")
Back to Journals » International Journal of Nanomedicine » Volume 19
Nanodelivery Optimization of IDO1 Inhibitors in Tumor Immunotherapy: Challenges and Strategies
Authors Jiang K, Wang Q , Chen XL, Wang X, Gu X, Feng S, Wu J , Shang H, Ba X, Zhang Y , Tang K
Received 30 January 2024
Accepted for publication 13 July 2024
Published 28 August 2024 Volume 2024:19 Pages 8847—8882
DOI https://doi.org/10.2147/IJN.S458086
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Kehua Jiang,1 Qing Wang,1 Xiao-Long Chen,1 Xiaodong Wang,1 Xiaoya Gu,1 Shuangshuang Feng,1 Jian Wu,2 Haojie Shang,2 Xiaozhuo Ba,2 Yanlong Zhang,1 Kun Tang2
1Department of Urology, Guizhou Provincial People’s Hospital, Guiyang, Guizhou, People’s Republic of China; 2Department of Urology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
Correspondence: Yanlong Zhang, Department of Urology, Guizhou Provincial People’s Hospital, Guiyang, Guizhou, People’s Republic of China, Email [email protected] Kun Tang, Department of Urology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1095, Jiefang Avenue, Qiaokou District, Wuhan, 430030, People’s Republic of China, Email [email protected]
Abstract: Tryptophan (Trp) metabolism plays a vital role in cancer immunity. Indoleamine 2.3-dioxygenase 1 (IDO1), is a crucial enzyme in the metabolic pathway by which Trp is degraded to kynurenine (Kyn). IDO1-mediated Trp metabolites can inhibit tumor immunity and facilitate immune evasion by cancer cells; thus, targeting IDO1 is a potential tumor immunotherapy strategy. Recently, numerous IDO1 inhibitors have been introduced into clinical trials as immunotherapeutic agents for cancer treatment. However, drawbacks such as low oral bioavailability, slow onset of action, and high toxicity are associated with these drugs. With the continuous development of nanotechnology, medicine is gradually entering an era of precision healthcare. Nanodrugs carried by inorganic, lipid, and polymer nanoparticles (NPs) have shown great potential for tumor therapy, providing new ways to overcome tumor diversity and improve therapeutic efficacy. Compared to traditional drugs, nanomedicines offer numerous significant advantages, including a prolonged half-life, low toxicity, targeted delivery, and responsive release. Moreover, based on the physicochemical properties of these nanomaterials (eg, photothermal, ultrasonic response, and chemocatalytic properties), various combination therapeutic strategies have been developed to synergize the effects of IDO1 inhibitors and enhance their anticancer efficacy. This review is an overview of the mechanism by which the Trp-IDO1-Kyn pathway acts in tumor immune escape. The classification of IDO1 inhibitors, their clinical applications, and barriers for translational development are discussed, the use of IDO1 inhibitor-based nanodrug delivery systems as combination therapy strategies is summarized, and the issues faced in their clinical application are elucidated. We expect that this review will provide guidance for the development of IDO1 inhibitor-based nanoparticle nanomedicines that can overcome the limitations of current treatments, improve the efficacy of cancer immunotherapy, and lead to new breakthroughs in the field of cancer immunotherapy.
Keywords: indoleamine 2, 3-dioxygenase 1, IDO1, IDO1 inhibitors, nanodrugs, nanodelivery, cancer immunotherapy, combination therapy
Introduction
Cancer, as a highly heterogeneous and complex systemic disease,1 has continuously increased in its global incidence and mortality rates in recent years. The relatively weak immunogenicity of cancer endows it with a “protective shield” during development, allowing it to evade recognition and attack by the immune system.2,3 The conventional treatment modalities for tumors; surgery, radiotherapy, and chemotherapy, are associated with several drawbacks: surgery may lead to trauma and the risk of recurrence, radiotherapy may induce severe toxic side effects and tolerance issues, and chemotherapy often results in adverse reaction events and drug resistance. In addition, these treatments have limited effectiveness in treating patients with advanced metastatic cancer. These factors render cancer one of the most challenging and difficult-to-cure diseases.
The emergence of cancer immunotherapy has brought new hope for cancer cure. This type of therapy can activate and train both the intrinsic and adaptive immune systems to recognize and remove cancer cells while building a longer-lasting anti-tumor immune memory.4 Compared with traditional cancer treatment strategies, cancer immunotherapy not only shows lower toxicity and fewer side effects, but also reduces the physical burden of patients. In addition, personalized treatment plans can be designed based on factors such as the patient’s immune status and tumor type, especially in advanced cancer patients for whom conventional treatment may not achieve the expected results. Cancer immunotherapy offers new treatment options for such patients.5 Several immunotherapy treatments have been clinically validated as effective against cancer, including the immune checkpoint blockade (ICB),6,7 chimeric antigen receptor T cell therapy,8–10 adoptive cell therapy,11,12 monoclonal antibodies,13 and tumor vaccines.14,15 Particularly significant progress has been made in the development of immune checkpoint blockers, such as PD-1/PD-L1 and CTLA-4, in recent decades, initiating a wave of tumor immunotherapy. However, it must be recognized that the therapeutic effects of these drugs are only effective in patients with clinically positive immune checkpoints. Therefore, it is necessary to search for a wider range of therapeutic targets or combination therapy strategies to improve the clinical efficacy and applicability.
Considering the physiological functions of proteins in the body, basic amino acids units play a critical role in tumor progression. Elevated tryptophan (Trp) catabolism is a common hallmark of the tumor microenvironment (TME) in the clinical manifestation of cancer.16 Therefore, targeting the rate-limiting enzyme Indoleamine 2.3-dioxygenase 1 (IDO1) for Trp degradation to modulate the TME and inhibit tumor progression is a highly promising therapeutic strategy. Although conventional IDO1 inhibitors have demonstrated significant anti-tumor efficacy in clinical practice, disadvantages such as low drug availability, obvious toxicity, and drug resistance are associated with each. Nanotechnology has become pivotal in addressing these challenges. Ideal nanoparticles (NPs) generally possess the following characteristics: (1) biocompatibility and low toxicity; (2) long circulation half-life; (3) stimuli-responsive release; (4) extended circulation of drugs in vivo; (5) targeting of the lesion site; and (6) the ability to deliver one or more reagents.17,18 In addition, it is worth noting that the vast majority of NPs have unique physicochemical properties, such as photothermal, sonication, and chemocatalytic properties. Based on these properties, several combination therapeutic strategies have been developed, including IDO1 inhibitor-based photodynamic therapy, photothermal therapy, and sonodynamic therapy. These therapeutic approaches have not only enhanced anti-cancer efficacy but also reduced the occurrence of drug resistance. In conclusion, the application of nanotechnology has led to new breakthroughs in immunotherapy for cancer. Here, we provide an overview of the biological function of IDO1 in tumor immunity, summarize the classification of IDO1 inhibitors and the latest clinical trial progress, and discuss the delivery barriers of IDO1 inhibitors and the application of IDO1 inhibitor-based NPs in cancer immunotherapy (Figure 1). Finally, the specific challenges of nanomedicine for future clinical applications are briefly discussed.
 |
Figure 1 Outline of this review on IDO1 inhibition for cancer immunotherapy. This review focuses on the role of IDO1 inhibitor nanodelivery in tumor immunotherapy and is divided into two aspects: synthesis and therapy. The synthesis part first categorizes IDO1 inhibitors and introduces the types of IDO1 inhibitor-based nanocarriers as well as target modification strategies. In the therapeutic aspect, the biological functions of IDO1 and its regulation of immune cells are mainly described. In addition, the applications of IDO1 inhibitors in the treatment of different types of tumors are enumerated, and the combined therapeutic strategies of IDO1 inhibitor-based nanoparticles are introduced. |
Biological Function of IDO1 in Tumor Immunity
The enzyme IDO1 enzyme is a key enzyme in Trp metabolism, and is also one of the main factors triggering abnormal Trp metabolism at tumor sites. Its metabolites have a significant impact in regulating tumor growth and shaping the tumor immune microenvironment. This section focuses on the biological functions of IDO1 and its role in tumor immune evasion.
Tryptophan Metabolism
Trp plays a crucial role in maintaining normal physiological activities, and both Trp and its metabolites can regulate cellular processes and coordinate the body in response to the environment. Trp has been found widely involved in the development and occurrence of diseases of the nervous, digestive, and immune systems.19 Trp metabolism primarily exerts its effects through three pathways: (1) the serotonin (5-HT) pathway that is catalyzed by tryptophan hydroxylase 1; (2) The Trp-Kynurenine (Kyn) pathway that is mediated by the rate-limiting enzymes IDO1, IDO2, and tryptophan 2.3-dioxygenase (TDO) 2; (3) the production pathway of indole-3-pyruvate(I3P), which is mediated by interleukin-4-induced-1.20
In the brain, Trp is primarily involved in regulating the central nervous system (CNS) through the 5-HT pathway, which regulates alterations in mood, anxiety, and cognition, and is an important neurotransmitter. Downstream metabolites of 5-HT have been shown to regulate circadian rhythmicity in animals.21 The metabolites of the Trp-IDO1-Kyn pathway; Kyn, anthranilic acid, and xanthurenic acid, can easily pass through the blood-brain barrier (BBB) easily and play a crucial role in regulating the CNS.22 Moreover, some of these compounds are excitotoxic to neurons and can lead to neuropathy. For example, the accumulation of quinolinic acid (QA) from Kyn that is catalyzed by kynurenine monooxygenase is associated with depressive disorders, schizophrenia, and neurodegenerative diseases such as Alzheimer’s disease.19,23 While the product nicotinamide adenine dinucleotide is further catalyzed by QA and the end product of the IDO1 pathway plays an important role in intracellular energy metabolism, it can influence numerous crucial cellular processes such as the metabolic pathways and immune cell function.24–26 The I3P pathway plays an immunomodulatory role by mediating aryl hydrocarbon receptor (AHR) activity, the activation of which recruits MDSCs and increases the infiltration of regulatory T cells (Tregs) infiltration into tumors, thus promoting tumor progress.27,28
IDO1 Mediation of Tumor Immune Escape
The Trp-IDO1-Kyn pathway is the main pathway for Trp degradation (accounting for more than 95%).29 Most Trp that is ingested by the body is metabolized into bioactive compounds such as Kyn through the Trp-IDO1-Kyn pathway, which is involved in inflammation, the immune response, and excitation; with only a small fraction used in anabolism.30 The activation and proliferation of T cells for tumor immunity are dependent on the essential amino acid Trp, which acts as a key enzyme in the initial stages of Trp degradation, catalyzing the degradation of Trp into Kyn. The two associated isoenzymes, TDO and IDO2, TDO is mainly expressed in liver tissue to maintain Trp homeostasis, while IDO2 is weakly expressed in extrahepatic tissues and some immune cells.31 Although IDO1 possesses broad substrate specificity compared to TDO and IDO2, it can catalyze L-Trp, D-Trp, and various indoleamine derivatives.32 IDO1 is mainly expressed in extrahepatic tissues, including the placenta, eye, brain, mucosa, and some immune cell subsets (eg, eosinophils, macrophage cells, and dendritic cells). In general, its expression in these tissues and cells is minimal. However, it can be significantly expressed in tissues and cells that are infected with pathogens or encounter inflammation,33 which may be related to its biological function. Early studies in mice demonstrated that IDO1 can regulate the maternal immune system during pregnancy and plays an important role during fetal development, leading to T cell dysfunction and protecting the fetus from the inflammatory response caused by xenoantigens.34,35 Dysfunction of the Kyn pathway, which is mediated by IDO1 and caused by inflammatory injury, can results in diseases of the CNS.36 Furthermore, reports have indicated high IDO1 expression in melanoma,37 triple-negative breast cancer,38 gastric cancer,39 and colorectal cancer.40 As shown in Figure 2, IDO1 is up-regulated by various signaling factors (eg, prostaglandin E2 (PGE2), IFN-γ, IL-6, and TGF-β) through one or more pathways in inflammatory and tumor tissues.33,41 Cyclooxygenase 2 (COX-2) and PGE2, which are dependent on MAPK signaling, can upregulate IDO1 expression via the PKC and PI3K pathways.42 In a study into pain and depression comorbidities, Mao et al found that IL-6 can induce IDO1 expression through the JAK/STAT pathway,43 while IL-6 can stimulate the upregulation of MDSCs via the STAT3/NF-κB pathway in breast cancer.44 INF-γ efficiently upregulates IDO1 by activating elements and the site within the IDO1 promoter region,33 while IFN-γ-JAK1-STAT1 signaling is activated by MUC1-C, driving the immunosuppressive IDO1 gene and leading to the dysfunction of CD8+ T cells. Thus, MUC1-1 plays a major role in tumor immune evasion.45 Other cytokines such as IFNα, TNF-α, IFNβ, PAMPs/DAMPs, some carcinogens, and antigens may also stimulate IDO1 upregulation.33
 |
Figure 2 Mechanism of action of IDO1 in the tumor microenvironment. ①IDO1 expression is regulated by a variety of signaling pathways, such as STAT/NF-κB, PI3K/Akt and JAK/STAT. These signaling pathways are in turn affected by a variety of signaling molecules such as IL-6, PGE, and IFN. ②Overexpression of IDO1 leads to tryptophan deficiency, which in turn increases the number of Treg cells and decreases the number of Teff cells, while inhibiting antigen presentation and DC cell maturation. In addition, it was able to inhibit DC cell and T cell proliferation by suppressing the mTOR pathway. |
IDO1 overexpression mainly mediates immune tolerance in the TME that is induced by three downstream signaling effectors; the two environmental sensing proteins general control nonderepressible 2 (GCN2) and the mammalian target of rapamycin (mTOR), and the ligand-dependent AHR (Figure 3).29 GCN2 is an amino acid-sensitive kinase that comprises serine and threonine residues. The overexpression of IDO1 results in local depletion and low Trp levels, increasing the level of uncharged tRNA and activating GCN2 to initiate an integrated stress response (ISR). This leads to phosphorylation of the translation promoter eukaryotic initiation factor 2a, resulting in translation and cell cycle arrest.46,47 Moreover, the ISR that is triggered by GCN2 can render cancer cells resistant to hypoxia-induced apoptosis, helping them adapt to the hypoxic stress environment and acquire drug resistance.48 GCN2 has also been found involved in TAM and MDSCs activation, promoting tumor development in the TME.49 In addition to its immunosuppressive effects, IDO1 counteracts the anti-vascular effects of IFN γ in MDSCs via IL-6, which is produced by ISR to promote the development of tumors.50 Another protein kinase that belongs to the PI3K-related serine/threonine kinase family, mTOR, is involved in the formation of two different complexes, mTORC1 and mTORC2, which can sense and integrate different nutritional and environmental factors to regulate organism growth and homeostasis.51 GCN2 and mTOR are interrelated and work together to regulate cellular metabolism during both amino acid deficiency and abundant conditions. However, few studies have investigated the mechanism by which IDO1-mediated mTOR regulation occurs in the TME. The activation of mTORC1 generally occurs when Trp is abundant, inhibiting the binding of the eukaryotic translation initiation factor-binding protein 4E-BP1 to the translation initiation factor elF4E by phosphorylating the ribosomal protein S6K1 kinase.52 Activated S6K1 reduces cell survival, 4E-BP1 promotes angiogenesis and cell cycle progression, and elF4E has been shown to have anti-apoptotic and transformation effects in vitro.53 IDO1 can inhibit mTORC1 through the BTK-IDO1-mTORC1 axis, blocking the Trp-sensitive inflammatory signaling pathway, and limiting the differentiation of inflammatory DCs in the monocyte line.54 Increases in the number of IDO1 pathway metabolites (except kynurenic acid) promotes cellular proliferation and resistance to apoptosis by rapidly activating PI3K-AKT signaling in the tumor epithelium.55 Additionally, IDO1 contains two tyrosine-based immunoreceptor inhibitory motifs (ITIMs) (Figure 4A), which can bind to the p110 and SHP-1 subunits of PI3K to trigger immunosuppressive responses.56 AHR is a ligand-activated transcription factor that is responsive to a range of compounds, including Kyn, halogenated aromatic hydrocarbons, indole derivatives, and certain flavonoids, and is involved in cell cycle, cell migration, immune function, and other cellular processes.29,57 Downstream metabolites of the IDO1 pathway (including Kyn and its derivatives) are weak AHR agonists; however, IDO1 overexpression increases the concentration of Kyn in the TME, resulting in continuous AHR activation. In contrast, local Trp deprivation increases AHR expression, enhancing its sensitivity to weak agonists and effectively leading to the conversion of immunogenic DC to its tolerogenic form, increasing Treg cell differentiation.58,59 This leads to an increase in the ratio of M1/M2 macrophages,60 promoting cancer immune evasion. Furthermore, IDO1 overexpression can increase the intratumoral invasiveness and regulation of MDSCs, leading to T cell dysfunction and the differentiation of inhibitory Tregs,61,62 which may be associated with the development of tumors and poor prognoses.
 |
Figure 3 Mechanism of IDO1 in immunosuppression and immune escape in tumor environments. IDO1 mediates immune evasion of tumors mainly through downstream signaling effectors GCN2, mTOR and aromatic hydrocarbon receptors. This leads to an increase in Tregs and MDSCs, polarization of M1 to M2-type macrophages, and inhibition of DC cell maturation, as well as NK cell dysfunction. |
 |
Figure 4 Structures of IDO1 and IDO1 inhibitors. (A) crystal structure of the IDO1 (PDB code: 2d0t). IDO1 contains a smaller N-terminal domain that contains the ITIM site and a larger C-terminal domain that compromises the heme binding pocket. (B) the binding sites of IDO1-Indoximod/Navoximod/Linrodostat/Epacadostat. Classification of IDO1 inhibitors, (C) type I, Trp-competitive;(D) type II, targeting heme-IDO1; (E) type III, targeting apo-IDO1; (F) type IV, others. |
Classification of IDO1 Inhibitors
As mentioned previously, IDO1 is a metabolic heme-containing enzyme that is involved in the Trp-IDO1-Kyn pathway. The enzyme comprises two domains; a large C-terminal domain that contains the heme-binding pocket, and a small N-terminal domain (NTD) that includes the ITIM site (Figure 4A). Common IDO1 inhibitors target heme-IDO1; however, other inhibitors have been observed (Figure 4B), including the four main types:(a) Trp-competitive (Type I; eg, indoximod), (b) targeting heme-IDO1, (Type II; eg, epacadostat), (c) targeting Apo-IDO1, heme-competitive (Type III; eg, linrodostat), and (d) others.63–65 The classification of these IDO1 inhibitors in this section allows better understanding of their mechanisms of action.
Trp-Competitive
Trp analogs are the earliest known IDO1 inhibitors that can block the IDO1-mediated degradation of Trp. Indoximod (1-MT, IND) (Figure 4C) is a typical example, but differs from other IDO1 inhibitors in that it acts through the IDO pathway to disarm the immunosuppressive effects produced by the IDO1 enzyme,66 whereas it is invalid in the APCs that knock out the IDO1 gene in mice.67 Regulation of the immune function in the TME occurs via two indoximod-associated mechanisms: (1) imitating Trp-abundant signals and (2) regulating AHR activity.68 On the one hand, indoximod can deactivate the mTORC1 inhibition that is caused by IDO1 overexpression as a Trp mimic; however, it cannot inhibit the activation of GCN2 that is caused by Trp deficiency, which works by activating MAP4K3/GLK1 kinase to restore the activity of mTORC1, thus relieving T cell autophagy and resuming T cell proliferation.69 In contrast, indoximod, which acts as a Kyn antagonist, regulates the AHR regulatory genes and blocks the downstream inhibition of Kyn in T cells.70 However, in cellular experiments targeting only tumors, indoximod not only induces the expression of IDO1 mRNA, but also leads to an increase in the Kyn content of cancer cells, inhibiting the proliferation of T cells.71 Results have suggested that using indoximod in combination with chemotherapeutic agents is associated with good antitumor activity.72 This may be due to the fact that chemotherapeutic agents induce immunogenic cell death (ICD) and enhance the immunogenicity of tumor cells, while IDO1 inhibitors renew the immunosuppressive effects in the tumor immune microenvironment that result from abnormalities in the IDO pathway. Therefore, combining indoximod with chemotherapeutic agents may be the best therapeutic strategy for treating tumors. To date, 24 clinical trials have been registered for the use of indoximod in mitigating tumor immunity, among which 15 have been completed, three are recruiting, and two have been withdrawn. The status of the remaining trial is unknown. In a Phase II clinical trial investigating the use of the checkpoint inhibitor pembrolizumab to treat advanced melanoma, the addition of indoximod led to an increase in the objective response rate (ORR) of patients increased from 43 to 51%, with the disease control rate (DCR) reaching 70%. Moreover, the combination demonstrated good tolerability, with minimal occurrence of grade 3/4 treatment-related adverse events, and the observed side effects were aligned with the anticipated profile of single-agent pembrolizumab,73 with the most common AEs including nausea, diarrhea, fatigue, and high blood pressure.74 In addition, D-1-MT, as a D-type stereoisomer of indoximod with IDO2 as a preferred target of action, is influenced by genetic polymorphisms, and is ineffective in populations lacking the IDO2 allele. This compelled us to emphasize the influence of SNP on the effectiveness of immunomodulators, with ethnic differences possibly impacting the therapeutic efficacy of DO1 inhibitors.75 Unlike D-1-MT, the L-type isoform reverses the IDO1 enzyme-mediated increase in Trp depletion and Kyn levels by inducing mitotic death and mitochondrial damage, inhibiting tumor proliferation, and acts as a chemotherapeutic agent in colon cancer. However, there are no reports of its use in clinical trials.76 In addition to the two isomers, the indoximod precursor NLG802 (Figure 4C) has shown robust efficacy, with widespread absorption and rapid conversion to indoximod in tested species, as demonstrated in preclinical trials. Notably, the oral bioavailability of NLG802 has been shown to surpass that of indoximod more than 5-fold when equivalent molar doses were compared.77 The indole-based compound, PF-06840003 (Figure 4C) has also shown good potency as a Trp mimic.78 In summary, Trp mimetic-type IDO1 inhibitors have diverse mechanisms of action, and an in-depth understanding of their different modes of action could help in the development of more active Trp mimetics. It is noteworthy that the different structures and actions of these drugs require attention in the future development and clinical application of such IDO1 inhibitors. Rational and effective therapeutic regimens that are tailored to the genetic polymorphisms, immune system characteristics, and tumor heterogeneity in patients should be designed to reduce the off-target effects and enhance the pharmacokinetic effects of the drugs.
Targeting Heme-IDO1
Another strategy for inhibiting IDO1 expression is to target heme, a coenzyme of IDO1. Epacadostat (Figure 4D) has been found to exhibit both selectivity and potency as an IDO1 inhibitor, reversing the immune system suppression of IDO1, promoting the growth of T and natural killer cells, and reducing Tregs transformation.79 Epacadostat has shown a significant IDO1 inhibition effect in human primary DC and Hela cells via IDO1 overexpression that is induced by IFN-γ, which combined with its effects in mice bearing IDO1-expressing Pan02 pancreatic carcinomas, syngeneic immunocompetent C57BL/6 mice with tumors, and CT26-tumors investigation, shows that epacadostat can affect the Kyn levels in tumors and tumor-draining lymph nodes (TDLNs) by inhibiting IDO1 activity, controlling tumor growth via lymphocyte-dependent mechanisms.80 The combination of epacadostat and pembrolizumab demonstrated considerable promise in a Phase I/II clinical trial for advanced melanoma, with an ORR of 56%,81 while in another randomized trial, epacadostat demonstrated consistent IDO1 inhibition that exceeded 80–90% throughout the dosing period, with seven of the 52 patients exhibiting stable conditions. However, adverse events such as fatigue, nausea, vomiting, abdominal pain, and diarrhea were observed in more than 20% of the patients.82 Epacadostat is generally well tolerated as a combination therapy and effectively normalizes Kyn levels. However, in a Phase III ECHO-301/KEYNOTE-252 study into advanced melanoma, the combination of epacadostat with pembrolizumab did not yield advancements in the progression-free survival for patients dealing with unresectable or metastatic melanoma.83 Additionally, the combined treatment strategy of epacadostat and pembrolizumab failed in a Phase II study into advanced sarcoma (NCT03414229).84 At present, the reason for the treatment failure of epacadostat remains unclear, with researchers uncertain as to whether the issue is solely attributable to inadequate dosing or the inherent characteristics of certain types of tumor immune microenvironments. The key to addressing the clinical application of epacadostat is therefore to evaluate an effective dose and identify more effective modes of action in the future.
Navoximod (NLG919) (Figure 4D) is also a heme-binding IDO1 inhibitor,85 PK/PD studies have shown that it has the potential to cross the BBB and inhibit Kyn levels in the brain in preclinical glioma models. Although no anti-tumor effects were observed for navoximod in a subcutaneous and in situ tumor model.86 When combined with ICBs, it has shown anti-tumor efficacy in B16F10 mouse models, enhanced the efficacy of the HGP100 anti-tumor vaccine in vivo, and may decrease the Treg-mediated inhibited immunity in the tumor host, enhancing the activity of DCs in tumors and TDLNs. When combined with chemotherapy, navoximod has been found to enable Teff to elicit an immune response to endogenous tumor antigens released via chemotherapy.87 The considerable challenges involved in the development of heme-targeted IDO1 inhibitors is evident, and navoximod is only used as an immune adjuvant at present. However, there is an urgent need to develop high affinity and potency drugs that target different heme sites. In addition, considering that heme also serves as a cofactor for hemoglobin, myoglobin, and peroxidase, the development of such IDO1 inhibitors must take into full consideration the impact of off-target effects. In addition to 4-phenyl imidazole (4PI) (Figure 4D), despite its weak activity, navoximod can also bind to ferric heme IDO1, hindering reductive enzymes reactivation, and inhibiting IDO1 functioning. The fact that chemical derivatives of this inhibitor have been shown able to bind to different active sites within heme63 indicates that different chemical modifications have a huge impact on the activity of the drug, and that probing the activity of the derivatives of heme-targeting drugs may render it possible to discover more affinity for this inhibitor.
Targeting Apo-IDO1
Linrodostat (BMS-986205) (Figure 4E) is a typical inhibitor of targeting Apo-IDO1, which competes with heme for IDO1 binding sites. Linrodostat has been positioned a best-in-class drug (IC50 ~1.1 nM), with better efficacy and selection specificity than both epacadostat (IC50 ~10 nM) and indoximod (IC50 ~7.7 μM). Linrodostat works primarily by occupying the binding sites of heme cofactors, irreversibly inhibiting IDO1 activity.88 Linrodostat has demonstrated significant in vivo pharmacokinetic properties in preclinical trials, inhibiting the production of Kyn when IDO1 is overexpressed in HEK293 and Hela cells as a result of IFN-γ stimulation, and restoring the proliferation of IDO1-overexpressed T cells. Linrodostat has been shown to decrease the Kyn levels in xenotransplantation human tumor models.89 In a Phase 1/2a clinical trial (NCT02658890) for advanced bladder cancer the combined linrodostat with nivolumab, 57% of 516 patients experienced treatment-related adverse events (grades 3‒4, 12%), primarily fatigue (15%) and nausea (12%). Treatment-related adverse events led to discontinuation in 19 patients (4%), and three patients (<1%) died due to treatment-related adverse events (myocarditis, Stevens‒Johnson syndrome, and hepatic failure). BMS-986242 is structurally similar to this drug, and although it also has good anticancer effects, both drugs contain easily oxidized and metabolized quinolines, which decrease the usability of the drug. Improving the chemical structure of this drug may therefore make it a highly promising IDO1 inhibitor.90 LY3381916 (Figure 4E) has the same mechanism of action as linrodostat can bind to apo-IDO1 but not heme-containing IDO1. In a Phase I a/b study (NCT03343613), pharmacodynamic assessment showed decreased Kyn levels in the tumor tissues of 68% of patients; this effect was more pronounced in extrahepatic than live tissues. However, this did not systematically translate into antitumor activity,91 and the use of this drug as an anticancer immune adjuvant in combination with chemotherapeutic agents may be a good therapeutic strategy.
Others
In addition to the above mentioned IDO1 inhibitors, several other inhibitors are available with other forms of action. For example, the 2-propanol analogue (Figure 4F) regulates the ITIM site in the NTD of IDO1, inhibiting its activity. Norharman (Figure 4F) is a naturally occurring quinone that weakly inhibits IDO1, and actually acts more as a substrate for NDQ1, which mediates intracellular redox reactions. Its main role is to exert an antitumor effect by generating reactive oxygen species (ROS) toxicity.63 Furthermore, other biological agents such as siRNA (Figure 4F),92 and shRNA (Figure 4F)93 also possess great potential for clinical transformation. Wang et al delivered IDO1 siRNA to both TDLNs and tumor tissues using nanotechnology, with results showing remarkable IDO1 downregulation in both TDLNs and tumor tissues.94 Although this approach is highly targeted and contributes to precision therapy, the delivery efficiency and stability may be slightly less efficient and the duration of its efficacy may be shorter than that of normal drugs, and there is potential for impact on normal cell functioning. Enhancing the efficacy of genetic engineering with nanocarriers may improve the delivery efficiency and target specific gene fragments.95,96
In summary, a wide variety of IDO1 inhibitors is available, and by understanding their chemical structures and different mechanisms of action, we can gain a deeper understanding of the role that IDO1 plays in cancer immunotherapy, reveal the multifaceted factors of disease development, and provide reference for the development of new drugs. At the same time, this understanding can also reduce the drug resistance that results from repeated treatment of the same target, prolonging the therapeutic effect and improving the likelihood of success. In addition, considering the limitations of the current monotherapy and individual differences, the combination of chemotherapeutic agents with immune checkpoint inhibitors may be an effective therapeutic option.
Development of IDO1 Inhibitors in Clinical Trials
IDO1 has become an important therapeutic target in tumor immunity owing to its immunosuppressive effect in the TME, and its inhibitors have been proven to induce anti-tumor immunity. Over the past decade, many IDO1 inhibitors and peptide vaccines have been developed for clinical trials (Table 1).
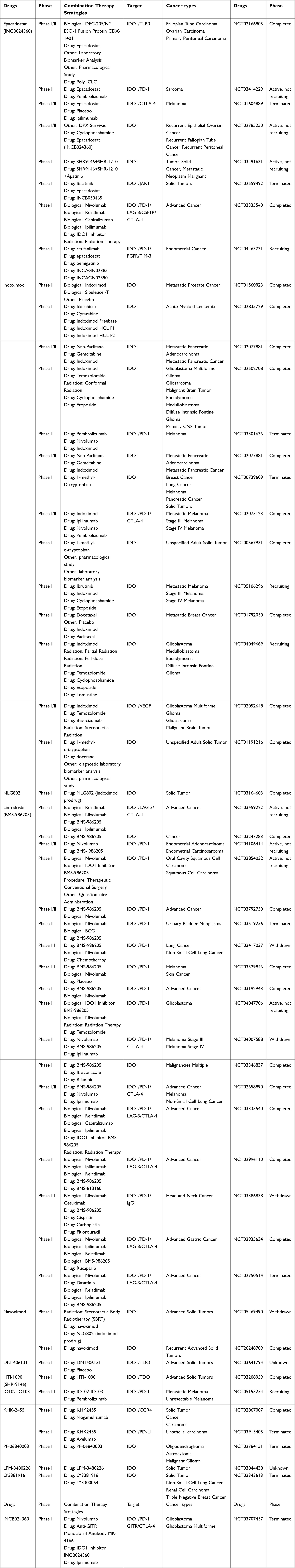 |
Table 1 IDO1 Inhibitors in Clinical Trials |
Overcoming Delivery Barriers for IDO1 Inhibitors Using Nanocarriers
At present, many IDO1 inhibitors are entering the stage of development and clinical trials; however, the toxicity, adverse effects, and low oral availability of IDO1 act as barriers to the clinical application of the IDO1 inhibitors. It is important to solve these problems if we are to achieve safe, efficient, and targeted delivery of these drugs. In this section, we explore how nanotechnology can be utilized to address the challenges of IDO1 inhibitor delivery, including aspects such as nanocarrier design, drug loading, half-life and drug release.
Nanocarrier Design
Although nanomaterials have been widely used in the delivery of anticancer drugs, many challenges remain in terms of design, including factors such as size, shape, and surface charge.97 To avoid the removal of nanomaterials by the renal filtration system, their hydrodynamic diameter is usually required to be greater than 6 nm, while, considering the enhanced permeability and retention (EPR) effect of tumors, NPs with a particle size of less than 200 nm are considered optimal for penetration into tumor tissue.98 It has been shown that shape also affects the accumulation of nanomaterials at the tumor site. For example, rod- and worm-shaped NPs are generally more likely to accumulate at the tumor site, mainly because their high aspect ratio and low surface curvature reduce the possibility of phagocytosis.97 Surface charge is also a key determinant in drug metabolism kinetics. In general, positively charged NPs are more likely to be recognized and phagocytosed by macrophages, whereas negatively charged NPs are more likely to be absorbed and cleared by organs such as the spleen and blood vessels. Therefore, NPs with neutral or slight negative charge may be the best choice for targeting tumor cells.99,100 In conclusion, the complex biological environment of the human body must be comprehensively considered and the physicochemical properties of nanodelivery systems understood when designing and selecting nanocarriers.
Drug Loading
Although nanomedicines can overcome many of the drawbacks of free drugs, such as poor solubility, high required dose and a short half-life, most currently-used NPs have low drug loading rates (<10%), and the excessive use of nanomaterials may lead to systemic toxicity during intravenous drug delivery.101,102 It is therefore necessary to select appropriate nanomaterial formulations based on the physicochemical properties of a drug. The vast majority of IDO1 inhibitors (eg, epacadostat, and navoximod) are lipid-soluble drugs, meaning that amphiphilic NPs,103–105 such as liposomes, copolymers, Janus particles, and lipid-polymer complexes are good choices for the delivery of these drugs. Lipidic drugs can be encapsulated within hydrophobic cores, while hydrophilic drugs can be attached to hydrophilic bilayers, increasing the solubility and bioavailability of the drugs without altering their molecular structures or pharmacological effects.106,107 In addition, inorganic NPs and metal-organic frameworks (MOFs) can also be used as drugs carriers that work by adsorption, encapsulation, or covalent modification; however, to enhance the adsorption and drug loading rate, the size, shape, and functional groups of the drug need to match the surface properties of the NPs when using this method.108,109 Solvent selection also has a significant impact on the drug loading rate, and it is important to ensure that the force between the NPs and the drug are effective for the extraction of drug from solvent. Such physicochemical properties must be carefully considered and evaluated when designing and selecting inorganic NPs and organometallic frameworks for use as drug delivery systems.
Half-Life
Nanoparticle stability is another factor that must be considered to ensure that IDO1 inhibitors are able to accumulate in sufficient quantities at the tumor site. After intravenous administration, particles need to avoid clearance by the liver, spleen, and macrophages, while remaining stable in the complex in vivo environment.98,110 Several modification strategies are commonly employed to extend the half-life of NPs, with examples including the addition of polysaccharides,111 polyethylene glycol (PEG) or polylactic acid-PEG copolymers,112 all of which can help NPs shield serum proteins from adsorption and thus avoid clearance by the body. Biomimetic nanotechnology is a widely used modification strategy that allows the host to mistake the NPs for its own components via camouflage with various biological structures such as erythrocyte membranes, macrophage membranes, and platelets.113,114 These biofilms can be extracted and wrapped around the surface of the NPs in a self-assembling manner, allowing them not only to evade recognition, but also to utilize the targeting or homing functions of the biofilms to reach the tumor site.115
Drug Release
The final step in the therapeutic phase, drug release, occurs when the NPs reach the tumor site or are taken up by tumor cells. Achieving controlled drug release is a necessary means of enhancing the efficacy of a drug, with pH-responsive (eg, polyethylene glycol-acrylic acid copolymer, 2-methyl imidazole),116 ROS-responsive (eg, cinnamaldehyde-thioacetal polymer, thioether-based polymer),117,118 and photosensitive materials (eg, polydopamine, Ce6, metal NPs)119 typically incorporated. Drugs that function at the subcellular level and supramolecular structures often require specific modifications to avoid degradation by lysosomes, with modifications including cationic polymers (eg, polyethyleneimine, poly-L-lysine) and membrane destabilizing peptides (eg, INF7, GALA).97 In addition, nucleic acid analogs require site-specific modification or binding to protein ligands to prevent degradation by nucleases.120
In summary, when designing nanodelivery systems for IDO1 inhibitors, due consideration should be given to designing formulations that can overcome delivery barriers. To provide a more detailed guidance strategy, the current recent advances in IDO1 inhibitor nanodelivery systems are summarized and the classification of nanodelivery systems, targeted modification strategies, and combination therapy strategies discussed in the following sections.
Recent Advances of IDO1 Inhibitor-Based Nanoparticles for Tumor Immunotherapy
Research on the design of nanodelivery systems can help advance the development of nanomedicines. As a guideline, the application of IDO1 inhibitor-based NPs in tumor immunotherapy is summarized (Table 2) and an in-depth analysis and discussion of different nanodelivery systems and targeted delivery strategies provided with the aim of comprehensively evaluating their potential in clinical applications.
 |
Table 2 Nanodelivery Based on IDO1 Inhibitors in Immunotherapy |
Classification of IDO1 Inhibitor-Based Nanoparticles
Lipidic NPs (LNPs)
Lipid NPs (LNPs) are tiny 10‒1000 nm phospholipid vesicles that are formed from a lipid bilayer and are usually composed of amphiphilic phospholipids such as phosphatidylcholine, phosphatidylethanolamine, and phosphatidylglycerol, with cholesterol as a stabilizer. LNPs can transport both hydrophobic and hydrophilic molecules; hydrophobic drugs are supported in hydrocarbon chains within the bilayer of liposomes, and hydrophilic drugs are encapsulated in the hydrophilic nucleus of liposomes.106,140 LNPs have many advantages as drug nanocarriers including (1) preventing degradation in vivo, (2) controlled drug release, (3) targeted drug delivery at the disease site, and (4) improved bioavailability. In addition, the liposome delivery system also acts as an effective vaccine adjuvant, delivering antigens (polypeptides, proteins, and nucleic acids) to antigen-presenting cells and activating the immune system. As nanocarriers, LNPs play a crucial role in the clinical treatment of human cardiovascular diseases, diabetes, and cancer.141,142 An increasing number of experiments have indicated the considerable potential of LNPs as delivery platforms for anti-tumor drugs. Nel et al developed a double-delivery liposome for tumor chemo-immunotherapy by injecting the anthraquinones mitoxantrone (MTO) into liposomes, and coupling the indoximod (IND) prodrug with the lipid bilayer (Figure 5). The results indicated that the MTO/IND vector is a robust ICD inducer that can stimulate the CT26 mouse model to develop a powerful immune response. Co-delivered IND prodrugs, as Trp mimics, can interfere with the immune metabolism by indirectly inhibiting the IDO1 pathway, enhancing the ICD response to MTO, reducing the number of Foxp3+ Tregs, producing sufficient cytotoxicity to kill cancer cells, and prolonging survival. In follow-up experiments, the efficacy of the NPs in EMT6, 4T1, and renal cancer models was also evaluated, in addition to the production of ICD markers, with results showing that the liposome delivery system can also generate a “hot” TME that intervenes in the tumor immune escape mechanism and inhibits the growth of tumors.121 Notably, in this study, liposomes were endowed with pH-dependent and reversible charge characteristics, which enabled better aggregation of the nanomedicine at the tumor site, enhancing the pharmacokinetics. However, adverse reactions to the chemotherapeutic agents occurred during the treatment, and adjustment of both dose and dosing interval of the drug may be required to improve the toxic side effects of the nanomedicine. Similarly, Liu et al constructed bifunctional liposomes using amphiphilic oxaliplatin (OXA) prodrug-coupled phospholipids, hydrophobic alkylated NLG919, and commercial liposomes as raw materials, which could release cytotoxic OXA into the reducing cytoplasm, induce ICD in cancer cells, and effectively delay Trp degradation to reversing the immunosuppressive nature of the TME through the NLG919-mediated inhibition of IDO1. Liposomes were found to significantly increase the intratumoral infiltration of CD8+ T cells and downregulate the immunosuppressive effect of Tregs in subcutaneous and orthotopic CT26 tumors, with significant anti-tumor effects. The results showed good biocompatibility and powerful therapeutic effects with great clinical transformation prospects.122 The properties of liposomes indicates that their full potential can be realized in the form of multidrug combination therapy strategies. In addition, optimizing intra-tumor drug release and ensuring the high-quality production of liposomes, as well as selecting the appropriate drug design based on the patient’s tumor type and immune status, are key issues that require urgent consideration in the clinical translation process.
 |
Figure 5 Design of liposomes for mitoxantrone and cholesterol indoximod prodrugs and efficacy in multiple solid tumors. (A) MTO/IND dual-delivery liposome reprogram the tumor microenvironment. (B) Schematic to outline the liposome synthesis steps. (C–E) Live-animal tumor growth curves, photographs of tumors (upper-right panel), and the quantitative IHC analysis of CRT, perforin, and NKp46 (lower right panel) from tumors harvested on day 23 in EMT6, RENCA, and 4T1 cancer models, respectively. *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001. Reprinted with permission from Mei KC, Liao YP, Jiang J et al. Liposomal Delivery of Mitoxantrone and a Cholesteryl Indoximod Prodrug Provides Effective Chemo-immunotherapy in Multiple Solid Tumors. ACS Nano. 2020;14(10):13,343–13,366. Copyright 2020. American Chemical Society.121 |
Polymeric NPs
Polymeric NPs, which are formed by the self-assembly of amphiphilic block copolymers and feature a hydrophilic outer layer and a hydrophobic inner core, are another promising nanomedicine delivery platform, with many types entering clinical trials.143,144 Polymeric NPs are typically prepared by emulsification, nano-precipitation and ionic gel methods. Therapeutic drugs are either encapsulated in the nanoparticle core or chemically coupled to the nanoparticle’s surface.109 Raw materials include polylactic acid,145 polysaccharides (cellulose, glucan, and hyaluronic acid),146 and cyclodextrin (CD).147 Polymeric NPs are mainly administered orally due to their structure and can be divided into polymers, micelles, and dendrimers.148 Compared with liposomes, the surface of polymeric NPs is more easily modified for the targeted delivery of anticancer drugs. Polymeric NPs possess good stability and drug loading rate, as well as improved PK characteristics, and drugs are released in a controlled manner,149 prolonging the circulation time of the drugs in vivo. Moreover, polymeric NPs can deliver plasmid DNA, small interfering RNA, proteins and vaccines, rendering them ideal drug delivery platforms.109 The copolymer PEG is a commonly used in applications, in which it serves as a delivery platform. Tang et al used a thin-film hydration method to prepare the DOX-supported polymeric micelle, Dox/PEG-Fmoc-NLG, and found that the PEG-Fmoc-NLG micellar administration of Dox may improve the immunity of the TME, significantly enhancing its anti-tumor activity. The delivery system was found to accumulate at the tumor site through passive targeting, with durable release characteristics, and showed significant anticancer effects, reversing the immunosuppression of the TME in A20 lymphoma cancer models via mediation by IDO1, and observably augmenting the ratio of CD4+ /CD8+ T cells, while also reducing the number of MDSCs and Tregs was also reduced.123 Similarly, Li et al developed a small particle sized PGEM micelle (~ 15 nm), which was co-loaded with the chemotherapy drug paclitaxel (PTX) and an IDO1 inhibitor; the micelle used copolymeric PGEM as a tumor penetration carrier and the redox-responsive GEM was coupled with POEG-co-PVD polymer (Figure 6). In contrast to the combination of traditional drugs, the vector was found able to improve the PK and biological distribution characteristics. The PGEM vector can also carry other anti-cancer drugs, such as curcumin and doxorubicin. The smaller particle size results in a longer cycle time, enhancing the permeability and retention, and contributing to effective tumor penetration. In vivo studies have shown that PGEM and PTX have synergistic chemotherapeutic effects, inducing the toxic death of cancer cells, and NLG919 has been used to enhance the immune activity of the TME. The inclusion of NLG919 has notably reduced the percentage of Tregs and increased the percentage of CD4+ T and CD8+ T cells. The PGEM copolymer has also demonstrated substantial anti-tumor potency in the PANC202 pancreatic cancer mouse models and the 4T1 and CT26 cancer mouse models, extending the survival rate of the mice.124 Nevertheless, the development of polymer NPs faces many challenges, including the complex formulation design and adequate characterization assays, and the need to address the impact of the complex human biological environment on the efficacy of nanomedicines in clinical applications. In addition, the micelle preparation process is relatively complex and requires the strict control of various parameters to ensure product quality, thus increasing the production cost and technical difficulty.
 |
Figure 6 Study of the mechanism of action of multifunctional PGEM NPs and therapeutic efficacy in vitro. (A) Schematic illustration of multi-functional PGEM NPs for co-delivery and intracellular GSH-responsive release of PTX, GEM and NLG919. Relative tumor volume changes (B), body weight (C), and survival rate (D) of the mice treated with various formulations. (E) CD4+T cells and CD8+T cells, as well as (F) regulatory T (Treg) cells. **: p < 0.01. Reprinted from Sun J, Wan Z, Chen Y et al. Triple drugs co-delivered by a small gemcitabine-based carrier for pancreatic cancer immunochemotherapy. Acta Biomater. 2020;106:289–300, Copyright 2020, with permission from Elsevier.124 |
Inorganic NPs
Inorganic NPs typically comprise metals, metal oxides, semiconductors, and magnetic materials. Compared to organic NPs, inorganic NPs have high quantum yields, low toxicities, long lifetimes, and high storage stabilities.150 Moreover, some metal-based NPs have characteristics such as photothermal effects, the Fenton reaction, an acid stimulation response, and easy surface modifications. The most common inorganic NPs include CeO2 NPs, selenium NPs, quantum dots, mesoporous silica, hollow carbon nanotubes, and mesoporous silica NPs (MSNPs).109,150 MSNPs are in particular, have been commonly used materials in biomedicine due to their excellent biodegradability, high porosity, and high surface area.151–153 Lu et al developed MSNPs that were encapsulated in a lipid bilayer with oxaliplatin (OX)+indoximod (IND) dual delivery capability (Figure 7). The OX was loaded with the pore-sized of MSNPs and the IND was encapsulated in a phospholipid bilayer. In the Kras-derived PDAC model, nano-carriers stimulate and interfere with immunosuppression by inducing the ICD to enhance innate and adaptive antitumor immunity and achieve synergistic immune effects. In this nanoparticle, OX induces ICD and increases the ratio of CD8+/Foxp3+ T cells at the tumor site to generate a systemic immune response, and its cooperation with IDO1 inhibitors not only improves efficacy but also promotes autophagy by activating the mTOR1 pathway. The antitumor effects of different dosing strategies were also examined, and comparison made to subcutaneous anti-tumor vaccination, local OX+IND-PL injection, and intravenous OX+IND-MSNP injection into orthotopic tumor-bearing B6/129 mice for delivery to an orthotopic PDAC site via intravenous biodistribution. Due to the formulation design of the NPs, the retention time and PK characteristics of the two drugs in the body were improved, which markedly elevated the concentration of the drug within the tumor and substantially enhanced its availability, showing a more significant anti-tumor effect.132 It is worth noting that the toxicity of metal NPs must be considered when using as carriers. Such NPs typically cause oxidative stress damage, cytotoxicity, and cellular dysfunction. Solubility, oxidation state, and soft and hard properties also have an impact on the cellular environment, which may result in difficulties with metabolism and lasting damage to the organism.120 Although the effectiveness of metal NPs in antitumor therapy is unquestionable, rigorous testing is required to ensure their safety profile before entering clinical trials. In addition, additional surface packaging and targeted modifications may be an effective means of attenuating their toxic side effects.
 |
Figure 7 Mechanism of anti-tumor immune action and anti-tumor efficacy of OX/IND-MSNP. (A) Schematic to illustrate how dual delivery of OX and IND may impact the anti-PDAC immune response. (B) KPC tumor growth curve after a single IT injection of the various drugs at a tumor size of 60–80 mm3. (C) Representative tumor images from each group after euthanizing the animal on day 31. (D) IHC depicting CD8 and Foxp3 biomarkers in the collected tumor tissue. (E) Flow cytometry determination of CD8/Tregs ratio, as described in D. (F) Flow cytometry analysis to determine CD91 expression in the population of CD45+/CD11b+/CD11c+ cells in the tumor tissue. (G) IHC to depict CRT and HMGB-1 expression in the collected tumor tissues. *: p<0.05; **: p<0.01. Reprinted from Lu J, Liu X, Liao YP et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat Commun. 2017;8(1):1811. Creative Commons.132 |
Metal Organic Frameworks (MOFs)
MOFs represent a growing category of porous hybrid materials that form via coordination bonds linking ions or clusters to organic ligands. They have been widely used in cancer therapy because of their high agent loading, tunable porosity, diverse compositions, controllable morphologies, and easy surface modifications.154 MOFs deliver drugs with prolonged circulation time in vivo, reducing the side effects.155 Moreover, certain NPs can respond to singular or multiple stimuli, both endogenous (eg, pH, ROS, and GSH) and exogenous (eg, ultrasonic, light, and magnetic fields), enabling controlled drug delivery.156 Zeolitic imidazolate framework-8 (ZIF-8) is a commonly used MOF for delivering anti-tumor drugs with excellent pH response.157 Kong et al prepared hyaluronic acid (HA)-modified ZIF-8 using one-pot synthesis for the co-delivery of Gem and D-1MT (Figure 8). The HA modifications helped target tumor sites in which CD44 was upregulated and they were easily taken up by OS cells, responding to the acidic TME to release the Gem and D-1MT. Gem has the ability to trigger apoptosis in OS cells and directly inhibits the energy of MDSCs by blocking DNA replication, whereas D-1MT reverses the immunosuppressive effect of the IDO1 pathway. The HA/ZIF-8@Gem/D-1-MT NPs decreased the toxicity of the combination drugs, with remarkable anti-tumor efficacy, and reactivated the immune system to eliminate the tumors. The design strategy for these NPs resulted in excellent application potential and flexibility.135 However, the clinical translation of MOF-based multifunctional nanoplatforms still faces many technical challenges, such as the complex preparation processes, difficulties in ligand selection, circulation stability and aggregation issues, long-term and acute toxicity assessment, and metabolism and degradation pathways. Only by solving these problems can comprehensive theoretical support be obtained.154,155,157 Nevertheless, MOF multifunctional nanoplatforms have a broad application prospect, and may promote the development of monotherapy to combination therapy, improving the anti-cancer effect.
 |
Figure 8 Preparation, Mechanism, and Immune Cell Regulation by HA/ZIF-8@Gem/D-1-MT NPs. (A) Illustration of the preparation of HA/ZIF-8@Gem/D-1-MT NPs and the mechanisms of the synergistic OS chemo-immunotherapy. The body weight (B) and tumor volumes (C) of K7M2 OS-bearing mice with different treatments. (D) OS tissues were obtained with BALB/c mice on day 21 after receiving different treatments. (E) Weights of isolated tumors and tumor inhibition ratio of K7M2 OS-bearing mice in different groups. (F-H) The percentage of M-MDSCs and G-MDSCs in tumor tissue. (I-K) The percentage of Th cells and CTLs in tumor tissue. Control, C; D-1-MT, M; Gem, G; ZIF-8@Gem/D-1-MT NPs, Z (GM); HA/ZIF-8@Gem/D-1-MT NPs, HZ (GM). ZIF-8@Gem/D-1-MT/Ce6 NPs, Z (GMC); HA/ZIF-8@Gem/D-1-MT/Ce6 NPs: HZ (GMC). *: p<0.05; ***: p<0.001. Reprinted from Fan Q, Zuo J, Tian H et al. Nanoengineering a metal-organic framework for osteosarcoma chemo-immunotherapy by modulating indoleamine-2,3-dioxygenase and myeloid-derived suppressor cells. J Exp Clin Cancer Res. 2022;41(1):162. Creative Commons.135 |
Biomimetic NPs
In addition to the NPs mentioned above, some biomimetic NPs such as cell membranes (ie, erythrocyte membranes, macrophage cell membranes, and neutrocyte membranes), biomacromolecules (ie, polysaccharides and proteins), and vesicles can be used as drug carriers. Proteins, a class of natural biomacromolecules, have recently been used as drug carriers to deliver anti-tumor pharmaceutical or diagnostic agents owing to the structural characteristics of their interior or surface.158 Compared to artificial NPs, they have superior biocompatibility, biodegradability, enhanced targeting capacity, and inherent non-immunogenicity. Albumin is an abundant protein that is found in human blood plasma and requires no additional modifications to achieve enhanced blood circulation and low immunogenicity.159 The potential of such molecules as nanocarriers has been fully explored as demonstrated by successful cancer models.136 Cell membrane-camouflaged NPs are a burgeoning category of nanocarriers that consist of synthetic NPs cloaked by a natural cell membrane and exhibit enhanced biocompatibility, immune evasion, and tumor-targeting capabilities. These particles excel at navigating intricate biological milieus,113,114,134 and have considerable potential for biomedical applications based on these characteristics. Tian et al developed a versatile biomimetic nanobullet comprising an erythrocyte membrane-coated thermally sensitive S-nitrosothiol (SNO) donor-pendant copolymer (PAAV-SNO) that was loaded with the IDO1 inhibitor 1-MT (Figure 9), integrating photothermal therapy (PTT) with immune activation to enhance the intratumoral infiltration of CD8+ cytotoxic T lymphocytes (CTLs), normalize tumor vessels, and alleviate tumor hypoxia. The immunosuppressive TME is thus comprehensively reprogrammed when using the method. The formulation demonstrated prolonged in vivo circulation, targeted delivery, improved biocompatibility, enhanced intratumoral accumulation, and controllable therapeutic release when facilitated by NIR-II laser irradiation. These characteristics indicate great potential for these NPs in clinical medicine.125 However, several translational hurdles still need to be overcome before clinical application, including the selection of cell sources, large-scale cell culture and quality control, and ensuring that cells are immunocompatible with the patient to avoid immunogenicity. Batch consistency and quality also needs to be ensured if genetically engineered modified or phenotypically differentiated cells are involved.113
 |
Figure 9 Drug release from erythrocyte membrane-camouflaged nanospheres and reprogramming of immunosuppressive TME for enhanced antitumor efficacy. (A) Schematic showing the structure and therapeutics releasing process of erythrocyte membrane-camouflaged nanobullets and (B) their capacities of reprogramming tumor immunosuppressive microenvironment. (C) Immunofluorescence images of pericyte coverage and Pimonidazole positive (as a marker for hypoxia, green) areas in 4T1 tumors after treatment as indicated. CD31+ endothelial cells are stained green, nucleuses are stained blue, and NG2+ pericytes are stained red. The cytokines of (D) INF-γ, (E) TNF-α, and (F) IL-6 in plasma and (G) the CD8+ T cells/Tregs ratio in tumor tissue in balb/c mice bearing 4T1 tumors following the indicated treatments. (H) Immunofluorescence staining for CD86 (hallmark of M1 phenotype, red), CD206 (hallmark of M2 phenotype, green), and nucleuses (blue), and IHC staining for PD-L1 of tumor tissue. *: p<0.05; **: p<0.01. Reprinted with permission from Yang Z, Gao D, Guo X et al. Fighting Immune Cold and Reprogramming Immunosuppressive Tumor Microenvironment with Red Blood Cell Membrane-Camouflaged Nanobullets. ACS Nano. 2020;14(12):17,442–17,457. Copyright 2020 American Chemical Society.125 |
Targeted Delivery Strategies
Although nanocarriers have significantly improved the pharmacokinetics of conventional drugs, they are still lacking in terms of precise delivery. Passive targeting that relies only on the EPR effect cannot achieve the desired therapeutic effect. Moreover, the EPR effect varies for different tumor types as well as different staging stages of the same tumor; therefore, more effective modification strategies are needed to improve targeting.100 Several targeted delivery strategies have been designed to address the characteristics of the TME, such as targeting high levels of ROS, receptor-mediated stimulus responsiveness, and redox responses.160 In addition, exogenous stimulus-activated drug delivery systems (such as magnetic fields, light, and ultrasound)161 improve the stability of drugs during blood circulation, allowing the drug to accumulate effectively at the lesion site, improving the efficacy, reducing the toxic side effects, and achieving highly controllable drug release in time and space.
Hyaluronic Acid (HA) Receptor-Mediated Targeting
HA is a natural anionic polysaccharide with good biocompatibility, non-immunogenicity, biodegradability, and active tumor-targeting ability.162–164 The targeting ability of HA-modified NPs is mainly due to interaction between HA and the CD44 receptor, which is a non-kinase transmembrane glycoprotein and is the most widely studied HA receptor.165 This important medium is highly expressed in breast cancer,166,167 gastric cancer,168 and colon cancer,169 among others. The CD44 receptor can regulate cell proliferation, migration, and adhesion by binding to HA, which is associated with tumor metastasis and anti-apoptosis.165,170 Based on the characteristics of HA, Fu et al developed HA-coated cationic albumin NPs (HNPs), which were loaded with hydrophobic celastrol and hydrophilic 1-MT to treat pancreatic cancer. Loading with HA helps HNPs to target tumor sites, and they are hydrolyzed into small drug molecules by hyaluronidase before penetrating deep tumor tissues via CD44 receptor-mediated accumulation and internalization. The results indicated that HNPs increase the ratio of CD4+/CD8+ T cells reversing TME immunosuppression and exhibiting remarkable antitumor efficacy in pancreatic cancer mouse models.137 Notably, the hyaluronic acid-targeted CD44 receptor is also highly expressed in inflammatory diseases, and a competitive adsorption effect may occur for tumor patients that also suffer from inflammatory diseases. Therefore, additional modifications or improved HA modifications are required to increase the drug delivery efficiency and reduce unwanted toxic side effects when using these NPs.
pH-Responsive Targeting Strategies
Acidic pH is an important characteristic of the TME. Compared to other tissues and organs, tumors demonstrate significantly lower pH values ranging from 6.0 to 7.0, whereas the pH of normal tissues is 7.4.171 Targeting this feature, pH-sensitive NPs play a crucial role in enhancing the therapeutic effects against cancer. This delivery strategy improves the drug release conditions and significantly reduces the toxicity of anticancer reagents in normal organs. CD is one building block that is used in stimulus-responsive NPs, and because of the dynamics and reversibility of its noncovalent interactions, it exhibits good shape-matching ability when binding with guest molecules.172 Zuo et al developed a tumor-targeting ferritin nanoparticle (FPBS@SN) with a pH-sensitive molecular switch by loading the chemotherapeutic drug sorafenib (SRF) and the IDO1 inhibitor NLG919 into a polymeric carrier that contained a benzimidazole (BM)-CD switch. Dissociation of the polymeric molecule allowed release of the SRF and NLG919 in a weakly acidic tumor environment. SRF can promote the expression of nuclear receptor coactivator 4, induce the degradation of ferritin and endogenous ferritin in NPs to release iron ions and promote tumor cell ferroptosis. NLG919 is responsible for blocking the Trp-Kyn pathway, inhibiting IDO1 enzyme activity, increasing Trp levels, and promoting T cell activation and proliferation. The NPs showed excellent antitumor and immune-activating effects by inducing ferroptosis and IDO1 inhibition in 4T1 mouse models, which is a potential strategy for cancer therapy.138
ROS-Responsive Targeting
Excessive ROS is a marker of tumors, and a double-edged sword. On the one hand, it can decrease the sensitivity of cancer to radiotherapy, lead to resistance against chemotherapy, and impact the potency of ROS-related therapies, such as PDT and SDT. However, the characteristics of excessive ROS can help researchers design targeted NPs for the TME. Moreover, enhanced ROS levels can induce cancer cell death via oxidative stress.173–175 Zong et al reported an ROS-responsive cinnamaldehyde (CA)-based poly(thioacetal) polymer (SANP1-MT) coupled with the IDO1 inhibitor 1-MT. This nanoparticle, combined with cancer immunotherapy, was found to reverse immunosuppression in the TME. Polythioacetals can be triggered by endogenous ROS, and CA lysis releases large amounts of ROS enhancing oxidative stress and inducing ICD. Simultaneously, 1-MT reduces Kyn levels. The results showed that SANP1-MT promoted DC maturation, induced cytotoxic T cell responses and CD8+CTL and NK cell intratumoral infiltration, and reduced the number of intratumoral MDSCs in the CT26 mouse models. This nanodelivery strategy demonstrated significant antitumor effects.118 However, this therapeutic strategy may be limited where inflammation and tumors coexist, and may even lead to further damage at the inflammation site. Therefore, careful evaluation of a patient’s medical condition is required when using these types of NPs.
Laser Stimuli‑responsive Targeting
In recent decades, photosensitizer-based photoactivation therapy has become a safe modality for tumor therapy for tumor therapy and has achieved broad clinical applications, with dermatologic indications the only issues observed. Favorable feedback and fewer adverse events have been observed when combined with chemotherapy or immunotherapy that targets and locally activates photosensitizers to treat tumors have been observed.176 Near-infrared (NIR) light is one of the most commonly used sources for phototherapy. Li et al reported a light-inducible nanocargo (LINC) that was co-assembled using the photosensitizer pheophidea, IDO1 inhibitor NLG919, and an OXA prodrug. Upon initial NIR light irradiation, ROS are produced, initiating the degradation of PEG and improving its permeability within the tumor tissues. The subsequent exposure to NIR light led to LINC demonstrating potent anti-immunogenic properties and an increase in the number of CTLs in the tumor. NLG919 improves the immune response effect of the TME by inhibiting IDO1 activity.126
Multistimulus Responsive Targeting
Ordinary NPs usually have only a single stimulus response or single tumor targeting; they cannot respond quickly to endogenous or exogenous stimuli in a complex internal environment to release anti-tumor drugs. As nanotechnology matures, the development of intelligent NPs with multiple responses that can deliver therapeutic drugs more accurately and quickly and thus possess considerable clinical transformation in receiving increasing attention.177 Wang et al reported a multi-responsive system that used MSNPs as the core for load PTX loading, with the IDO1 inhibitor 1-MT encapsulated in polymers as immune activators in the forms of a shell (β-CD-PEI/Ge1MT). This nanosystem (DOX@GMTMSNs) accumulated at the tumor site, where the NPs responded to the matrix metalloproteinase rich TME by releasing 1-MT extracellularly, followed by intracellular DOX release that was triggered by the highly acidic and redox lysosomal environment. In terms of mechanism, DOX-induced ICD promotes Teff cell infiltration and the maturation of DCs. However, 1-MT suppresses IDO1 activity, reducing the number of Tregs and reversing the immunosuppression of the TME. In short, the nanodrug delivery systems delivered drugs to their target cells precisely and exhibited a significant ability to inhibit primary tumor growth, increase the metastatic foci, and prolong animal survival.133 Of course, it is also possible to combine the combination design with other tumor characteristics for targeted modification. Despite the complexity of the development process, there is no doubt that these designs can effectively improve the anticancer efficacy of nanomedicines.
Combination Therapy Based on IDO1 Inhibitor Nanoparticles
Currently, the efficacy of IDO1 monotherapy is limited, and it is impossible to prevent tumor immune escape by blocking the Trp-IDO1-Kyn pathway alone. In clinical trials, IDO1 inhibitors are often used in combination with radiotherapy, chemotherapy, sonodynamic therapy (SDT), phototherapy, and ICB to exert significant anti-tumor effects.
Radiotherapy
Radiotherapy is one of the most important methods for the treatment of malignant tumors. Although breakthroughs have been made in recent decades, the therapeutic effect of this method still depends on immunosuppression of the TME. Therefore, combining radiotherapy with immunotherapy has become the most promising therapeutic approach for clinical applications, as it can reduce drug toxicity while ensuring a therapeutic effect.178,179 Wang et al reported a class of pH-responsive acidic IDO-modulated NPs (AIM NPs), which were based on calcium carbonate (CaCO3) NPs as delivery carriers, with IDO1 inhibitor 4PI was coated onto the surface of the CaCO3, and enhanced the radiotherapy efficacy. The AIM NPs effectively altered the intratumoral acidic environment, overcoming the radio resistance of the tumor. The NPs further enhanced X-ray-induced cell death by inducing an increase in the intracellular ROS levels. Moreover, the rapid release of 4PI in response to the acidic conditions suppressed the Kyn pathway, activating the anti-tumor immunity, and enhancing the effectiveness of radiotherapy for irradiating metastatic and highly immunogenic tumors in CT26 and 4T1 tumor models.180 Despite the efficacy of this combination strategy, several limitations were associated with this study. The retention and metabolism of NPs were not monitored in vivo, and long-term safety was not assessed. In addition, only a small animal model was used as the study subject, and it is unclear whether the same efficacy will occur in the complex biological environment of the human body. Perhaps large animals or patient tumor cells could be considered as study subjects to further validate the clinical translational value of the NPs.
Chemotherapy
Chemotherapy aims to destroy tumor cells by interfering with the normal metabolism and destroying the DNA structures and functions of cancer cells. Regardless of the route of administration, it is a systemic treatment that can eradicate tumors and limit antitumor proliferation and metastasis effects via the blood circulation. However, it also causes irreversible damage to normal organs, and an increase in the use of chemotherapy drugs is often accompanied by tumor drug resistance.181 The emergence of nanotechnology has rendered the combination of chemotherapy and immunotherapy effective. Jiang et al created NPs (DOX/IND@NPs) based on prodrugs to co-deliver immune activators and chemotherapeutic agents. DOX contributes to the augmentation of tumor immunogenicity by initiating damage-associated molecular pattern development and enhancing the number of CD8+ T cells in the TME. The IDO1 inhibitor IND has been found to decrease the number of Tregs, MDSCs, TAM, and other immunosuppressive cells, further enhancing the DOX efficacy and CD8+/Treg ratio and exhibiting significant antitumor efficacy in 4T1-cell-implanted BALB/c mice.127 Another study reported a binary cooperative prodrug nanoparticle with activation triggered specifically by the tumor microenvironment, which is a reduction-activated homodimer that co-delivers the OXA prodrug with the immunosuppressive TME regulator IDO1 inhibitor NLG919. OXA can trigger ICD to promote the intratumoral accumulation of CTLs and elicit antitumor immunity; meanwhile, NLG919 reverses IDO1-mediated immunosuppression and inhibits the intratumoral infiltration of Tregs, synergistically modulating the immune TME and regressing tumor development in 4T1 and CT26 cancer models.128 In summary, the co-delivery of IDO1 inhibitors with chemotherapeutic drugs is a therapeutic strategy with high potential and application value. Optimal dose ratios of chemotherapeutic agents and IDO1 inhibitors need to be determined in subsequent experiments to further minimize toxic side effects and improve the anticancer efficacy.
Phototherapy (PDT/PTT)
Phototherapy has rapidly developed into a cancer therapy in recent decades. Two main available approaches are considered: photodynamic therapy (PDT) and photothermal therapy (PTT). Lasers can be employed to accurately target and remove cancer cells with spatiotemporal precision, either through the generation of ROS or by increasing the temperature. These approaches not only overcome chemotherapy resistance and reduce off-target toxicity but also increase tumor permeability and intratumoral drug delivery.182 A novel nanoplatform, a cluster-bomb-like nanoplatform (CPIM) that employs a combination of size-transforming and transcytosis strategies has recently been reported. This nanoplatform is responsive to elevated ROS levels in the TME and is designed to release drug-loaded “bomblets” that contain IR780 and indoximod within a cc structure, enhancing the EPR effect in tumors. Notably, when exposed to NIR irradiation, IR780 induces ROS generation, further augmenting the ROS responsiveness, CPIM can be used for PDT, PTT, and the IDO1 inhibition effect of 1-MT, inducing ICD to enhance tumor immunogenicity and remodel the TME and exhibiting significant tumor-killing effect and tumor penetration in B16F10-tumor bearing mice.129 Similarly, Qin et al developed a nanosystem (NLG919/ IR780 micelles) that was triggered by a NIR laser, with in vivo antitumor studies revealing that when combined with PTT, it could effectively suppress primary tumors and tumor margin growth in a 4T1 cancer model.130 However, the complex preparation of phototherapy NPs needs to be noted, as well as the availability, cost, temperature, and dosimetry requirements of the light source equipment. The choice between PDT and PTT depends on several factors: for example, the easy activation of photosensitizers that is associated with PDT renders it more effective for well-vascularized superficial site lesions, such as the skin, esophagus, and bladder, while PTT is more suitable for deeper tumors, especially when using NIR light sources. In addition, systemic photosensitization and lack of thermal limitation are safety issues that must be considered when using phototherapy. These safety concerns may be mitigated by choosing an appropriate treatment that is based on the condition and tumor characteristics of a patient. Nonetheless, phototherapy has important clinical translational value and, in combination with other treatment modalities, may provide patients with a more personalized and effective treatment plan.
Sonodynamic Therapy (SDT)
SDT has emerged in the last decade and is relatively safe. Compared to phototherapy, SDT can enact deeper tissue penetration, excellent spatiotemporal selectivity, no phototoxicity, and fewer side effects.183,184 In particular, the application of nanoparticle-based sonosensitizers enhances the SDT efficacy and targeting specificity, and extends the internal circulation.185,186 SDT plays a role in cancer therapy by generating ROS and singlet oxygen (1O2), leading to tumor cell death.184 Luo et al developed a small-molecule self-assembling nanoparticle (HB-NLG8189@MPCM) that was coated with macrophage cell membranes (MPCMs) to combine SDT with the IDO1 inhibitor NLG8189. It preferentially accumulates within tumors to reduce systemic toxicity via the surface membrane proteins of macrophages. In addition, hemoglobin (HB), a natural metal porphyrin, is utilized as the acoustic sensitizer to efficiently induce ICD by generating ROS upon the application of ultrasound, enhancing the immunogenicity in the tumor. NLG8189 activates the immune response by inhibiting IDO1 activation and reversing the immunosuppression in the TME.139 Overall, the NPs are formulated to be biocompatible and safe; however, their effectiveness in the complex environment of the human body has not been established and in vivo therapeutic effects cannot be monitored in real time. It is recommended that large animal or patient-derived tumor cell effects are tested prior to clinical translation. In addition, SDT is still in its infancy, with unclear mechanisms of action other than inducing ROS cytotoxic death, and the few applications of acoustic-sensitive materials with complex design and synthesis processes may hinder its clinical translation.186,187 Therefore, SDT still requires continuous exploration and optimization before application.
Immune Checkpoint Blockade (ICB)
ICB is a potential therapeutic strategy. Immune checkpoints can maintain self-tolerance and prevent autoimmune damage to normal tissues by binding receptors to ligands; however, tumor cells can harness this mechanism to achieve immune escape and resist immune attacks. Under the dysfunction of the immune system that is caused by tumor cells, ICB can activate not only T cells, but also innate and adaptive immunity, and its emergence has revolutionized the field of cancer treatment.6,188 In contrast to chemotherapy, ICI monotherapy exhibits superior clinical efficacy; however, most patients still cannot get good feedback and the efficacy is limited.189,190 Therefore, the use of ICI in combination with other therapies may be a good strategy. Li et al developed a self-assembled nickel-doped IDO1 peptide drug (NLG-RGD NI) that contained a polypeptide with targeting properties, and released NLG919 and aPD-L1 in an environment of acidic pH and ester catalysis. These NPs have a long-lasting inhibitory effect on IDO1 inhibitors in intratumoral cells, with fewer systemic side effects. The results showed that the IDO1 inhibitors led to a significant increase in the proportion of CD4+ and CD8+T cells when combined with aPD-L1, enhancing the NK cell response, reducing the accumulation of immunosuppressive Treg cells, and exhibiting effective anti-tumor efficacy in the subcutaneous Pan02 and orthotopic Pan02 tumor models.131 Therefore, IDO1 inhibitors are expected to play a crucial role in cancer therapy as excellent partners for immune checkpoint inhibitors. However, for clinical translational development, appropriate checkpoint inhibitors should be selected based on the immunologic characteristics of the patient and the expression levels of tumor immune checkpoints, and any adverse reactions and tolerance to specific inhibitors should be assessed to ensure safe and effective treatment.
Conclusion and Future Perspectives
Immunotherapy has become a popular target for research in the field of tumor treatment, and numerous immune checkpoint inhibitors and immunomodulators have been put into clinical trials, including IDO1 inhibitors. The failure of the Phase III clinical trial of the IDO1 inhibitor epacadostat in the treatment of malignant melanoma had a negative impact on the development of IDO1 inhibitors. However, its therapeutic value in some types of cancers cannot be denied. At present, the successful clinical application of IDO1 inhibitors is facing several problems: (1) The existing IDO1 inhibitors have numerous mechanisms of action; however, the effect is slightly insufficient, and researchers need to continue to improve and discover more effective action sites for targeting IDO1 enzymes. (2) From the biological function of IDO1 and the mechanism of action of existing IDO1 inhibitors, there may be other metabolic compensation mechanisms in tumors that weaken the effect of IDO1 inhibitors. Therefore, combination with chemotherapy, radiotherapy, phototherapy and other therapies should be considered, and the selection of an appropriate combination therapy strategy evaluated based on the patient’s physical condition as well as tumor characteristics and other conditions. (3) Current studies are limited to small animal models and animal-derived cell lines, and it is unclear whether they are effective in the complex biological environment of the human body. Therefore, it is recommended that IDO1 inhibitor-based therapeutic strategies are transferred to large animal or patient-derived tumor cell lines or xenografts. In addition, there are many issues associated with the development and treatment with IDO1 inhibitor-based NPs, including possible long-term toxicity and unmonitored in vivo anti-cancer efficacy. Multifunctional modification of NPs that are currently in clinical use (eg, liposomal NPs, polymeric NPs, albumin) may be able to address these issues. However, at the technical level, the complicated nanoparticle preparation process, high production cost, and difficulty in large-scale production still exist. These issues need to be coordinated and solved by research institutions, enterprises and governmental departments to accelerate the development of nanotechnology and products.
In summary, the research and development of IDO1 inhibitors and IDO1 inhibitor-based nanomedicines still face many challenges; however, the deepening scientific research and continuous progress of science and technology means that this direction is expected to play an important role in the field of tumor therapy in the future. We anticipate that this review will help in the development and clinical translation of nanomedicines, ultimately resulting in precision medicine that can better benefit society.
Abbreviations
Trp, Tryptophan; IDO1, indoleamine 2.3-dioxygenase 1; Kyn, kynurenine; APCs, antigen-presenting cells; DAMP, danger-associated molecular patterns; ICB, immune checkpoint blockade; TME, tumor microenvironment; TDO, tryptophan 2.3-dioxygenase; I3P, indole-3-pyruvate; IL4I1, interleukin-4-induced-1; CNS, central nervous system; BBB, blood-brain barrier; AHR, aryl hydrocarbon receptor; Tregs, regulatory T cells; COX-2, cyclooxygenase 2; PGE2, prostaglandin E2; GCN2, general control nonderepressible 2; mTOR, mammalian target of rapamycin; ISR, integrated stress response; ITIM, immunoreceptor inhibitory motifs; 1-MT, IND, Indoximod; ORR, objective response rate; DCR, disease control rate; TDLNs, tumor-draining lymph node; 4-PI, 4-phenyl imidazole; LNPs, lipid nanoparticles; MTO, mitoxantrone; ICD, immunogenetic cell death; PEG, poly ethylene glycol; PTX, paclitaxel; MSNP, mesoporous silica nanoparticles; OX, OXA, oxaliplatin; MOFs, metal organic frameworks; ZIF-8, zeolitic imidazolate framework-8; HA, hyaluronic acid; CNPs, cell membrane camouflaged nanoparticles; SNO, S-nitrosothiols; PTT, photothermal therapy; CTLs, cytotoxic T lymphocytes; ROS, reactive oxygen species; GSH, glutathione; HNPs, HA-coated cationic albumin nanoparticles; CLT, celastrol; SRF, sorafenib; CA, cinnamaldehyde; LINC, light-inducible nanocargo; MSNPs, mesoporous silica nanoparticles; SDT, sonodynamic therapy; AIM NPs, acidic IDO modulate nanoparticles; BCNP, binary cooperative prodrug nanoparticle; PDT, photodynamic therapy; NIR, Near Infrared; 1O2, singlet oxygen; MPCMs, macrophage cell membranes; HB, hemoglobin; EPR, the effect of permeability and retention; QA, quinolinic acid; TAM, tumor-associated macrophages; MDSCs: myeloid-derived suppressor cells; CD, cyclodextrin; GEM, Gemcitabine ICI, immune checkpoint inhibitors.
Acknowledgments
We thank all of the participants of this project. This study was funded by National Natural Science Foundation of China (No.82060462, No.82270804), The Science and Technology Foundation of Guizhou Province (Number: [2020]1Y303), Science and Technology Plan Project of Guizhou Province (Number: [2019]5405), Foundation of Health and Family Planning Commission of Guizhou Province (Number: gzwjkj2019-1-127) and the Doctoral Foundation of Guizhou Provincial People’s Hospital (GZSYBS [2018]02). The funding agencies and donors had no role in any aspect of this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer. 2021;21(6):345–359. doi:10.1038/s41568-021-00347-z
2. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21(5):298–312. doi:10.1038/s41568-021-00339-z
3. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi:10.1038/ni1102-991
4. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi:10.1126/science.1203486
5. Gotwals P, Cameron S, Cipolletta D, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. 2017;17(5):286–301. doi:10.1038/nrc.2017.17
6. Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol. 2021;16:223–249. doi:10.1146/annurev-pathol-042020-042741
7. Allison J, Allison J. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–214. doi:10.1016/j.cell.2015.03.030
8. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell. 2020;38(4):473–488. doi:10.1016/j.ccell.2020.07.005
9. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. 2019;290(1):127–147. doi:10.1111/imr.12772
10. Huang R, Li X, He Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86. doi:10.1186/s13045-020-00910-5
11. Rosenberg SA, Restifo N. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. doi:10.1126/science.aaa4967
12. Met Ö, Jensen KM, Chamberlain CA, et al. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 2019;41(1):49–58. doi:10.1007/s00281-018-0703-z
13. Weiner LM, Dhodapkar MV, Ferrone S. Monoclonal antibodies for cancer immunotherapy. Lancet. 2009;373(9668):1033–1040. doi:10.1016/S0140-6736(09)60251-8
14. Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18(1):128. doi:10.1186/s12943-019-1055-6
15. Wu H, Fu X, Zhai Y, et al. Development of Effective Tumor Vaccine Strategies Based on Immune Response Cascade Reactions. Adv Healthc Mater. 2021;10(13):e2100299. doi:10.1002/adhm.202100299
16. Lemos H, Huang L, Prendergast GC, et al. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer. 2019;19(3):162–175. doi:10.1038/s41568-019-0106-z
17. Liu J, Zhang R, Xu Z. Nanoparticle-Based Nanomedicines to Promote Cancer Immunotherapy: recent Advances and Future Directions. Small. 2019;15(32):e1900262. doi:10.1002/smll.201900262
18. Patra JK, Das G, Fraceto LF, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnology. 2018;16(1):71. doi:10.1186/s12951-018-0392-8
19. Platten M, Nollen EAA, Röhrig UF, et al. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019;18(5):379–401. doi:10.1038/s41573-019-0016-5
20. Seo SK, Kwon B. Immune regulation through tryptophan metabolism. Exp Mol Med. 2023;55(7):1371–1379. doi:10.1038/s12276-023-01028-7
21. Kałużna-Czaplińska J, Gątarek P, Chirumbolo S, et al. How important is tryptophan in human health? Crit Rev Food Sci Nutr. 2019;59(1):72–88. doi:10.1080/10408398.2017.1357534
22. Cervenka I, Agudelo LZ, Ruas JL. Kynurenines: tryptophan’s metabolites in exercise, inflammation, and mental health. Science. 2017;357(6349):6349. doi:10.1126/science.aaf9794
23. Schwarcz R, Stone TW. The kynurenine pathway and the brain: challenges, controversies and promises. Neuropharmacology. 2017;112(Pt B):237–247. doi:10.1016/j.neuropharm.2016.08.003
24. Covarrubias AJ, Perrone R, Grozio A, et al. NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021;22(2):119–141. doi:10.1038/s41580-020-00313-x
25. Cantó C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: a Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22(1):31–53. doi:10.1016/j.cmet.2015.05.023
26. Liu C, Vyas A, Kassab MA, et al. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017;45(14):8129–8141. doi:10.1093/nar/gkx565
27. Sadik A, Somarribas Patterson LF, Öztürk S, et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell. 2020;182(5):1252–1270.e34. doi:10.1016/j.cell.2020.07.038
28. Wang Z, Li T, Mao C, et al. IL4I1-driven AHR signature: a new avenue for cancer therapy. Signal Transduct Target Ther. 2021;6(1):118. doi:10.1038/s41392-021-00529-z
29. Fujiwara Y, Kato S, Nesline MK, et al. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer Treat Rev. 2022;110:102461. doi:10.1016/j.ctrv.2022.102461
30. Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci. 2013;34(2):136–143. doi:10.1016/j.tips.2012.09.006
31. Prendergast GC, Mondal A, Dey S, et al. Inflammatory Reprogramming with IDO1 Inhibitors: turning Immunologically Unresponsive ‘Cold’ Tumors ‘Hot’. Trends Cancer. 2018;4(1):38–58. doi:10.1016/j.trecan.2017.11.005
32. Zhang Y, Hu Z, Zhang J, et al. Dual-target inhibitors of indoleamine 2, 3 dioxygenase 1 (Ido1): a promising direction in cancer immunotherapy. Eur J Med Chem. 2022;238:114524. doi:10.1016/j.ejmech.2022.114524
33. Tang K, Wu Y-H, Song Y, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol. 2021;14(1):68. doi:10.1186/s13045-021-01080-8
34. Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. doi:10.1126/science.281.5380.1191
35. Munn DH, Shafizadeh E, Attwood JT, et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–1372. doi:10.1084/jem.189.9.1363
36. Xue C, Li G, Zheng Q, et al. Tryptophan metabolism in health and disease. Cell Metab. 2023;35(8):1304–1326. doi:10.1016/j.cmet.2023.06.004
37. Torre E, Pinton G, Lombardi G, et al. Melanoma Cells Inhibit iNKT Cell Functions via PGE2 and IDO1. Cancers. 2023;15(13):3498. doi:10.3390/cancers15133498
38. Wei JL, Wu SY, Yang YS, et al. GCH1 induces immunosuppression through metabolic reprogramming and IDO1 upregulation in triple-negative breast cancer. J Immunother Cancer. 2021;9(7):1.
39. Xiang Z, Li J, Song S, et al. A positive feedback between IDO1 metabolite and COL12A1 via MAPK pathway to promote gastric cancer metastasis. J Exp Clin Cancer Res. 2019;38(1):314. doi:10.1186/s13046-019-1318-5
40. Chen B, Alvarado DM, Iticovici M, et al. Interferon-Induced IDO1 Mediates Radiation Resistance and Is a Therapeutic Target in Colorectal Cancer. Cancer Immunol Res. 2020;8(4):451–464. doi:10.1158/2326-6066.CIR-19-0282
41. Liu M, Wang X, Wang L, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11(1):100. doi:10.1186/s13045-018-0644-y
42. Hennequart M, Pilotte L, Cane S, et al. Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol Res. 2017;5(8):695–709. doi:10.1158/2326-6066.CIR-16-0400
43. Kim H, Chen L, Lim G, et al. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J Clin Invest. 2012;122(8):2940–2954. doi:10.1172/JCI61884
44. Yu J, Wang Y, Yan F, et al. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014;193(5):2574–2586. doi:10.4049/jimmunol.1400833
45. Yamashita N, Long M, Fushimi A, et al. MUC1-C integrates activation of the IFN-γ pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J Immunother Cancer. 2021;9(1):e002115. doi:10.1136/jitc-2020-002115
46. Revelo XS, Winer S, Winer DA. Starving Intestinal Inflammation with the Amino Acid Sensor GCN2. Cell Metab. 2016;23(5):763–765. doi:10.1016/j.cmet.2016.04.020
47. Cordova RA, Misra J, Amin PH, et al. GCN2 eIF2 kinase promotes prostate cancer by maintaining amino acid homeostasis. Elife. 2022;2022:11.
48. Tian X, Zhang S, Zhou L, et al. Targeting the Integrated Stress Response in Cancer Therapy. Front Pharmacol. 2021;12:747837. doi:10.3389/fphar.2021.747837
49. Halaby MJ, Hezaveh K, Lamorte S, et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci Immunol. 2019;4(42). doi:10.1126/sciimmunol.aax8189
50. Dey S, Mondal A, DuHadaway JB, et al. IDO1 Signaling through GCN2 in a Subpopulation of Gr-1(+) Cells Shifts the IFNγ/IL6 Balance to Promote Neovascularization. Cancer Immunol Res. 2021;9(5):514–528. doi:10.1158/2326-6066.CIR-20-0226
51. Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18(12):744–757. doi:10.1038/s41568-018-0074-8
52. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–976. doi:10.1016/j.cell.2017.02.004
53. Ediriweera MK, Tennekoon KH, Samarakoon SR. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: biological and therapeutic significance. Semin Cancer Biol. 2019;59:147–160. doi:10.1016/j.semcancer.2019.05.012
54. Sharma MD, Pacholczyk R, Shi H, et al. Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity. 2021;54(10):2354–2371.e8. doi:10.1016/j.immuni.2021.09.005
55. Bishnupuri KS, Alvarado DM, Khouri AN, et al. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019;79(6):1138–1150. doi:10.1158/0008-5472.CAN-18-0668
56. Salminen A. Role of indoleamine 2,3-dioxygenase 1 (IDO1) and kynurenine pathway in the regulation of the aging process. Ageing Res Rev. 2022;75:101573. doi:10.1016/j.arr.2022.101573
57. Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14(12):801–814. doi:10.1038/nrc3846
58. Solvay M, Holfelder P, Klaessens S, et al. Tryptophan depletion sensitizes the AHR pathway by increasing AHR expression and GCN2/LAT1-mediated kynurenine uptake, and potentiates induction of regulatory T lymphocytes. J Immunother Cancer. 2023;11(6). doi:10.1136/jitc-2023-006728
59. Gutiérrez-Vázquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity. 2018;48(1):19–33. doi:10.1016/j.immuni.2017.12.012
60. Yang X, Liu H, Ye T, et al. AhR activation attenuates calcium oxalate nephrocalcinosis by diminishing M1 macrophage polarization and promoting M2 macrophage polarization. Theranostics. 2020;10(26):12011–12025. doi:10.7150/thno.51144
61. Kenski JCN, Huang X, Vredevoogd DW, et al. An adverse tumor-protective effect of IDO1 inhibition. Cell Rep Med. 2023;4(2):100941. doi:10.1016/j.xcrm.2023.100941
62. Jitschin R, Braun M, Büttner M, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood. 2014;124(5):750–760. doi:10.1182/blood-2013-12-546416
63. Feng X, Liao D, Liu D, et al. Development of Indoleamine 2,3-Dioxygenase 1 Inhibitors for Cancer Therapy and Beyond: a Recent Perspective. J Med Chem. 2020;63(24):15115–15139. doi:10.1021/acs.jmedchem.0c00925
64. Röhrig UF, Reynaud A, Majjigapu SR, et al. Inhibition Mechanisms of Indoleamine 2,3-Dioxygenase 1 (IDO1). J Med Chem. 2019;62(19):8784–8795.
65. Hennes E, et al. Cell-Based Identification of New IDO1 Modulator Chemotypes. Angew Chem Int Ed Engl. 2021;60(18):9869–9874. doi:10.1002/anie.202016004
66. Fox E, Oliver T, Rowe M, et al. Indoximod: an Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front Oncol. 2018;8:370. doi:10.3389/fonc.2018.00370
67. Hou DY, Muller AJ, Sharma MD, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67(2):792–801. doi:10.1158/0008-5472.CAN-06-2925
68. Brincks EL, Adams J, Wang L, et al. Indoximod opposes the immunosuppressive effects mediated by IDO and TDO via modulation of AhR function and activation of mTORC1. Oncotarget. 2020;11(25):2438–2461. doi:10.18632/oncotarget.27646
69. Metz R, Rust S, DuHadaway JB, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1(9):1460–1468. doi:10.4161/onci.21716
70. Moyer BJ, Rojas IY, Murray IA, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors activate the aryl hydrocarbon receptor. Toxicol Appl Pharmacol. 2017;323:74–80. doi:10.1016/j.taap.2017.03.012
71. Opitz CA, Litzenburger UM, Opitz U, et al. The indoleamine-2,3-dioxygenase (IDO) inhibitor 1-methyl-D-tryptophan upregulates IDO1 in human cancer cells. PLoS One. 2011;6(5):e19823. doi:10.1371/journal.pone.0019823
72. Muller AJ, DuHadaway JB, Donover PS, et al. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–319. doi:10.1038/nm1196
73. Zakharia Y, McWilliams RR, Rixe O, et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J Immunother Cancer. 2021;9(6):e002057. doi:10.1136/jitc-2020-002057
74. Soliman HH, Jackson E, Neuger T, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5(18):8136–8146. doi:10.18632/oncotarget.2357
75. Metz R, DuHadaway JB, Kamasani U, et al. Novel Tryptophan Catabolic Enzyme IDO2 Is the Preferred Biochemical Target of the Antitumor Indoleamine 2,3-Dioxygenase Inhibitory Compound d −1-Methyl-Tryptophan. Cancer Res. 2007;67(15):7082–7087. doi:10.1158/0008-5472.CAN-07-1872
76. Liu X, Zhou W, Zhang X, et al. 1-L-MT, an IDO inhibitor, prevented colitis-associated cancer by inducing CDC20 inhibition-mediated mitotic death of colon cancer cells. Int, J, Cancer. 2018;143(6):1516–1529. doi:10.1002/ijc.31417
77. Kumar S, Jaipuri FA, Waldo JP, et al. Discovery of indoximod prodrugs and characterization of clinical candidate NLG802. Eur J Med Chem. 2020;198:112373. doi:10.1016/j.ejmech.2020.112373
78. Crosignani S, Bingham P, Bottemanne P, et al. Discovery of a Novel and Selective Indoleamine 2,3-Dioxygenase (IDO-1) Inhibitor 3-(5-Fluoro-1 H -indol-3-yl)pyrrolidine-2,5-dione (EOS200271/PF-06840003) and Its Characterization as a Potential Clinical Candidate. J Med Chem. 2017;60(23):9617–9629. doi:10.1021/acs.jmedchem.7b00974
79. Liu X, Shin N, Koblish HK, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115(17):3520–3530. doi:10.1182/blood-2009-09-246124
80. Koblish HK, Hansbury MJ, Bowman KJ, et al. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol Cancer Ther. 2010;9(2):489–498. doi:10.1158/1535-7163.MCT-09-0628
81. Mitchell TC, Hamid O, Smith DC, et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J Clin Oncol. 2018;36(32):3223–3230. doi:10.1200/JCO.2018.78.9602
82. Beatty GL, O’Dwyer PJ, Clark J, et al. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin Cancer Res. 2017;23(13):3269–3276. doi:10.1158/1078-0432.CCR-16-2272
83. Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a Phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–1097. doi:10.1016/S1470-2045(19)30274-8
84. Kelly CM, Qin L-X, Whiting KA, et al. A Phase II Study of Epacadostat and Pembrolizumab in Patients with Advanced Sarcoma. Clin Cancer Res. 2023;29(11):2043–2051. doi:10.1158/1078-0432.CCR-22-3911
85. Kumar S, Waldo JP, Jaipuri FA, et al. Discovery of Clinical Candidate (1 R,4 r)-4-((R)-2-((S)-6-Fluoro-5 H -imidazo[5,1- a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan-1-ol (Navoximod), a Potent and Selective Inhibitor of Indoleamine 2,3-Dioxygenase 1. J Med Chem. 2019;62(14):6705–6733. doi:10.1021/acs.jmedchem.9b00662
86. Kesarwani P, Prabhu A, Kant S, et al. Tryptophan Metabolism Contributes to Radiation-Induced Immune Checkpoint Reactivation in Glioblastoma. Clin Cancer Res. 2018;24(15):3632–3643. doi:10.1158/1078-0432.CCR-18-0041
87. Jung KH, LoRusso P, Burris H, et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin Cancer Res. 2019;25(11):3220–3228. doi:10.1158/1078-0432.CCR-18-2740
88. Pham KN, Yeh SR. Mapping the Binding Trajectory of a Suicide Inhibitor in Human Indoleamine 2,3-Dioxygenase 1. J Am Chem Soc. 2018;140(44):14538–14541. doi:10.1021/jacs.8b07994
89. Balog A, Lin T-A, Maley D, et al. Preclinical Characterization of Linrodostat Mesylate, a Novel, Potent, and Selective Oral Indoleamine 2,3-Dioxygenase 1 Inhibitor. Mol Cancer Ther. 2021;20(3):467–476. doi:10.1158/1535-7163.MCT-20-0251
90. Luke JJ, Tabernero J, Joshua A, et al. BMS-986205, an indoleamine 2, 3-dioxygenase 1 inhibitor (IDO1i), in combination with nivolumab (nivo): updated safety across all tumor cohorts and efficacy in advanced bladder cancer (advBC). J Clin Oncol. 2019;37(7):358. doi:10.1200/JCO.2019.37.7_suppl.358
91. Kotecki N, Vuagnat P, O’Neil BH, et al. A Phase I Study of an IDO-1 Inhibitor (LY3381916) as Monotherapy and in Combination With an Anti-PD-L1 Antibody (LY3300054) in Patients With Advanced Cancer. J Immunother. 2021;44(7):264–275. doi:10.1097/CJI.0000000000000368
92. Endo R, Nakamura T, Kawakami K, et al. The silencing of indoleamine 2,3-dioxygenase 1 (IDO1) in dendritic cells by siRNA-loaded lipid nanoparticles enhances cell-based cancer immunotherapy. Sci Rep. 2019;9(1):11335. doi:10.1038/s41598-019-47799-w
93. Blache CA, Manuel ER, Kaltcheva TI, et al. Systemic Delivery of Salmonella typhimurium Transformed with IDO shRNA Enhances Intratumoral Vector Colonization and Suppresses Tumor Growth. Cancer Res. 2012;72(24):6447–6456. doi:10.1158/0008-5472.CAN-12-0193
94. Huang H, Jiang C-T, Shen S, et al. Nanoenabled Reversal of IDO1-Mediated Immunosuppression Synergizes with Immunogenic Chemotherapy for Improved Cancer Therapy. Nano Lett. 2019;19(8):5356–5365. doi:10.1021/acs.nanolett.9b01807
95. Amreddy N, Babu A, Muralidharan R, et al. Recent Advances in Nanoparticle-Based Cancer Drug and Gene Delivery. Adv Cancer Res. 2018;137:115–170.
96. Yoo YJ, Lee CH, Park SH, et al. Nanoparticle-based delivery strategies of multifaceted immunomodulatory RNA for cancer immunotherapy. J Control Release. 2022;343:564–583. doi:10.1016/j.jconrel.2022.01.047
97. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. doi:10.1038/nbt.3330
98. Fan D, Cao Y, Cao M, et al. Nanomedicine in cancer therapy. Signal Transduct Target Ther. 2023;8(1):293. doi:10.1038/s41392-023-01536-y
99. Zhang P, Chen D, Li L, et al. Charge reversal nano-systems for tumor therapy. J Nanobiotechnology. 2022;20(1):31. doi:10.1186/s12951-021-01221-8
100. Fang J, Islam W, Maeda H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv Drug Deliv Rev. 2020;157:142–160. doi:10.1016/j.addr.2020.06.005
101. Shen S, Wu Y, Liu Y, et al. High drug-loading nanomedicines: progress, current status, and prospects. Int J Nanomed. 2017;12:4085–4109. doi:10.2147/IJN.S132780
102. Yang W, Wang L, Mettenbrink EM, et al. Nanoparticle Toxicology. Annu Rev Pharmacol Toxicol. 2021;61(1):269–289. doi:10.1146/annurev-pharmtox-032320-110338
103. Liu Z, McClements DJ, Shi A, et al. Janus particles: a review of their applications in food and medicine. Crit Rev Food Sci Nutr. 2023;63(29):10093–10104. doi:10.1080/10408398.2022.2067831
104. Hwang D, Ramsey JD, Kabanov AV. Polymeric micelles for the delivery of poorly soluble drugs: from nanoformulation to clinical approval. Adv Drug Deliv Rev. 2020;156:80–118. doi:10.1016/j.addr.2020.09.009
105. Teixeira MC, Carbone C, Souto EB. Beyond liposomes: recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog Lipid Res. 2017;68:1–11. doi:10.1016/j.plipres.2017.07.001
106. Large DE, Abdelmessih RG, Fink EA, et al. Liposome composition in drug delivery design, synthesis, characterization, and clinical application. Adv Drug Deliv Rev. 2021;176:113851. doi:10.1016/j.addr.2021.113851
107. Nsairat H, Khater D, Sayed U, et al. Liposomes: structure, composition, types, and clinical applications. Heliyon. 2022;8(5):e09394. doi:10.1016/j.heliyon.2022.e09394
108. Baeza A, Ruiz-Molina D, Vallet-Regí M. Recent advances in porous nanoparticles for drug delivery in antitumoral applications: inorganic nanoparticles and nanoscale metal-organic frameworks. Expert Opin Drug Deliv. 2017;14(6):783–796. doi:10.1080/17425247.2016.1229298
109. Mitchell MJ, Billingsley MM, Haley RM, et al. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
110. Gunawan C, Lim M, Marquis CP, et al. Nanoparticle-protein Corona complexes govern the biological fates and functions of nanoparticles. J Mater Chem B. 2014;2(15):2060–2083. doi:10.1039/c3tb21526a
111. Seidi F, Jenjob R, Phakkeeree T, et al. Saccharides, oligosaccharides, and polysaccharides nanoparticles for biomedical applications. J Control Release. 2018;284:188–212. doi:10.1016/j.jconrel.2018.06.026
112. Suk JS, Xu Q, Kim N, et al. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(Pt A):28–51. doi:10.1016/j.addr.2015.09.012
113. Fang RH, Gao W, Zhang L. Targeting drugs to tumours using cell membrane-coated nanoparticles. Nat Rev Clin Oncol. 2023;20(1):33–48. doi:10.1038/s41571-022-00699-x
114. Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. J Control Release. 2015;220(Pt B):600–607. doi:10.1016/j.jconrel.2015.07.019
115. Oroojalian F, Beygi M, Baradaran B, et al. Immune Cell Membrane-Coated Biomimetic Nanoparticles for Targeted Cancer Therapy. Small. 2021;17(12):e2006484. doi:10.1002/smll.202006484
116. Ding H, Tan P, Fu S, et al. Preparation and application of pH-responsive drug delivery systems. J Control Release. 2022;348:206–238. doi:10.1016/j.jconrel.2022.05.056
117. Criado-Gonzalez M, Mecerreyes D. Thioether-based ROS responsive polymers for biomedical applications. J Mater Chem B. 2022;10(37):7206–7221. doi:10.1039/D2TB00615D
118. Tu Y, Xiao X, Dong Y, et al. Cinnamaldehyde-based poly(thioacetal): a ROS-awakened self-amplifying degradable polymer for enhanced cancer immunotherapy. Biomaterials. 2022;289:121795. doi:10.1016/j.biomaterials.2022.121795
119. Guo R, Wang S, Zhao L, et al. Engineered nanomaterials for synergistic photo-immunotherapy. Biomaterials. 2022;282:121425. doi:10.1016/j.biomaterials.2022.121425
120. Paunovska K, Loughrey D, Dahlman JE. Drug delivery systems for RNA therapeutics. Nat Rev Genet. 2022;23(5):265–280. doi:10.1038/s41576-021-00439-4
121. Mei KC, Liao Y-P, Jiang J, et al. Liposomal Delivery of Mitoxantrone and a Cholesteryl Indoximod Prodrug Provides Effective Chemo-immunotherapy in Multiple Solid Tumors. ACS Nano. 2020;14(10):13343–13366. doi:10.1021/acsnano.0c05194
122. Shen F, Feng L, Zhu Y, et al. Oxaliplatin-/NLG919 prodrugs-constructed liposomes for effective chemo-immunotherapy of colorectal cancer. Biomaterials. 2020;255:120190. doi:10.1016/j.biomaterials.2020.120190
123. Zhai Q, Chen Y, Xu J, et al. Lymphoma Immunochemotherapy: targeted Delivery of Doxorubicin via a Dual Functional Nanocarrier. Mol Pharm. 2017;14(11):3888–3895. doi:10.1021/acs.molpharmaceut.7b00606
124. Sun J, Wan Z, Chen Y, et al. Triple drugs co-delivered by a small gemcitabine-based carrier for pancreatic cancer immunochemotherapy. Acta Biomater. 2020;106:289–300. doi:10.1016/j.actbio.2020.01.039
125. Yang Z, Gao D, Guo X, et al. Fighting Immune Cold and Reprogramming Immunosuppressive Tumor Microenvironment with Red Blood Cell Membrane-Camouflaged Nanobullets. ACS Nano. 2020;14(12):17442–17457. doi:10.1021/acsnano.0c07721
126. Feng B, Hou B, Xu Z, et al. Self-Amplified Drug Delivery with Light-Inducible Nanocargoes to Enhance Cancer Immunotherapy. Adv Mater. 2019;31(40):e1902960. doi:10.1002/adma.201902960
127. Zang X, Song J, Yi X, et al. Polymeric indoximod based prodrug nanoparticles with doxorubicin entrapment for inducing immunogenic cell death and improving the immunotherapy of breast cancer. J Mater Chem B. 2022;10(12):2019–2027. doi:10.1039/D2TB00197G
128. Feng B, Zhou F, Hou B, et al. Binary Cooperative Prodrug Nanoparticles Improve Immunotherapy by Synergistically Modulating Immune Tumor Microenvironment. Adv Mater. 2018;30(38):e1803001. doi:10.1002/adma.201803001
129. Zhang Y, Du X, Liu S, et al. NIR-triggerable ROS-responsive cluster-bomb-like nanoplatform for enhanced tumor penetration, phototherapy efficiency and antitumor immunity. Biomaterials. 2021;278:121135. doi:10.1016/j.biomaterials.2021.121135
130. Peng J, Xiao Y, Li W, et al. Photosensitizer Micelles Together with IDO Inhibitor Enhance Cancer Photothermal Therapy and Immunotherapy. Adv Sci (Weinh). 2018;5(5):1700891. doi:10.1002/advs.201700891
131. Han X, Cheng K, Xu Y, et al. Modularly Designed Peptide Nanoprodrug Augments Antitumor Immunity of PD-L1 Checkpoint Blockade by Targeting Indoleamine 2,3-Dioxygenase. J Am Chem Soc. 2020;142(5):2490–2496. doi:10.1021/jacs.9b12232
132. Lu J, Liu X, Liao Y-P, et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat Commun. 2017;8(1):1811. doi:10.1038/s41467-017-01651-9
133. Li Q, Liu J, Fan H, et al. IDO-inhibitor potentiated immunogenic chemotherapy abolishes primary tumor growth and eradicates metastatic lesions by targeting distinct compartments within tumor microenvironment. Biomaterials. 2021;269:120388. doi:10.1016/j.biomaterials.2020.120388
134. Fang RH, Kroll AV, Gao W, et al. Cell Membrane Coating Nanotechnology. Adv Mater. 2018;30(23):e1706759. doi:10.1002/adma.201706759
135. Fan Q, Zuo J, Tian H, et al. Nanoengineering a metal-organic framework for osteosarcoma chemo-immunotherapy by modulating indoleamine-2,3-dioxygenase and myeloid-derived suppressor cells. J Exp Clin Cancer Res. 2022;41(1):162. doi:10.1186/s13046-022-02372-8
136. Hu Z, Zheng B, Xu J, et al. An albumin-bound drug conjugate of paclitaxel and indoleamine-2,3-dioxygenase inhibitor for enhanced cancer chemo-immunotherapy. Nanotechnology. 2020;31(29):295101. doi:10.1088/1361-6528/ab824d
137. Hu Y, Chen X, Xu Y, et al. Hierarchical assembly of hyaluronan coated albumin nanoparticles for pancreatic cancer chemoimmunotherapy. Nanoscale. 2019;11(35):16476–16487. doi:10.1039/C9NR03684A
138. Zuo T, Fang T, Zhang J, et al. pH-Sensitive Molecular-Switch-Containing Polymer Nanoparticle for Breast Cancer Therapy with Ferritinophagy-Cascade Ferroptosis and Tumor Immune Activation. Adv Healthc Mater. 2021;10(21):e2100683. doi:10.1002/adhm.202100683
139. Xie F, Liu Z, Wang P, et al. Self-Delivering Nanodrugs Developed via Small-Molecule-Directed Assembly and Macrophage Cloaking for Sonodynamic-Augmented Immunotherapy. Adv Healthc Mater. 2022;11(16):e2102770. doi:10.1002/adhm.202102770
140. Tenchov R, Bird R, Curtze AE, et al. Lipid Nanoparticles─From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano. 2021;15(11):16982–17015. doi:10.1021/acsnano.1c04996
141. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi:10.1038/nrd1632
142. Shah S, Dhawan V, Holm R, et al. Liposomes: advancements and innovation in the manufacturing process. Adv Drug Deliv Rev. 2020;154-155:102–122. doi:10.1016/j.addr.2020.07.002
143. Haider M, Zaki KZ, El Hamshary MR, et al. Polymeric nanocarriers: a promising tool for early diagnosis and efficient treatment of colorectal cancer. J Adv Res. 2022;39:237–255. doi:10.1016/j.jare.2021.11.008
144. Ghosh B, Biswas S. Polymeric micelles in cancer therapy: state of the art. J Control Release. 2021;332:127–147. doi:10.1016/j.jconrel.2021.02.016
145. Farah S, Anderson DG, Langer R. Physical and mechanical properties of PLA, and their functions in widespread applications - A comprehensive review. Adv Drug Deliv Rev. 2016;107:367–392. doi:10.1016/j.addr.2016.06.012
146. Plucinski A, Lyu Z, Schmidt B. Polysaccharide nanoparticles: from fabrication to applications. J Mater Chem B. 2021;9(35):7030–7062. doi:10.1039/D1TB00628B
147. Bose R, Jayawant M, Raut R, et al. Cyclodextrin nanoparticles in targeted cancer theranostics. Front Pharmacol. 2023;14:1218867. doi:10.3389/fphar.2023.1218867
148. Pridgen EM, Alexis F, Farokhzad OC. Polymeric nanoparticle technologies for oral drug delivery. Clin Gastroenterol Hepatol. 2014;12(10):1605–1610. doi:10.1016/j.cgh.2014.06.018
149. Yokoyama M. Polymeric micelles as a new drug carrier system and their required considerations for clinical trials. Expert Opin Drug Deliv. 2010;7(2):145–158. doi:10.1517/17425240903436479
150. Khan MA, Singh D, Ahmad A, et al. Revisiting inorganic nanoparticles as promising therapeutic agents: a paradigm shift in oncological theranostics. Eur J Pharm Sci. 2021;164:105892. doi:10.1016/j.ejps.2021.105892
151. Sun M, Wang T, Li L, et al. The Application of Inorganic Nanoparticles in Molecular Targeted Cancer Therapy: EGFR Targeting. Front Pharmacol. 2021;12:702445. doi:10.3389/fphar.2021.702445
152. Ryoo R. Birth of a class of nanomaterial. Nature. 2019;575(7781):40–41. doi:10.1038/d41586-019-02835-7
153. Kankala RK, Han Y-H, Na J, et al. Nanoarchitectured Structure and Surface Biofunctionality of Mesoporous Silica Nanoparticles. Adv Mater. 2020;32(23):e1907035. doi:10.1002/adma.201907035
154. Wu M-X, Yang Y-W. Metal–Organic Framework (MOF)-Based Drug/Cargo Delivery and Cancer Therapy. Adv Mater. 2017;29(23). doi:10.1002/adma.201606134
155. Yang J, Dai D, Zhang X, et al. Multifunctional metal-organic framework (MOF)-based nanoplatforms for cancer therapy: from single to combination therapy. Theranostics. 2023;13(1):295–323. doi:10.7150/thno.80687
156. Cai W, Wang J, Chu C, et al. Metal-Organic Framework-Based Stimuli-Responsive Systems for Drug Delivery. Adv Sci. 2019;6(1):1801526. doi:10.1002/advs.201801526
157. Liu J, Huang J, Zhang L, et al. Multifunctional metal-organic framework heterostructures for enhanced cancer therapy. Chem Soc Rev. 2021;50(2):1188–1218. doi:10.1039/d0cs00178c
158. Kianfar E. Protein nanoparticles in drug delivery: animal protein, plant proteins and protein cages, albumin nanoparticles. J Nanobiotechnology. 2021;19(1):159. doi:10.1186/s12951-021-00896-3
159. Zhang N, Mei K, Guan P, et al. Protein-Based Artificial Nanosystems in Cancer Therapy. Small. 2020;16(23):e1907256. doi:10.1002/smll.201907256
160. Xie F, Wang M, Chen Q, et al. Endogenous stimuli-responsive nanoparticles for cancer therapy: from bench to bedside. Pharmacol Res. 2022;186:106522. doi:10.1016/j.phrs.2022.106522
161. Tian H, Zhang T, Qin S, et al. Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies. J Hematol Oncol. 2022;15(1):132. doi:10.1186/s13045-022-01320-5
162. Li M, Sun J, Zhang W, et al. Drug delivery systems based on CD44-targeted glycosaminoglycans for cancer therapy. Carbohydr Polym. 2021;251:117103. doi:10.1016/j.carbpol.2020.117103
163. Luo Z, Dai Y, Gao H. Development and application of hyaluronic acid in tumor targeting drug delivery. Acta Pharm Sin B. 2019;9(6):1099–1112. doi:10.1016/j.apsb.2019.06.004
164. Kim H, Jeong H, Han S, et al. Hyaluronate and its derivatives for customized biomedical applications. Biomaterials. 2017;123:155–171. doi:10.1016/j.biomaterials.2017.01.029
165. Chen C, Zhao S, Karnad A, et al. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018;11(1):64. doi:10.1186/s13045-018-0605-5
166. Brown RL, Reinke LM, Damerow MS, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121(3):1064–1074. doi:10.1172/JCI44540
167. Yae T, Tsuchihashi K, Ishimoto T, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3(1):883. doi:10.1038/ncomms1892
168. Lau WM, Teng E, Chong HS, et al. CD44v8-10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res. 2014;74(9):2630–2641. doi:10.1158/0008-5472.CAN-13-2309
169. Todaro M, Gaggianesi M, Catalano V, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14(3):342–356. doi:10.1016/j.stem.2014.01.009
170. Weng X, Maxwell-Warburton S, Hasib A, et al. The membrane receptor CD44: novel insights into metabolism. Trends Endocrinol Metab. 2022;33(5):318–332. doi:10.1016/j.tem.2022.02.002
171. Liu J, Huang Y, Kumar A, et al. pH-sensitive nano-systems for drug delivery in cancer therapy. Biotechnol Adv. 2014;32(4):693–710. doi:10.1016/j.biotechadv.2013.11.009
172. Zhang YM, Liu YH, Liu Y. Cyclodextrin-Based Multistimuli-Responsive Supramolecular Assemblies and Their Biological Functions. Adv Mater. 2020;32(3):e1806158. doi:10.1002/adma.201806158
173. Glass SB, Gonzalez-Fajardo L, Beringhs AO, et al. Redox Potential and ROS-Mediated Nanomedicines for Improving Cancer Therapy. Antioxid Redox Signal. 2019;30(5):747–761. doi:10.1089/ars.2017.7370
174. Li Y, Yang J, Sun X. Reactive Oxygen Species-Based Nanomaterials for Cancer Therapy. Front Chem. 2021;9:650587. doi:10.3389/fchem.2021.650587
175. Zhou W, Jia Y, Liu Y, et al. Tumor Microenvironment-Based Stimuli-Responsive Nanoparticles for Controlled Release of Drugs in Cancer Therapy. Pharmaceutics. 2022;14(11):2346. doi:10.3390/pharmaceutics14112346
176. Li X, Lovell JF, Yoon J, et al. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat Rev Clin Oncol. 2020;17(11):657–674. doi:10.1038/s41571-020-0410-2
177. Zhang J, Lin Y, Lin Z, et al. Stimuli-Responsive Nanoparticles for Controlled Drug Delivery in Synergistic Cancer Immunotherapy. Adv Sci (Weinh). 2022;9(5):e2103444. doi:10.1002/advs.202103444
178. Arina A, Gutiontov SI, Weichselbaum RR. Radiotherapy and Immunotherapy for Cancer: from ”Systemic” to ”Multisite”. Clin Cancer Res. 2020;26(12):2777–2782. doi:10.1158/1078-0432.CCR-19-2034
179. Durante M, Reppingen N, Held KD. Immunologically augmented cancer treatment using modern radiotherapy. Trends Mol Med. 2013;19(9):565–582. doi:10.1016/j.molmed.2013.05.007
180. Wang C, Dong Z, Hao Y, et al. Coordination Polymer-Coated CaCO 3 Reinforces Radiotherapy by Reprogramming the Immunosuppressive Metabolic Microenvironment. Adv Mater. 2022;34(3):e2106520. doi:10.1002/adma.202106520
181. Thomas A, Pommier Y. Small cell lung cancer: time to revisit DNA-damaging chemotherapy. Sci Transl Med. 2016;8(346):346fs12. doi:10.1126/scitranslmed.aaf6282
182. Overchuk M, Weersink RA, Wilson BC, et al. Photodynamic and Photothermal Therapies: synergy Opportunities for Nanomedicine. ACS Nano. 2023;17(9):7979–8003. doi:10.1021/acsnano.3c00891
183. Liang S, Deng X, Ma P, et al. Recent Advances in Nanomaterial-Assisted Combinational Sonodynamic Cancer Therapy. Adv Mater. 2020;32(47):e2003214. doi:10.1002/adma.202003214
184. Pan X, Wang H, Wang S, et al. Sonodynamic therapy (SDT): a novel strategy for cancer nanotheranostics. Sci China Life Sci. 2018;61(4):415–426. doi:10.1007/s11427-017-9262-x
185. Yang Y, Huang J, Liu M, et al. Emerging Sonodynamic Therapy-Based Nanomedicines for Cancer Immunotherapy. Adv Sci. 2023;10(2):e2204365. doi:10.1002/advs.202204365
186. Liang S, Yao J, Liu D, et al. Harnessing Nanomaterials for Cancer Sonodynamic Immunotherapy. Adv Mater. 2023;35(33):e2211130. doi:10.1002/adma.202211130
187. Liang J, Qiao X, Qiu L, et al. Engineering Versatile Nanomedicines for Ultrasonic Tumor Immunotherapy. Adv Sci. 2024;11(3):e2305392. doi:10.1002/advs.202305392
188. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet. 2021;398(10304):1002–1014. doi:10.1016/S0140-6736(21)01206-X
189. Yi M, Zheng X, Niu M, et al. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21(1):28. doi:10.1186/s12943-021-01489-2
190. Doroshow DB, Bhalla S, Beasley MB, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345–362. doi:10.1038/s41571-021-00473-5
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.