilia A, Sierra-Davidson K, et al. Defining clinically useful biomarkers of immune checkpoint inhibitors in solid tumours. Nat Rev Cancer. 2024;24(7):498–512. doi:10.1038/s41568-024-00705-7
222. Trujillo-Alonso V, Pratt EC, Zong H, et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat Nanotechnol. 2019;14(6):616. doi:10.1038/s41565-019-0406-1
223. Zaimy MA, Saffarzadeh N, Mohammadi A, et al. New methods in the diagnosis of cancer and gene therapy of cancer based on nanoparticles. Cancer Gene Ther. 2017;24(6):233–243. doi:10.1038/cgt.2017.16
224. Zhang J, Hu Y, Yang J, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. 2022;609(7926):369–374. doi:10.1038/s41586-022-05140-y
225. Nawaz W, Huang B, Xu S, et al. AAV-mediated in vivo CAR gene therapy for targeting human T-cell leukemia. Blood Cancer J. 2021;11(6):119. doi:10.1038/s41408-021-00508-1
226. Jitschin R, Hofmann AD, Bruns H, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014;123(17):2663–2672. doi:10.1182/blood-2013-10-532200
227. Mattes K, Vellenga E, Schepers H. Differential redox-regulation and mitochondrial dynamics in normal and leukemic hematopoietic stem cells: a potential window for leukemia therapy. Crit Rev Oncol Hematol. 2019;144:102814. doi:10.1016/j.critrevonc.2019.102814
228. Buccarelli M, D’Alessandris QG, Matarrese P, et al. Elesclomol-induced increase of mitochondrial reactive oxygen species impairs glioblastoma stem-like cell survival and tumor growth. J Exp Clin Cancer Res CR. 2021;40:228. doi:10.1186/s13046-021-02031-4
229. Wang C, Shao L, Pan C, et al. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res Ther. 2019;10:175. doi:10.1186/s13287-019-1265-2
230. Mantovani G, Macciò A, Madeddu C, et al. The impact of different antioxidant agents alone or in combination on reactive oxygen species, antioxidant enzymes and cytokines in a series of advanced cancer patients at different sites: correlation with disease progression. Free Radic Res. 2003;37(2):213–223. doi:10.1080/10715760303849
231. Liu Y, Yuang G, Chen X, Liu J. Near-infrared band responsive ROS regulator selectively inhibits breast cancer cells by programming combination phototherapy. J Mater Chem B. 2023;11(6):1356–1364. doi:10.1039/D2TB02508F
232. Liu ITT, Kesselheim AS, Cliff ERS. Clinical benefit and regulatory outcomes of cancer drugs receiving accelerated approval. JAMA. 2024;331(17):1471–1479. doi:10.1001/jama.2024.2396
233. Commissioner O of the FDA approves first injectable treatment for HIV pre-exposure prevention. FDA. 2024. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-injectable-treatment-hiv-pre-exposure-prevention.
234. Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66(6):3271–3277. doi:10.1158/0008-5472.CAN-05-4007
235. Krauss AC, Gao X, Li L, et al. FDA approval summary: (daunorubicin and cytarabine) liposome for injection for the treatment of adults with high-risk acute myeloid leukemia. Clin Cancer Res off J Am Assoc Cancer Res. 2019;25(9):2685–2690. doi:10.1158/1078-0432.CCR-18-2990
236. Adynovate. Safety and Utilization Review; 2019.
237. Leucht S, Crippa A, Siafis S, Patel MX, Orsini N, Davis JM. Dose-response meta-analysis of antipsychotic drugs for acute schizophrenia. Am J Psychiatry. 2020;177(4):342–353. doi:10.1176/appi.ajp.2019.19010034
238. Boey A, Ho HK. All roads lead to the liver: metal nanoparticles and their implications for liver health. Small Weinh Bergstr Ger. 2020;16(21):e2000153. doi:10.1002/smll.202000153
239. Shehata AM, Salem FMS, El-Saied EM, El-Rahman SS A, Mahmoud MY, Noshy PA. Evaluation of the ameliorative effect of zinc nanoparticles against silver nanoparticle–induced toxicity in liver and kidney of rats. Biol Trace Elem Res. 2022;200(3):1201–1211. doi:10.1007/s12011-021-02713-2
240. Maurer-Jones MA, Christenson JR, Haynes CL. TiO2 nanoparticle-induced ROS correlates with modulated immune cell function. J Nanopart Res. 2012;14(12):1291. doi:10.1007/s11051-012-1291-9
241. Hu D, Huang Z, Li W, et al. Macrophage membrane-cloaked ros-responsive albumin nanoplatforms for targeted delivery of curcumin to alleviate acute liver injury. Mol Pharm. 2025;22(2):771–786. doi:10.1021/acs.molpharmaceut.4c00808
242. Xiao J, Cao H, Guo S, et al. Long-term administration of low-dose selenium nanoparticles with different sizes aggravated atherosclerotic lesions and exhibited toxicity in apolipoprotein E-deficient mice. Chem Biol Interact. 2021;347:109601. doi:10.1016/j.cbi.2021.109601
243. Xu W, Zhou M, Guo Z, et al. Impact of macroporous silica nanoparticles at sub-50nm on bio-behaviors and biosafety in drug-resistant cancer models. Colloids Surf B Biointerfaces. 2021;206:111912. doi:10.1016/j.colsurfb.2021.111912
244. Bajpai A, Desai NN, Pandey S, Shukla C, Datta B, Basu S. Nanoparticle-mediated routing of antibiotics into mitochondria in cancer cells. ACS Appl Bio Mater. 2021. doi:10.1021/acsabm.1c00527
245. Han Y, Dong J, Zhang L, et al. Biocompatible and size-dependent melanin-like nanocapsules for efficient therapy in hyperoxia-induced acute lung injury. Biomaterials. 2025;318:123169. doi:10.1016/j.biomaterials.2025.123169
246. Yang Z, Ren X, Li L, et al. Trojan-horse inspired nanoblaster: x-ray triggered spot attack on radio-resistant cancer through radiodynamic therapy. Biomaterials. 2025;313:122814. doi:10.1016/j.biomaterials.2024.122814
247. Arms L, Smith DW, Flynn J, et al. Advantages and limitations of current techniques for analyzing the biodistribution of nanoparticles. Front Pharmacol. 2018;9:802. doi:10.3389/fphar.2018.00802
248. Kamel AG, Sabet S, El-Shibiny A. Potential mitochondrial ROS-mediated damage induced by chitosan nanoparticles bee venom-loaded on cancer cell lines. Int J Biol Macromol. 2024;279(Pt 4):135362. doi:10.1016/j.ijbiomac.2024.135362
249. Banstola A, Poudel K, Pathak S, et al. Hypoxia-mediated ROS amplification triggers mitochondria-mediated apoptotic cell death via PD-L1/ROS-responsive, dual-targeted, Drug-Laden Thioketal Nanoparticles. ACS Appl Mater Interfaces. 2021;13(19):22955–22969. doi:10.1021/acsami.1c03594
250. Healy S, Henderson ET, Ke S, et al. Spatial analysis of nanoparticle distribution in human breast xenografts reveals nanoparticles targeted to cancer cells localized with tumor-associated stromal cells. Nanotheranostics. 2023;7(4):393–411. doi:10.7150/ntno.84255
251. Johnson K K, Kopecky C, Koshy P, et al. Theranostic activity of ceria-based nanoparticles toward parental and metastatic melanoma: 2D vs 3D models. ACS Biomater Sci Eng. 2023;9(2):1053–1065. doi:10.1021/acsbiomaterials.2c01258
252. Fang J, Islam W, Maeda H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv Drug Deliv Rev. 2020;157:142–160. doi:10.1016/j.addr.2020.06.005
253. Robbins D, Zhao Y. Manganese superoxide dismutase in cancer prevention. Antioxid Redox Signal. 2014;20(10):1628. doi:10.1089/ars.2013.5297
254. Jomova K, Alomar SY, Alwasel SH, Nepovimova E, Kuca K, Valko M. Several lines of antioxidant defense against oxidative stress: antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch Toxicol. 2024;98(5):1323–1367. doi:10.1007/s00204-024-03696-4
255. An X, Yu W, Liu J, Tang D, Yang L, Chen X. Oxidative cell death in cancer: mechanisms and therapeutic opportunities. Cell Death Dis. 2024;15(8):556. doi:10.1038/s41419-024-06939-5
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Multifunctional Nanoparticles Codelivering Doxorubicin and Amorphous Calcium Carbonate Preloaded with Indocyanine Green for Enhanced Chemo-Photothermal Cancer Therapy

Yu J, Wang L, Xie X, Zhu W, Lei Z, Lv L, Yu H, Xu J, Ren J
International Journal of Nanomedicine 2023, 18:323-337
Published Date: 18 January 2023